

Abstract
Dengue virus has emerged as a global health threat to over one-third of humankind. As a positive-strand RNA virus, dengue virus relies on the host cell metabolism for its translation, replication, and egress. Therefore, a better understanding of the host cell metabolic pathways required for dengue virus infection offers the opportunity to develop new approaches for therapeutic intervention. In a recently described screen of known drugs and bioactive molecules, we observed that methotrexate and floxuridine inhibited dengue virus infections at low micromolar concentrations. Here, we demonstrate that all serotypes of dengue virus, as well as West Nile virus, are highly sensitive to both methotrexate and floxuridine, whereas other RNA viruses (Sindbis virus and vesicular stomatitis virus) are not. Interestingly, flavivirus replication was restored by folinic acid, a thymidine precursor, in the presence of methotrexate and by thymidine in the presence of floxuridine, suggesting an unexpected role for thymidine in flavivirus replication. Since thymidine is not incorporated into RNA genomes, it is likely that increased thymidine production is indirectly involved in flavivirus replication. A possible mechanism is suggested by the finding that p53 inhibition restored dengue virus replication in the presence of floxuridine, consistent with thymidine-less stress triggering p53-mediated antiflavivirus effects in infected cells. Our data reveal thymidine synthesis pathways as new and unexpected therapeutic targets for antiflaviviral drug development.
INTRODUCTION
Dengue virus (DENV) poses a significant human health risk for approximately 40% of the world's population (http://www.who.int/mediacentre/factsheets/fs117/en/). DENV occurs as four serotypes, with increasing and widespread circulation of all serotypes considered a contributing factor for the rising incidence of dengue disease. A particularly severe complication is dengue hemorrhagic fever (DHF), which results in approximately 22,000 deaths annually and of which the incidence is currently on the rise (http://www.who.int/csr/disease/dengue/impact/en/index.html). However, even nonfatal, self-limiting DENV infection can result in a severe, painful disease that is also known as “break-bone fever.” No vaccine or antiviral therapeutics are licensed to address DENV infections, and treatment is currently limited to supportive care.
Many studies are ongoing with the hopes of identifying effective novel anti-DENV therapeutics and/or potential therapeutic targets. Several viral factors have been explored as therapeutic targets, including the viral protease, helicase, and RNA-dependent RNA polymerase (reviewed in reference 1). As an alternative to directly targeting viral proteins, it is conceivable to target host cell metabolic pathways that are required for viral replication. Flaviviruses, small RNA viruses that include DENV as well as West Nile virus (WNV), yellow fever virus, and Japanese encephalitis virus, are highly dependent on the host cell to provide support for viral entry, replication, assembly, and egress. Examples of such support include host cholesterol biosynthesis, required for flavivirus entry, and the host kinases c-Src and c-Yes, required for DENV and WNV maturation and egress, respectively (2–6). Three DENV-specific therapeutics are currently in clinical trials (clinicaltrials.gov): celgosivir, which interferes with the folding of DENV NS1 protein and triggers the host unfolded protein response (7), and chloroquine and anti-Rh0-D antibodies, which block viral entry and increase platelet counts, respectively, during DHF (8, 9). Of note, all three therapeutics affect host cell processes and are not directed solely against viral factors.
To identify additional host cell pathways that are required for DENV replication, we previously described a high-content cell-based screen of a library of bioactive small molecules, including established drugs (10). Here, we further characterize two of the most effective small-molecule inhibitors of DENV identified in that screen, the antimetabolites methotrexate (MTX) and floxuridine (FUDR). We demonstrate that these compounds inhibit the replication of multiple flaviviruses in multiple cell types. Our results reveal an unanticipated sensitivity of flaviviruses to the inhibition of thymidine synthesis, suggesting that these cellular pathways contain viable targets for the development of antivirals that interfere with host-virus interactions.
MATERIALS AND METHODS
Cells and viruses.
Low-passage-number HEK293 cells were obtained from Microbix, Vero and HeLa cells were obtained from the ATCC, Huh7 cells were obtained from F. Chisari (Scripps Research Institute), and the BHK-DENV replicon was obtained from M. Diamond (Washington University, St. Louis, MO). HEK293 cells were cultured in minimal essential medium Eagle (Cellgro) supplemented with nonessential amino acids, l-glutamine, 10% heat-inactivated fetal bovine serum (FBS), and antibiotics (100 units/ml penicillin and 100 g/ml streptomycin). All other cell types described were cultured in Dulbecco's modification of Eagle's medium (Cellgro) supplemented with 10% heat-inactivated FBS and antibiotics (100 units/ml penicillin and 100 g/ml streptomycin).
DENV2 (strain New Guinea C), DENV3 (strain H87), DENV4 (strain H241), and Sindbis virus (strain AR-339) were obtained from the ATCC. DENV1 (strain West-Pac) was obtained from R. Putnak (Walter Reed Army Institute of Research). DENV2 (strain S221) was obtained from S. Shresta (La Jolla Institute). All DENV serotypes and strains as well as WNV (strain 385-99) (11) were grown, and titers were determined, as previously described (10). Herpes simplex virus 1 (HSV-1; strain F1) was obtained from A. Hill (Oregon Health and Science University). Vaccinia virus (VACV; strain Western Reserve) was obtained from M. Slifka (Oregon Health and Science University). Vesicular stomatitis virus (VSV; strain Indiana) was obtained from J. Hiscott (VGTI Florida) (12).
Immunofluorescence assay (IFA).
HEK293 cells were infected with DENV2 at a multiplicity of infection (MOI) of 0.5 PFU/cell in cell medium containing 10 μM MTX, 10 μM FUDR, or 1% dimethyl sulfoxide (DMSO) (vol/vol). Cells were fixed 48 h postinfection (p.i.) and stained with a monoclonal antibody specific for DENV/WNV envelope protein (clone 4G2; ATCC), an Alexa Fluor 488-conjugated goat anti-mouse secondary antibody (Santa Cruz), and 4′,6-diamidino-2-phenylindole (DAPI).
Dose-response curves.
HEK293 cells were infected with DENV2 at an MOI of 3 PFU/cell in cell medium containing increasing concentrations of MTX or FUDR (in a constant 1% DMSO). Supernatants were collected at 48 h p.i., and titers were determined on Vero cells. Fifty percent inhibitory concentration (IC50) values were calculated using Prism software (GraphPad).
Cell viability.
HEK293 cells were incubated in cell medium containing increasing concentrations of MTX or FUDR in a constant 1% (vol/vol) DMSO. After 48 h, Cell Titer Glo reagent (Promega) was added and luminescence was measured per the manufacturer's instructions. Fifty percent cytotoxic concentration (CC50) values were calculated using Prism software. In some experiments (where indicated), Cell Titer Fluor reagent (Promega) was substituted for Cell Titer Glo to measure cell viability.
Huh7, Vero, and HeLa infections.
Cells were infected with DENV2 at an MOI of 3 PFU/cell in cell medium containing 10 μM MTX, 10 μM FUDR, or 1% (vol/vol) DMSO as a control. Supernatants were collected at 48 h postinfection, and titers were determined on Vero cells.
Flavivirus multistep growth curves.
HEK293 cells were infected with DENV or WNV at an MOI of 0.1 PFU/cell in cell medium containing 1 μM MTX, 1 μM FUDR, or 1% (vol/vol) DMSO as a control. Supernatants were collected at indicated times postinfection, and titers were determined on Vero cells.
HSV-1, vaccinia virus, Sindbis virus, and VSV infections.
HEK293 cells were infected with each virus at an MOI of 3 PFU/cell in cell medium containing increasing concentrations of MTX or FUDR in a constant 1% (vol/vol) DMSO. Supernatants were collected at 24 h (Sindbis virus and VSV) or 48 h (HSV-1 and VACV) postinfection and assayed for infectious virus by a standard plaque assay on Vero cells.
DENV replicon experiments.
BHK cells harboring a DENV replicon expressing a Renilla luciferase reporter were treated with increasing concentrations of ribavirin, MTX, or FUDR (or DMSO as a negative control). At 48 h posttreatment, cells were lysed and Renilla luciferase activity was measured using the dual-luciferase reporter assay (Promega).
Folinic acid treatment.
HEK293 cells were infected with DENV2 at an MOI of 3 PFU/cell in cell medium containing 1 μM MTX and increasing concentrations of folinic acid (leucovorin). Supernatants were collected, and infectious virus was quantified as described above.
Thymidine/uridine treatment.
HEK293 cells were infected with DENV2 at an MOI of 3 PFU/cell in the presence of 1 μM FUDR and increasing concentrations of uridine or thymidine. Supernatants were collected at 48 h p.i., and titers were determined on Vero cells.
Pifithrin-α treatment.
HEK293 cells were infected with DENV2 at an MOI of 3 PFU/cell in the presence of 1 μM FUDR and/or 10 μM pifithrin-α. Supernatants were collected at 48 h p.i., and titers were determined on Vero cells.
Mouse studies.
All mouse studies were approved by the OHSU West Campus Institutional Animal Use and Care Committee. The AG129 mouse model of DENV infection has been previously characterized (13). AG129 mice were obtained from M. Slifka (Oregon Health and Science University) and bred at the Vaccine and Gene Therapy Institute. Subcutaneous osmotic pumps (Alzet) were implanted into the mice to deliver MTX, FUDR, or 50% (vol/vol) DMSO vehicle alone at a constant rate. Mice were infected intraperitoneally (i.p.) with the S221 strain of DENV2. C57BL/6 mice, purchased from Jackson Laboratories, were utilized as a mouse model of WNV infection. Mice were injected i.p. with MTX, FUDR, or 10% (vol/vol) DMSO vehicle alone and then infected subcutaneously (s.c.) with WNV at a 90% lethal dose (LD90) 24 h later. Mice received additional injections of drugs or the vehicle control 24 h p.i. and then every 48 h thereafter.
RESULTS
MTX and FUDR inhibit DENV replication.
In the high-throughput screen previously described (10), as well as in confirmatory experiments in a 96-well plate format, immunofluorescent detection of DENV envelope protein was used to assess virus replication. At a concentration of 10 μM, both MTX and FUDR strongly inhibited the accumulation of DENV envelope protein in infected cells at 48 h p.i. (Fig. 1A). Both MTX and FUDR inhibited DENV replication in a dose-dependent manner with IC50 values of 0.09 and 0.06 μM, respectively (Table 1). We further confirmed the inhibitory effects of MTX and FUDR by performing a dose-response experiment on HEK293 cells and quantifying virus released into the supernatant 48 h p.i. (Fig. 1B). Using this assay, viral release into the supernatant was reduced by approximately 3 logs at concentrations above 2 μM with an IC50 of 0.17 μM for MTX and 0.22 μM for FUDR (Table 1).
Fig 1.

Characterization of the anti-DENV efficacy of MTX and FUDR. (A) HEK293 cells were treated with DMSO only, 10 μM MTX, or 10 μM FUDR and infected with DENV2 at an MOI of 0.5 PFU/cell. Cells were analyzed 48 h p.i. by immunofluorescence for the presence of DENV-Env protein. (B) HEK293 cells were treated with increasing doses of MTX or FUDR as indicated and infected with DENV at an MOI of 3 PFU/cell. Supernatant was collected 48 h p.i., and the titers of infectious virions were determined. (C) HEK293 cells were treated with increasing doses of MTX or FUDR as indicated, and cell viability was assessed after 48 h using Cell Titer Glo reagent.
Table 1.
IC50 and CC50 values for MTX and FUDR in HEK293 cells
| Treatment | Concn (μM) |
||
|---|---|---|---|
| IC50 (IFA) | IC50 (plaque assay) | CC50 (Cell Titer Glo) | |
| MTX | 0.09 | 0.17 | 12.42 |
| FUDR | 0.06 | 0.22 | >100 |
At 10 μM, the concentration used in the primary screen, little reduction of cell nuclei was observed (Fig. 1A), suggesting that the anti-DENV effect was not due to cytotoxicity. In addition, effects of the compounds on cell viability were determined using Cell Titer Glo (Promega) (Fig. 1C). While the CC50 value of FUDR was more than 100 μM, the CC50 value for MTX was 12.42 μM. For both compounds, the IC50 values were thus at least 10-fold lower than the CC50, indicating a therapeutic index within a range appropriate for further consideration.
DENV inhibition is cell type independent.
Every immortalized cell line possesses its own physiological properties, with factors such as species origin, cell lineage, and immortalization method contributing to potential differences. To determine whether the anti-DENV2 properties of MTX and FUDR were confined to the HEK293 cells used in our screen, we tested the molecules in several different cell lines. As with HEK293 cells, both MTX and FUDR reduced the viral titer in supernatants of infected Huh7 (human hepatocarcinoma) and Vero (African green monkey kidney epithelial) cells at 48 h p.i. (Fig. 2A).
Fig 2.
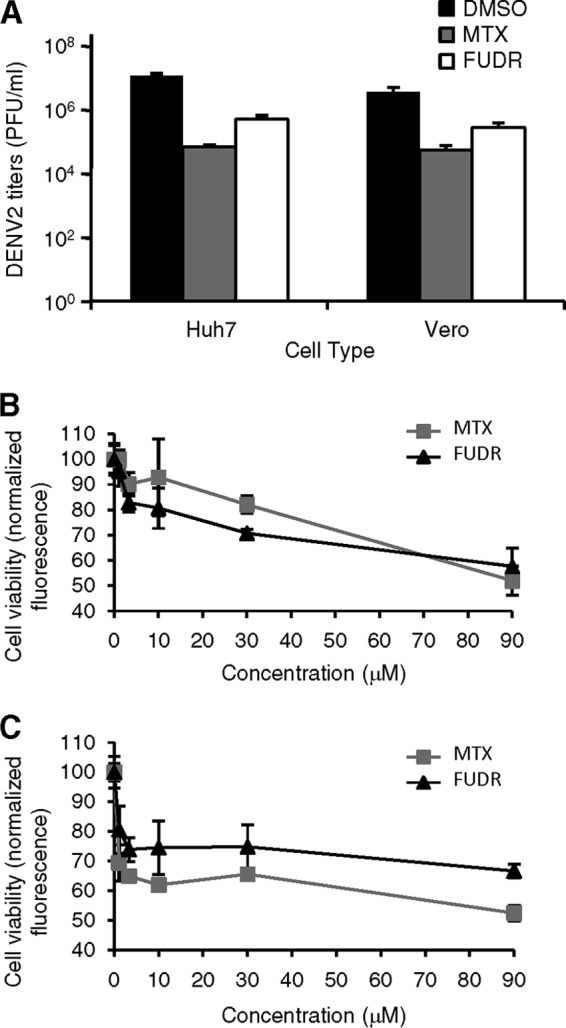
Anti-DENV efficacy is not HEK293 specific. (A) Huh7 or Vero cells were treated with DMSO only, 10 μM MTX, or 10 μM FUDR and infected with DENV2 at an MOI of 3 PFU/cell. Supernatants were collected 48 h p.i., and titers of infectious virions were determined. (B and C) Huh7 (B) or Vero (C) cells were treated with increasing doses of MTX or FUDR as indicated, and cell viability was assessed after 48 h using the Cell Titer Fluor reagent.
The CC50 values of MTX and FUDR were calculated for Huh7 and Vero cells using the Cell Titer Fluor reagent (Fig. 2B and C; Table 2). As with HEK293 cells, the IC50 values were approximately 10-fold or more lower than the CC50 (Table 2).
Table 2.
IC50 and CC50 values for MTX and FUDR in Huh7 and Vero cells
| Cell line | Treatment | Concn (μM) |
|
|---|---|---|---|
| IC50 (plaque assay) | CC50 (Cell Titer Fluor) | ||
| Huh7 | MTX | 1 | >90 |
| FUDR | 15 | >90 | |
| Vero | MTX | 0.2 | >90 |
| FUDR | 0.1 | >90 | |
MTX and FUDR are effective against multiple flaviviruses and DNA viruses but not against all RNA viruses.
Numerous flaviviruses, including strains from all four serotypes of DENV, are capable of causing severe human disease. To determine if MTX and/or FUDR would inhibit multiple members of the flavivirus family, we conducted a multistep growth curve analysis of the replication of DENV1, DENV2, DENV3, DENV4, and WNV in the presence of 1 μM (each) antimetabolite. With each virus tested, both MTX and FUDR reduced viral titers by 1 to 2 logs (Fig. 3). Consistent with this observation, it was previously reported that MTX inhibits the replication of another flavivirus, yellow fever virus, vaccine strain 17D (14).
Fig 3.

MTX and FUDR have activity against multiple flaviviruses. HEK293 cells were treated with DMSO vehicle, 1 μM MTX, or 1 μM FUDR and infected with representative strains of each DENV serotype as well as WNV at an MOI of 0.1 PFU/cell. Samples of the supernatant were taken at the indicated times p.i., and titers of infectious virions were determined.
To determine if the inhibition of viral replication by these compounds is flavivirus specific, we further examined whether MTX and FUDR inhibit the replication of HSV-1, VACV, Sindbis virus, or VSV using plaque assays. At 10 μM, both MTX and FUDR inhibited the DNA viruses tested, with the titer of HSV-1 reduced by several orders of magnitude and VACV by about 1 log (Fig. 4A and B). These findings agree with previous reports demonstrating that each drug inhibits DNA viruses (15–18). Of interest, neither drug affected the replication of either Sindbis virus or VSV, both RNA viruses (Fig. 4C and D).
Fig 4.
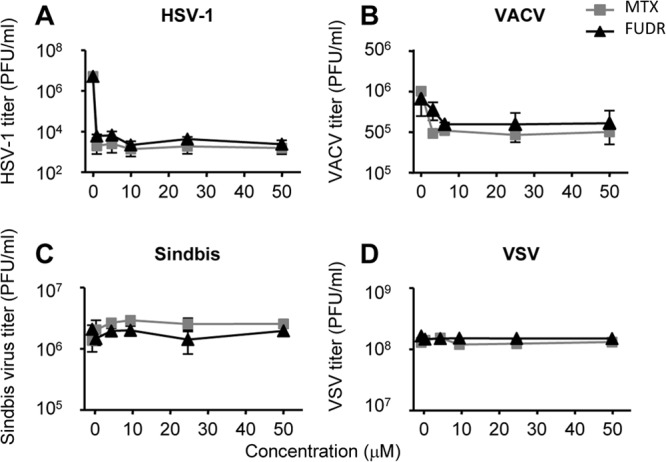
MTX and FUDR are not effective against all viruses. HEK293 cells were treated with increasing doses of MTX or FUDR as indicated and infected with HSV-1, VACV, Sindbis virus, or VSV at an MOI of 3 PFU/cell. Supernatants were collected at 24 h (Sindbis virus and VSV) or 48 h (HSV-1 and VACV), and titers of infectious virions were determined.
Mechanisms of DENV inhibition.
Both MTX and FUDR are widely used therapeutics for the treatment of certain cancers. As such, the antimetabolitic mechanisms causing interference with the life cycle of cancer cells have been characterized for both compounds (Fig. 5A and B) (19, 20). We hypothesized that as antimetabolites, both molecules likely inhibited flavivirus replication by interfering with nucleic acid synthesis and thus genomic replication. To test this hypothesis, we utilized BHK cells that stably express a replicon with the nonstructural proteins of DENV along with luciferase as a read-out (21). When these cells were treated with MTX, FUDR, or the positive-control ribavirin, luciferase levels were decreased (Fig. 5C), suggesting that both compounds affect genome replication and/or gene expression of DENV.
Fig 5.

Mechanisms of anti-DENV activity of MTX and FUDR. (A) MTX blocks the enzyme DHFR upstream of the synthesis of thymidine, purines, and methionine. (B) FUDR is metabolized into products that inhibit thymidylate synthase or are incorporated into RNA as a uracil analog. (C) BHK cells stably maintaining a DENV replicon expressing a luciferase reporter were treated with DMSO only or increasing doses of ribavirin, MTX, or FUDR. After 48 h, luciferase was quantified via luminescence. (D) HEK293 cells were treated with combinations of MTX (1 μM) and folinic acid (10 μg/ml) as indicated and infected with DENV2 at an MOI of 3 PFU/cell. Supernatants were collected 48 h p.i., and titers of infectious virions were determined. (E) HEK293 cells were treated with FUDR and increasing concentrations of uridine as indicated and infected with DENV2 at an MOI of 3 PFU/cell. Supernatants were collected 48 h p.i., and titers of infectious virions were determined. (F) HEK293 cells were treated with combinations of FUDR (1 μM) and thymidine (10 μg/ml) as indicated and infected with DENV2 at an MOI of 3 PFU/cell. Supernatants were collected 48 h p.i., and titers of infectious virions were determined. (G) HEK293 cells were treated with DMSO only, FUDR, or 5FU and infected with DENV2 at an MOI of 3 PFU/cell. Supernatants were collected 48 h p.i., and titers of infectious virions were determined. (H) HEK293 cells were treated with increasing doses of 5FU as indicated and infected with DENV2 at an MOI of 3 PFU/cell. Supernatants were collected 48 h p.i., and titers of infectious virions were determined.
MTX is an inhibitor of the enzyme dihydrofolate reductase (DHFR) (Fig. 5A) (20). To determine if the reduction of DENV2 replication is dependent on DHFR inhibition, we treated HEK293 cells with MTX in the presence or absence of folinic acid (leucovorin). Folinic acid can readily be converted to tetrahydrofolate independently of DHFR, bypassing the effects of blocking the enzyme (18). Indeed, DENV2 levels in the presence of both MTX and folinic acid were comparable to those levels in the absence of MTX (Fig. 5D), demonstrating the involvement of DHFR in the anti-DENV2 activity of MTX.
FUDR functions against cancer using two mechanisms, by acting directly as a uracil analog and by inhibiting thymidylate synthase (Fig. 5B) (19, 22). If the antiviral activity of FUDR was due to it functioning as a nucleoside analog, we reasoned that the addition of exogenous uridine would be able to compete with FUDR, thereby reducing its efficacy. However, the addition of various doses of uridine was unable to reduce the effects of FUDR on DENV2 titers (Fig. 5E). We then tested the ability of thymidine added exogenously, thus bypassing the requirement for thymidylate synthase (23), to counteract the inhibitory effects of FUDR on DENV2 replication. Indeed, DENV2 replication in the presence of both FUDR and thymidine was comparable to that observed in the absence of FUDR (Fig. 5F). These data suggest that FUDR interferes with DENV2 replication through the inhibition of thymidylate synthase.
5-Fluorouracil (5FU), an intermediate of FUDR along the pathway toward incorporation into RNA (Fig. 5B), is also commercially available and used as an antimetabolitic drug in the treatment of certain cancers. We tested the ability of 5FU to inhibit DENV2 replication in HEK293 cells at concentrations known to be effective for FUDR. At both 1 and 10 μM, 5FU did not inhibit DENV2 replication (Fig. 5G). Further, a dose-response curve demonstrated no significant effect on DENV2 replication levels at concentrations up to 100 μM 5FU (Fig. 5H). This result further supports our conclusion that FUDR does not inhibit flavivirus replication through interference with RNA metabolism.
The anti-DENV activity of FUDR is p53 dependent.
As an RNA virus, DENV2 does not directly incorporate deoxythymidine into its genome, thus suggesting that any antiviral effects of thymidylate synthase inhibition would be indirect. One possible mechanism for this antiviral effect is the p53 pathway, as thymidine deprivation results in the accumulation of p53 and the expression of p53-regulated genes (24–26). Activation of p53 has been shown as possessing antiviral properties against a broad range of viruses in vitro and in vivo. The antiviral activity of p53 has been attributed both to its inhibition of the cell cycle and contribution to virus-induced apoptosis and to its ability to induce antiviral, interferon (IFN)-stimulated genes (27–29). Interestingly, the accumulation of p53 following FUDR treatment could be prevented through the addition of exogenous thymidine (26), potentially explaining the FUDR-overriding effects that we observed using exogenous thymidine (Fig. 5F).
HEK293 cells, utilized in the initial high-throughput screen as well as the majority of subsequent experiments, contain the adenovirus E1B protein, capable of reducing but not totally eliminating p53 activity (30). In contrast, p53 is efficiently degraded in HeLa cells due to the human papillomavirus E6 protein (31) and cannot be detected (32), making this cell line essentially a “functional knockout” of p53. We therefore treated HeLa cells with FUDR and infected them with DENV2. Unlike the other diverse cell lines tested (Fig. 2), comparable levels of DENV were measured in the supernatant of cells infected in the presence and in the absence of FUDR (Fig. 6A).
Fig 6.
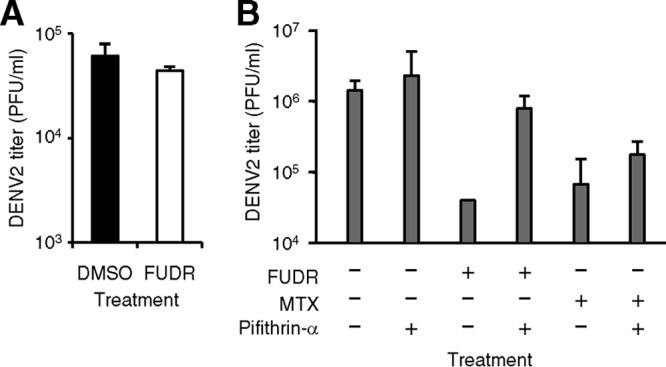
FUDR-mediated DENV inhibition is p53 dependent. (A) HeLa cells were treated with DMSO only or 10 μM FUDR and infected with DENV2 at an MOI of 3 PFU/cell. Supernatants were collected 48 h p.i., and titers of infectious virions were determined. (B) HEK293 cells were treated with combinations of FUDR (1 μM), MTX (1 μM), and pifithrin-α (10 μM) as indicated and infected with DENV2. Supernatants were collected 48 h p.i., and titers of infectious virions were determined.
The lack of FUDR efficacy in HeLa cells cannot definitively be attributed to p53, as there are a number of differences between this cell line and the others used in this study. To directly address the role of p53 in FUDR activity, we utilized pifithrin-α, a known chemical inhibitor of p53 (33). HEK293 cells were infected with DENV2 in the presence of FUDR and/or pifithrin-α. After 48 h, cells treated with FUDR alone demonstrated a reduced amount of DENV2 in the supernatant (as previously demonstrated) whereas cells treated with pifithrin-α and the same dose of FUDR did not (Fig. 6B). Therefore, functional p53 must be present for FUDR to reduce DENV2 replication. In contrast to the findings with FUDR, pifithrin-α did not restore DENV replication levels in the presence of MTX (Fig. 6B), suggesting that in addition to depletion of thymidine, MTX affects DENV replication by its known effects on purine or methionine synthesis.
MTX and FUDR do not prolong survival in murine models of flaviviral infection.
To test the efficacy of MTX and FUDR in vivo, we utilized the AG129 mouse model of DENV infection. AG129 mice were implanted with subcutaneous osmotic pumps that released a constant level of MTX, FUDR, or DMSO alone as a control. They were then infected with the S221 mouse-adapted strain of DENV2. In this experiment, all of the mice succumbed to infection rapidly with no enhancement of survival observed in the mice receiving MTX or FUDR (data not shown). In addition, uninfected mice that received MTX demonstrated dramatically enhanced weight loss (data not shown), a result of the toxicity of the high concentrations (20 mg/kg of body weight or greater) of MTX used in these experiments.
We next tested the efficacy of MTX and FUDR in the murine model of WNV infection. C57BL/6 mice were infected subcutaneously with WNV. They were additionally given i.p. injections of MTX, FUDR, or DMSO alone on the day prior to infection as well as days 1, 3, 5, and 7 postinfection. As with DENV, neither MTX nor FUDR significantly prolonged survival in these mice at the doses tested (Fig. 7).
Fig 7.
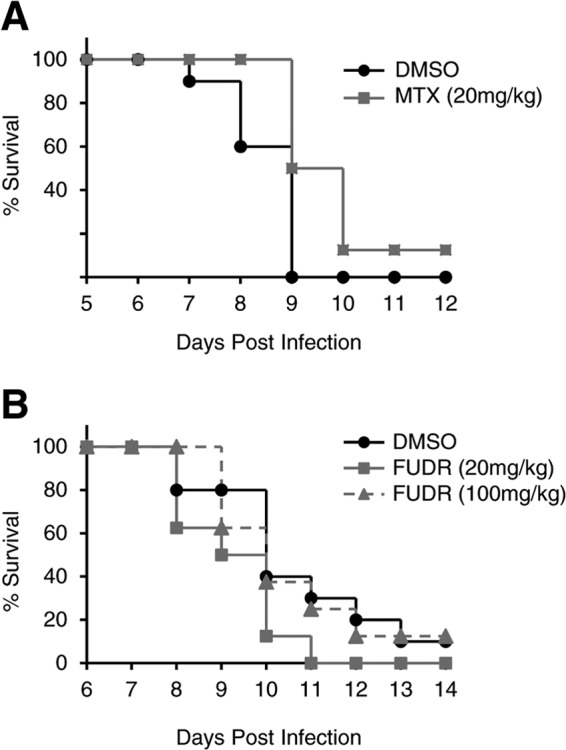
MTX and FUDR do not prolong survival of WNV-infected mice. C57BL/6 mice were infected s.c. with an LD90 of WNV and injected i.p. daily beginning on day −1 with 10% DMSO or 20 mg/kg MTX (A) or injected i.p. every other day beginning on day −1 with 10% DMSO, 20 mg/kg FUDR, or 100 mg/kg FUDR (B).
DISCUSSION
We report here the surprising finding that flaviviruses are highly susceptible to inhibitors of thymidine synthesis. FUDR is metabolized in the cell to a product that inhibits thymidine synthase, and the addition of exogenous thymidine was able to overcome the inhibition of DENV replication. MTX inhibits DHFR, upstream of thymidine synthesis. While exogenous thymidine alone did not rescue DENV replication in the presence of MTX, other critical molecules such as purines are also downstream of DHFR, suggesting that multiple critical metabolites are affected by DHFR inhibition. This conclusion is also supported by the inability of pifithrin-α to restore DENV replication in the presence of MTX. As RNA viruses, flaviviruses do not directly incorporate deoxythymidine into their genome. Nonetheless, the profound changes in the host cell initiated by thymidine-less stress have a profound effect on the ability of the flaviviruses, but not the other RNA viruses tested, to replicate their genome. This sensitivity suggests that components of the thymidine synthesis pathway could represent therapeutic targets.
The profound impact of thymidine depletion on DENV replication has not been observed previously and was unexpected given the viral RNA genome. Our results thus demonstrate the power of systematically evaluating known drugs against different viral agents by high-content screening of appropriate libraries (10). In contrast to screening small molecules having unexplored mechanisms of action, the molecular targets of approved drugs are generally known, thus implicating specific pathways in viral propagation. In this case, thymidine synthesis emerged as a significant process supporting DENV replication.
MTX and FUDR were unable to inhibit DENV or WNV replication in vivo. Other recent studies have documented in vitro inhibition of flavivirus replication by antimetabolites which were also unable to prolong survival in in vivo models of infection (34, 35). A few antimetabolites have successfully prolonged survival but also exhibited severe toxicity (36, 37). Similarly, we observed that MTX was toxic at high concentrations. A third antimetabolite, mercaptopurine, was identified in our initial high-throughput screen as possessing anti-DENV2 efficacy. While unrelated to thymidine synthesis, this molecule is related to another thiopurine that was recently identified as inhibitory of WNV replication in vitro (34). However, in follow-up evaluations, mercaptopurine demonstrated high toxicity to HEK293 cells (data not shown), and we therefore did not include it in further studies.
The studies here support the potential of antimetabolites as anti-DENV therapeutics but also illustrate the major challenges associated with drugs targeting host cell mechanisms. For instance, it is possible that in vivo, thymidine is less limiting than in tissue culture. It has been hypothesized for other antimetabolites that have failed to demonstrate in vivo efficacy that the animals' diet is sufficient to overcome any drug-induced deficiency (35). It is also conceivable that some nondividing cell types infected with DENV are less susceptible to the antimetabolites used than are the rapidly dividing cells used in tissue culture. Both MTX and FUDR have shown efficacy in vivo as anticancer drugs. As such, these drugs preferentially affect cells undergoing rapid replication. In addition to cancer cells, these drugs also affect rapidly dividing immune cells so that a beneficial effect of MTX and FUDR on infected cells may have been counteracted by a reduced immune response to DENV. In vivo, FUDR could conceivably be preferentially metabolized to the RNA incorporation pathway rather than the thymidylate synthase inhibition pathway (Fig. 5B), which our results have suggested is ineffective at inhibiting flaviviral replication (Fig. 5E, G, and H). The previous finding that FUDR was preferentially converted to 5FU in a murine tumor in vivo supports this idea, although in this study, FUDR inhibition of thymidylate synthase was also observed (22). It is possible that these limitations can be overcome by inhibiting several metabolic pathways simultaneously or by developing more-active and less-toxic inhibitors that more specifically eliminate or induce host cell pathways that modulate viral replication. Although MTX is widely used in humans, we observed significant toxicity at high concentrations in the form of dramatic weight loss in mice implanted with MTX-containing subcutaneous osmotic pumps. Thus, along with potential bioavailability issues, the adverse side effects of these drugs may outweigh desired antiviral effects.
Nonetheless, our results clearly uncover a previously unappreciated susceptibility of flaviviruses for the inhibition of thymidine synthesis. Interestingly, it has been recently demonstrated that excess thymidine, which leads to stalling of the cell cycle in S phase, strongly increased DENV production in insect cells due to acceleration of replication and assembly (38). In contrast, the same treatment did not affect DENV production in mammalian cells. Thus, it seems that excess thymidine does not affect DENV in mammalian cells whereas depletion of thymidine drastically inhibits DENV replication at a postentry step. Our data suggest that thymidine depletion likely does not affect viral replication by depletion of nucleotides but via an indirect pathway that might involve activation of p53. While p53 is mostly known for its role as a tumor suppressor and in the induction of apoptosis, an emerging body of literature supports a role of p53 in inducing an antiviral transcriptional program. Several studies have demonstrated p53-mediated induction of interferon (IFN)-stimulated genes that inhibit viral replication in vitro and in vivo (27–29). However, we did not observe direct innate immune gene stimulation by FUDR using reporter cell lines encoding luciferase driven by either the interferon-sensitive response element or the NF-κB transcriptional response element (data not shown), suggesting that IFN or NF-κB activation is not the primary mechanism by which FUDR acts. It is thus likely that p53 activation prevents DENV replication by a different mechanism that could involve modulation of cell cycle or proapoptotic genes. It is in this capacity that p53 likely influences the replication of flaviviruses, which are highly dependent on host cell proteins and processes for their own replication. Further studies are needed to dissect the role of p53 in the absence of thymidine in more detail.
DENV currently poses a risk for over 40% of the world's population. The increasing range of mosquito vectors has raised concerns that this estimate could drastically increase in the future. However, there is currently no therapeutic treatment available for DENV-related diseases. Here, we demonstrate that two antimetabolites well established in the treatment of cancers, MTX and FUDR, are able to inhibit DENV replication in vitro. While treatment of flavivirus-infected mice with either drug did not prolong survival, this study suggests that components of the thymidine synthesis pathway represent potential therapeutic targets.
ACKNOWLEDGMENTS
This work was supported by the NIAID Pacific Northwest Regional Center of Excellence for Biodefense and Emerging Infectious Diseases Research (grant U54 AI 081680) and National Center for Research Resources support for the Oregon National Primate Research Center (grant 8P51 OD011092-54). M.A.F. and J.L.S. were supported by OHSU training grants T32 AI074494 and T32 AI07472, respectively. The HTS Core Facility is partially supported by William H. Goodwin and Alice Goodwin and the Commonwealth Foundation for Cancer Research, the Experimental Therapeutics Center of the Memorial Sloan-Kettering Cancer Center, the William Randolph Hearst Fund in Experimental Therapeutics, and the Lillian S. Wells Foundation and by NIH/NCI Cancer Center support grant 5 P30 CA008748-44.
Footnotes
Published ahead of print 3 July 2013
REFERENCES
- 1.Noble CG, Chen YL, Dong H, Gu F, Lim SP, Schul W, Wang QY, Shi PY. 2010. Strategies for development of dengue virus inhibitors. Antiviral Res. 85:450–462 [DOI] [PubMed] [Google Scholar]
- 2.Chu JJ, Yang PL. 2007. c-Src protein kinase inhibitors block assembly and maturation of dengue virus. Proc. Natl. Acad. Sci. U. S. A. 104:3520–3525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hirsch AJ, Medigeshi GR, Meyers HL, DeFilippis V, Fruh K, Briese T, Lipkin WI, Nelson JA. 2005. The Src family kinase c-Yes is required for maturation of West Nile virus particles. J. Virol. 79:11943–11951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lee CJ, Lin HR, Liao CL, Lin YL. 2008. Cholesterol effectively blocks entry of flavivirus. J. Virol. 82:6470–6480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Medigeshi GR, Hirsch AJ, Streblow DN, Nikolich-Zugich J, Nelson JA. 2008. West Nile virus entry requires cholesterol-rich membrane microdomains and is independent of alphavbeta3 integrin. J. Virol. 82:5212–5219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rothwell C, Lebreton A, Young Ng C, Lim JY, Liu W, Vasudevan S, Labow M, Gu F, Gaither LA. 2009. Cholesterol biosynthesis modulation regulates dengue viral replication. Virology 389:8–19 [DOI] [PubMed] [Google Scholar]
- 7.Rathore AP, Paradkar PN, Watanabe S, Tan KH, Sung C, Connolly JE, Low J, Ooi EE, Vasudevan SG. 2011. Celgosivir treatment misfolds dengue virus NS1 protein, induces cellular pro-survival genes and protects against lethal challenge mouse model. Antiviral Res. 92:453–460 [DOI] [PubMed] [Google Scholar]
- 8.de Castro RA, de Castro JA, Barez MY, Frias MV, Dixit J, Genereux M. 2007. Thrombocytopenia associated with dengue hemorrhagic fever responds to intravenous administration of anti-D (Rh(0)-D) immune globulin. Am. J. Trop. Med. Hyg. 76:737–742 [PubMed] [Google Scholar]
- 9.Tricou V, Minh NN, Van TP, Lee SJ, Farrar J, Wills B, Tran HT, Simmons CP. 2010. A randomized controlled trial of chloroquine for the treatment of dengue in Vietnamese adults. PLoS Negl. Trop. Dis. 4:e785. 10.1371/journal.pntd.0000785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shum D, Smith JL, Hirsch AJ, Bhinder B, Radu C, Stein DA, Nelson JA, Fruh K, Djaballah H. 2010. High-content assay to identify inhibitors of dengue virus infection. Assay Drug Dev. Technol. 8:553–570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xiao SY, Guzman H, Zhang H, Travassos da Rosa AP, Tesh RB. 2001. West Nile virus infection in the golden hamster (Mesocricetus auratus): a model for West Nile encephalitis. Emerg. Infect. Dis. 7:714–721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sharif-Askari E, Nakhaei P, Oliere S, Tumilasci V, Hernandez E, Wilkinson P, Lin R, Bell J, Hiscott J. 2007. Bax-dependent mitochondrial membrane permeabilization enhances IRF3-mediated innate immune response during VSV infection. Virology 365:20–33 [DOI] [PubMed] [Google Scholar]
- 13.Johnson AJ, Roehrig JT. 1999. New mouse model for dengue virus vaccine testing. J. Virol. 73:783–786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Neyts J, Meerbach A, McKenna P, De Clercq E. 1996. Use of the yellow fever virus vaccine strain 17D for the study of strategies for the treatment of yellow fever virus infections. Antiviral Res. 30:125–132 [DOI] [PubMed] [Google Scholar]
- 15.Drew WL, Love R. 1968. Production of herpes simplex virus by HeLa cells treated with 5-fluoro-2′-deoxyuridine. Am. J. Pathol. 53:169–182 [PMC free article] [PubMed] [Google Scholar]
- 16.Goodheart CR, Filbert JE, McAllister RM. 1963. Human cytomegalovirus. Effects of 5-fluorodeoxyuridine on viral synthesis and cytopathology. Virology 21:530–532 [Google Scholar]
- 17.Loh PC, Payne FE. 1965. Effect of 5-fluoro-2′-deoxyuridine on the synthesis of vaccinia virus. Virology 25:575–584 [DOI] [PubMed] [Google Scholar]
- 18.Shanley JD, Debs RJ. 1989. The folate antagonist, methotrexate, is a potent inhibitor of murine and human cytomegalovirus in vitro. Antiviral Res. 11:99–106 [DOI] [PubMed] [Google Scholar]
- 19.Avendano C, Menendez JC. 2008. Medicinal chemistry of anticancer drugs. Elsevier, Amsterdam, The Netherlands [Google Scholar]
- 20.Jackson RC, Grindey GB. 1985. The biochemical basis of methotrexate cytotoxicity, p 289–315 In Sirotnak FM, Burchall JJ, Ensminger WD, Montgomery JA. (ed), Folate antagonists as therapeutic agents, vol 1 Academic Press, New York, NY [Google Scholar]
- 21.Xie X, Wang QY, Xu HY, Qing M, Kramer L, Yuan Z, Shi PY. 2011. Inhibition of dengue virus by targeting viral NS4B protein. J. Virol. 85:11183–11195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.van Laar JA, van der Wilt CL, Rustum YM, Noordhuis P, Smid K, Pinedo HM, Peters GJ. 1996. Therapeutic efficacy of fluoropyrimidines depends on the duration of thymidylate synthase inhibition in the murine colon 26-B carcinoma tumor model. Clin. Cancer Res. 2:1327–1333 [PubMed] [Google Scholar]
- 23.Peters GJ, van der Wilt CL, van Triest B, Codacci-Pisanelli G, Johnston PG, van Groeningen CJ, Pinedo HM. 1995. Thymidylate synthase and drug resistance. Eur. J. Cancer 31A:1299–1305 [DOI] [PubMed] [Google Scholar]
- 24.Harwood FG, Frazier MW, Krajewski S, Reed JC, Houghton JA. 1996. Acute and delayed apoptosis induced by thymidine deprivation correlates with expression of p53 and p53-regulated genes in colon carcinoma cells. Oncogene 12:2057–2067 [PubMed] [Google Scholar]
- 25.Ju J, Pedersen-Lane J, Maley F, Chu E. 1999. Regulation of p53 expression by thymidylate synthase. Proc. Natl. Acad. Sci. U. S. A. 96:3769–3774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Munoz-Pinedo C, Oliver FJ, Lopez-Rivas A. 2001. Apoptosis of haematopoietic cells upon thymidylate synthase inhibition is independent of p53 accumulation and CD95-CD95 ligand interaction. Biochem. J. 353:101–108 [PMC free article] [PubMed] [Google Scholar]
- 27.Munoz-Fontela C, Macip S, Martinez-Sobrido L, Brown L, Ashour J, Garcia-Sastre A, Lee SW, Aaronson SA. 2008. Transcriptional role of p53 in interferon-mediated antiviral immunity. J. Exp. Med. 205:1929–1938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Munoz-Fontela C, Pazos M, Delgado I, Murk W, Mungamuri SK, Lee SW, Garcia-Sastre A, Moran TM, Aaronson SA. 2011. p53 serves as a host antiviral factor that enhances innate and adaptive immune responses to influenza A virus. J. Immunol. 187:6428–6436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Takaoka A, Hayakawa S, Yanai H, Stoiber D, Negishi H, Kikuchi H, Sasaki S, Imai K, Shibue T, Honda K, Taniguchi T. 2003. Integration of interferon-alpha/beta signalling to p53 responses in tumour suppression and antiviral defence. Nature 424:516–523 [DOI] [PubMed] [Google Scholar]
- 30.Liu Y, Colosimo AL, Yang XJ, Liao D. 2000. Adenovirus E1B 55-kilodalton oncoprotein inhibits p53 acetylation by PCAF. Mol. Cell. Biol. 20:5540–5553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Scheffner M, Werness BA, Huibregtse JM, Levine AJ, Howley PM. 1990. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell 63:1129–1136 [DOI] [PubMed] [Google Scholar]
- 32.Matlashewski G, Banks L, Pim D, Crawford L. 1986. Analysis of human p53 proteins and mRNA levels in normal and transformed cells. Eur. J. Biochem. 154:665–672 [DOI] [PubMed] [Google Scholar]
- 33.Komarov PG, Komarova EA, Kondratov RV, Christov-Tselkov K, Coon JS, Chernov MV, Gudkov AV. 1999. A chemical inhibitor of p53 that protects mice from the side effects of cancer therapy. Science 285:1733–1737 [DOI] [PubMed] [Google Scholar]
- 34.Lim PY, Keating JA, Hoover S, Striker R, Bernard KA. 2011. A thiopurine drug inhibits West Nile virus production in cell culture, but not in mice. PLoS One 6:e26697. 10.1371/journal.pone.0026697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang QY, Bushell S, Qing M, Xu HY, Bonavia A, Nunes S, Zhou J, Poh MK, Florez de Sessions P, Niyomrattanakit P, Dong H, Hoffmaster K, Goh A, Nilar S, Schul W, Jones S, Kramer L, Compton T, Shi PY. 2011. Inhibition of dengue virus through suppression of host pyrimidine biosynthesis. J. Virol. 85:6548–6556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen YL, Yin Z, Lakshminarayana SB, Qing M, Schul W, Duraiswamy J, Kondreddi RR, Goh A, Xu HY, Yip A, Liu B, Weaver M, Dartois V, Keller TH, Shi PY. 2010. Inhibition of dengue virus by an ester prodrug of an adenosine analog. Antimicrob. Agents Chemother. 54:3255–3261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yin Z, Chen YL, Schul W, Wang QY, Gu F, Duraiswamy J, Kondreddi RR, Niyomrattanakit P, Lakshminarayana SB, Goh A, Xu HY, Liu W, Liu B, Lim JY, Ng CY, Qing M, Lim CC, Yip A, Wang G, Chan WL, Tan HP, Lin K, Zhang B, Zou G, Bernard KA, Garrett C, Beltz K, Dong M, Weaver M, He H, Pichota A, Dartois V, Keller TH, Shi PY. 2009. An adenosine nucleoside inhibitor of dengue virus. Proc. Natl. Acad. Sci. U. S. A. 106:20435–20439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Helt AM, Harris E. 2005. S-phase-dependent enhancement of dengue virus 2 replication in mosquito cells, but not in human cells. J. Virol. 79:13218–13230 [DOI] [PMC free article] [PubMed] [Google Scholar]