

Abstract
Highly active antiretroviral therapy (HAART) is able to suppress human immunodeficiency virus type 1 (HIV-1) to undetectable levels in the majority of patients, but eradication has not been achieved because latent viral reservoirs persist, particularly in resting CD4+ T lymphocytes. It is generally understood that HIV-1 does not efficiently infect resting CD4+ T cells, and latent infection in those cells may arise when infected CD4+ T lymphoblasts return to resting state. In this study, we found that stimulation by endothelial cells can render resting CD4+ T cells permissible for direct HIV infection, including both productive and latent infection. These stimulated T cells remain largely phenotypically unactivated and show a lower death rate than activated T cells, which promotes the survival of infected cells. The stimulation by endothelial cells does not involve interleukin 7 (IL-7), IL-15, CCL19, or CCL21. Endothelial cells line the lymphatic vessels in the lymphoid tissues and have frequent interactions with T cells in vivo. Our study proposes a new mechanism for infection of resting CD4+ T cells in vivo and a new mechanism for latent infection in resting CD4+ T cells.
INTRODUCTION
Highly active antiretroviral therapy (HAART) is able to suppress human immunodeficiency virus type 1 (HIV-1) to undetectable levels in the majority of patients (1–3). Although the suppression can be maintained for many years, eradication has not been achieved with HAART alone (4) because latent viral reservoirs persist in patients despite successful treatment (reviewed in reference 5). The most prominent and extensively characterized latent reservoir exists in resting CD4+ T lymphocytes (6–8). CD4+ T cells are the major target cells for HIV. In productive infection, upon entry the virus goes through reverse transcription, integration, virus gene expression, and new virus assembly and budding. However, in latent infection, specifically in postintegration latency, the virus expresses no or minimal levels of viral transcripts from the integrated proviruses (9). Since viral antigens are not expressed in latent infection, latently infected cells suffer no cytopathic effects of viral proteins and are not recognized or targeted by cytotoxic T cells. Moreover, antiretroviral drugs target active viral replication and have no effect on latent proviruses that have integrated into the host cell genome. Therefore, when viral latency is established in resting CD4+ T cells, such latently infected cells persist in HIV-positive (HIV+) patients for a long time with minimal decay (10). As a result of the fact that resting CD4+ T cells (especially memory T cells) have a naturally long life span in vivo, latent HIV infection in such cells forms a very stable viral reservoir and poses a huge barrier to viral eradication. The integrated provirus is maintained in the latent state by several means, including the lack of necessary host transcription factors, chromatin structure, and integration location that prevent or minimize the expression of the provirus (reviewed in reference 11). However, if the latently infected resting T cell encounters its antigen and becomes activated or is exposed to certain cytokines or chemokines, the latent virus also goes into productive infection and starts producing new virions (7, 12). Such activation events are likely the major source of viral rebound after treatment interruption. Thus, the latent reservoir ensures viral persistence and renders lifelong therapy necessary to control HIV diseases in patients. In order to advance our current strategy for treating HIV diseases, understanding of the latent reservoirs, particularly the reservoir in resting CD4+ T cells, is critical.
Since the discovery of the most stable latent HIV reservoir in resting (especially memory) CD4+ T cells in 1997, much has been learned about latency maintenance and reactivation of latent proviruses (reviewed in reference 11). However, how this latent reservoir is formed is still quite unclear.
Based primarily on in vitro evidence, it is generally understood that HIV can replicate only in activated CD4+ T cells (13–17). In resting T cells, the virus can enter the cell but either cannot complete reverse transcription (16) or can complete reverse transcription at a much lower efficiency but cannot integrate its cDNA into the host genome (18, 19). On the other hand, when activated T cells are infected, productive infection usually results. Such productive infection typically results in virus-induced cytopathic effects and/or elimination by CD8+ T cells; thus, these infected cells usually do not have a chance to return to resting memory state. This poses a difficulty in explaining how resting T cells that harbor latent integrated provirus are formed. One favored explanation in the field is that if an activated T cell is infected by HIV during its transition to a resting memory T cell, the virus becomes stably integrated into the host cell genome but cannot produce new virus, which generates postintegration viral latency (20, 21).
However, more recent evidence, especially evidence from in vivo or ex vivo studies, has demonstrated that resting CD4+ T cells are productively infected in vivo or can be infected directly ex vivo (22–26). Both naive and memory CD4+ T cells isolated from patients were found to harbor integrated DNA (25); in situ hybridization in lymphoid tissues in infected individuals showed that resting CD4+ T cells contain viral RNA (27), and direct infection of resting CD4+ T cells in intact lymphoid tissue ex vivo resulted in productive infection in those cells (22, 28). One of the studies found that resting CD4+ T cells support HIV replication in lymphoid tissue (tonsil) explants, whereas purified tonsillar resting CD4+ T cells did not support HIV replication (29). Such results suggest that isolated resting T cells may not be permissive for HIV replication, but resting T cells residing in lymphoid tissues may well be infected both productively and latently in vivo. The in vivo lymphoid tissue microenvironment plays a crucial role in inducing productive infection in resting CD4+ T cells and probably in the establishment of latent reservoir in those cells as well.
Identifying the specific role the lymphoid tissue microenvironment plays in HIV infection of resting CD4+ T cells is extremely important in understanding latent reservoir formation in such cells. However, not much is known about the mechanisms involved in rendering resting T cells permissive for HIV replication. A few studies have found soluble factors that were involved: proinflammatory cytokines, such as interleukin 2 (IL-2), IL-4, IL-7, and IL-15 (30), and chemokines CCL19 and CCL21 (31). In addition, two studies by Choi et al. in 2005 (32, 33) showed that interactions between endothelial cells and T cells rendered them permissive for HIV replication while continuing to exhibit a resting phenotype, and such interactions required cell-cell contact and were dependent upon major histocompatibility complex (MHC) class II and CD58 on the endothelial cells. Endothelial cells express both classes I and II MHC as well as costimulatory molecules and are considered antigen-presenting cells. They line the lymphatic vessels in the lymphoid tissues and have constant interactions with T cells trafficking through them. Endothelial cells may contribute significantly to HIV infection of resting CD4+ T cells as well as latency formation in these cells. In this study, we used a pseudotyped reporter virus capable of only single-round infection to further elucidate the roles endothelial cells play in inducing productive and latent infection of resting CD4+ T cells.
MATERIALS AND METHODS
Endothelial cells and in vitro infection assay.
Human umbilical vein endothelial cells (HUVEC or EC) were purchased from PromoCell (Germany) and cultured in M199 medium supplemented with l-glutamine (Invitrogen) and 20% fetal bovine serum (FBS). Endothelial cell growth factors (BD Biosciences) were added fresh every 3 days to the final concentration of 50 μg/ml. Where indicated, endothelial cells were pretreated with gamma interferon (IFN-γ; 50 ng/ml) (Invitrogen) for 3 days prior to the addition of resting T cells. Endothelial cells were plated to 50% confluence the day before or 100% confluence the same day, and up to 1 million resting T cells were cocultured with EC per well of a 24-well plate, or up to 5 million T cells were cultured per well in a 6-well plate. Resting T cells were cocultured with EC for 1 day in RPMI 1640 plus 10% FBS plus 1% penicillin-streptomycin (without EC growth factor or IFN-γ) prior to overnight infection or, in a few experiments, spinoculation (2 h at 1,200 × g at room temperature). The cocultures were maintained in the same medium for the duration of the experiments. Expressions of green fluorescent protein (GFP) and T-cell activation markers were examined on various days postinfection using flow cytometry. Antibodies for various activation markers were all purchased from BD Biosciences. For experiments on latent infections, flow cytometric sorting was also done at various days postinfection. After sorting, the GFP− cells were cultured with or without phorbol myristate acetate (PMA) (10 ng/ml) plus ionomycin (1 μg/ml) (both from Sigma) and raltegravir (3.3 μM) (Selleck) for 2 days before flow cytometric analysis of GFP expression.
Virus stock production.
NL43-dE-GFP pseudotyped reporter virus was generated by cotransfecting HEK293T cells with a plasmid encoding NL43-dE-GFP and a plasmid encoding the HIV-1 envelope (pWE-CXCR4) using Lipofectamine 2000 (Invitrogen) or TrueFect (United BioSystems) according to the manufacturer's instructions. Supernatants were collected after 72 h and filtered through a 0.22-μm membrane to clear cell debris. Virus particles were either pelleted at 25,000 × g with a 10% volume of 20% sucrose in the bottom for 2 h at 4°C or concentrated using a Lenti-X concentrator (Clontech) by following the manufacturer's instructions. NL43-dNef-GFP virus was generated similarly by transfecting HEK293T cells with a plasmid encoding NL43-dNef-GFP. The Bcl-2 lentiviral vectors were generated similarly by cotransfecting HEK293T cells with a plasmid encoding EB-FLV, vesicular stomatitis virus G protein (VSV-G) envelope (pVSVG), and a packaging vector, pC-Help, using Lipofectamine 2000 (Invitrogen) by following the manufacturer's instructions. pC-Help (34) was a gift from Jakob Reiser (Louisiana State University Health Sciences Center, New Orleans, LA). The modified pseudotyped reporter virus pNL4-3-Δ6-drEGFP was generated by transfection with a plasmid encoding pNL4-3-Δ6-drEGFP (35), HIV-1 envelope (pWE-CXCR4), and pC-Help.
Separation of various T cell populations and T cell activation in vitro.
Human peripheral blood mononuclear cells (PBMC) were obtained from HIV-negative (HIV−) blood by centrifugation through a Ficoll-Hypaque density gradient at 335 × g for 40 min. Total CD4+ T cells were purified from PBMC using Miltenyi microbeads (negative depletion kit for isolating CD4+ T cells). Resting CD4+ T cells were isolated by adding biotin-labeled anti-CD25 and anti-HLA-DR antibodies to the Miltenyi depletion cocktail mix and subsequently increasing the amount of antibiotin microbeads added. To activate T cells in vitro using anti-CD3 and anti-CD28 bodies (BD Biosciences), we coated tissue culture plates with anti-CD3 antibodies and then added anti-CD28 antibodies with T cells and IL-2 (PeproTech) in media (35). In order to separate memory and naive T cells, purified resting CD4+ T cells were stained with CD45RO-allophycocyanin (CD45RO-APC) and CD45RA-phycoerythrin (CD45RA-PE) (BD Biosciences) and sorted using flow cytometry for RO+ RA− memory T cells and RO− RA+ naive T cells. When separating effector and central memory T (TEM and TCM, respectively) cells, purified resting CD4+ T cells were stained with CCR7-PE and CD45RA-APC (BD Biosciences) and sorted for CCR7+ RA− central memory T cells, CCR7− RA− effector memory T cells, and CCR7+ RA+ naive T cells.
Cell cycle analysis with DNA or RNA staining.
The procedures for cell cycle analysis have been described previously (36). Briefly, 2 × 105 cells were obtained from each culture well, washed with 1× phosphate-buffered saline (PBS), and resuspended in 100 μl of nucleic acid staining solution (NASS) with a pH of 4.8 (0.15 M NaCl in 0.1 M phosphate-citrate buffer containing 5 mM sodium EDTA and 0.5% bovine serum albumin [BSA]) and 1 μl of 2% saponin (Sigma). Twenty microliters of 20-μg/ml 7-amino-actinomycin D (7AAD; Life Technology) was added to each sample to stain for DNA, and cells were incubated at room temperature for 20 min in the dark. Samples were washed and then resuspended in 100 μl of NASS and 20 μl of 20-μg/ml actinomycin D (AD; Sigma). Cells were incubated on ice for 5 min in the dark, and then 5 μl of a fresh 1:10 dilution of 1-mg/ml pyronin Y (PY; Sigma) was added to each sample to stain for RNA. Cells were then incubated for 10 min on ice in the dark. Samples were analyzed on a BD FACSCalibur flow cytometer using the CellQuest Pro program (Becton, Dickinson). In some cultures, sodium butyrate (5 mM; Sigma) was used to arrest CD3- or CD28-activated T cells at G1a to serve as a control. Sodium butyrate was added to the cultures at the time of activation, and samples were stained for cell cycle analysis the next day.
Long-lived primary T cell latency model (generation of latently infected B2T cells).
The procedures to generate long-lived primary T cells were described in reference 35. Briefly, CD4+ T cells were purified from PBMC using Miltenyi microbeads (negative depletion kit for isolating CD4+ T cells) and activated with anti-CD3 and anti-CD28 antibodies (BD Biosciences) and IL-2 for 3 days before they were transduced by a lentiviral vector carrying the Bcl-2 gene (EB-FLV) to promote survival. The cells were cultured in the absence of stimuli or cytokines for 3 to 4 weeks. The resulting cell population (designated B2T cells) was small, bore few to no activation markers, and was not proliferating. Such cells were then activated using anti-CD3 and anti-CD28 antibodies, infected with a modified pseudotyped reporter virus, pNL4-3-Δ6-drEGFP, that expresses GFP, cultured for 4 weeks, and sorted for GFP− populations, which contain uninfected and latently infected cells.
HIV+ patient samples and modified viral outgrowth assay to determine infectious units per million cells (IUPM).
HIV+ patients were on highly active antiretroviral therapy (HAART), and the viral load had been suppressed below the detection limit for at least 6 months. Purified resting CD4+ T cells were activated with anti-CD3 or -CD28 (CD3/28) antibodies (BD Biosciences) or cocultured with EC in duplicated 5-fold limiting dilutions, and p24 was measured in culture supernatants 14 days later using a p24 enzyme-linked immunosorbent assay (ELISA) kit (PerkinElmer). No allogeneic lymphoblasts were added. IUPM were determined statistically based on the percentage of positive culture supernatants at each dilution (described in reference 7).
Detection of CCL19, CCL21, IL-7, and IL-15 using ELISA.
Supernatants from cell culture wells were collected and frozen at −80°C. ELISA kits for CCL19, CCL21, IL-7, and IL-15 were purchased from RayBiotech, and experiments were performed according to the manufacturer's instructions. Two hundred microliters of supernatant was used from each sample in duplicates. A human breast cancer cell line, MCF7, was obtained from the ATCC.
RESULTS
General scheme of EC and CD4+ T cell coculture and infection.
We first established the endothelial cell-T cell coculture system and infected the resting CD4+ T cells with a reporter HIV in which a portion of the envelope gene was replaced with enhanced GFP (EGFP) (NL43-dE-GFP [37]), as illustrated in Fig. 1A. Human umbilical vein endothelial cells (EC) were first treated with IFN-γ for 3 days to induce the expression of MHC class II in EC in vitro (in vivo EC do express MHC class II) (Fig. 1B), as the expression of MHC class II on EC is thought to enhance HIV infection of resting T cells (32, 33). Resting CD4+ T cells were purified from healthy donors and cocultured with endothelial cells treated with IFN-γ (EC+) or endothelial cells without IFN-γ treatment (EC−) for 1 day. Then the T cells, in coculture with endothelial cells in RPMI medium plus 10% FBS without EC growth factors or IFN-γ, were infected overnight with NL43-dE-GFP, a pseudotyped reporter HIV expressing GFP and carrying an HIV Env that uses CXCR4 as a coreceptor for entry. Viral infection, as indicated by GFP expression, was then measured on various days postinfection using flow cytometry. T cells were distinguished from EC by forward and side scatter gating, as they were much smaller and less granular than EC. We found no infection of EC, consistent with the results of Choi et al. (32, 33). We also infected resting T cells alone (R) as well as anti-CD3/CD28 antibody (CD3/28)-activated T cells (ACT) as controls. Activated T cells were infected at day 2 postactivation. In some experiments, GFP+ and/or GFP− cells were sorted at various days postinfection for cell cycle or latency analysis.
Fig 1.

Viral infection of resting CD4+ T cells cocultured with endothelial cells. (A) General scheme of EC and CD4+ T cell coculture and infection. (B) Expression of HLA-DR in EC− and EC+ cells. FITC, fluorescein isothiocyanate. (C) Kinetics of infection. Endothelial cells treated with IFN-γ or without IFN-γ treatment were cocultured with resting CD4+ T cells and infected with a reporter virus expressing GFP. Expression of GFP was measured on days 3 to 8 postinfection. Resting T cells alone and CD3/28-activated T cells were included as controls. (D) Death rates of infected resting CD4+ T cells alone or in EC cocultures. Measurements were taken 5 days postinfection. Death rates were calculated as the number of dead cells over the number of total cells. Panels C and D show the means ± standard errors of quadruplicate and duplicate samples, respectively, and are representative of at least three experiments. *, P < 0.05, Student t test.
Kinetics of viral infection in resting CD4+ T cells cocultured with endothelial cells.
First we compared viral infection kinetics in endothelial-cell-stimulated resting T cells with those in CD3/28-activated T cells. As shown in Fig. 1C, viral infection in endothelial-cell-stimulated resting T cells had slower kinetics than infection in CD3/28-activated T cells. In CD3/28-activated T cells, viral gene expression, represented by GFP expression, peaked at day 3 (or day 2 in some similar experiments [data not shown]), and in endothelial-cell-stimulated T cells, the peak did not typically come until at least day 5. After day 5, there was a stable proportion of GFP+ T cells in endothelial-cell-stimulated cells, whereas in CD3/28-activated T cells, there was a decline of GFP+ cells after day 2 or 3, probably due to preferential death of infected cells. The seemingly stable proportion of GFP-expressing cells over time was likely the combination of cell death in some infected cells (decrease in GFP+) and the emergence of new GFP-expressing cells (increase of GFP+), because in some experiments we sorted GFP− cell populations and continued culturing them, and we observed an increase in the GFP+ population over time (data not shown). The slower kinetics of infection in endothelial-cell-stimulated T cells were similar to those in resting T cells alone (Fig. 1C), even though the total infection rates were much higher in endothelial-cell-stimulated T cells. The overall infection rates in endothelial-cell-stimulated resting T cells were even higher than the infection rates in CD3/28-activated T cells.
Lastly, there was substantial infection in T cells cocultured with EC−, even though it was typically a bit lower than in those cocultured with EC+.
Resting T cells cocultured with EC have lower death rates than activated T cells.
Since we observed a decline of GFP+ cells after day 2 or 3 in CD3/28-activated T cells and a stable proportion of GFP+ T cells in EC cocultures, we then examined whether cell death rates were generally lower in EC cocultures. We followed the general infection scheme in Fig. 1A and examined cell viability via flow cytometry on day 5 postinfection. We gated for live cells and dead or dying cells separately based on forward and side scatter patterns and calculated death rates by dividing the number of dead cells by the number of total cells. As shown in Fig. 1D, resting T cells cocultured with EC only had a third to half of the death rates in CD3/28-activated T cells. Death rates from EC−-stimulated T cells were lower than those from EC+-stimulated T cells and were similar to those from resting T cells alone.
Resting T cells cocultured with EC can be productively infected by HIV while remaining in the resting state.
Since we found untreated EC to be similar to IFN-γ-treated EC in promoting productive infection of resting CD4+ T cells, we performed additional experiments to examine the interaction of T cells with EC that had not been stimulated with IFN-γ. Choi et al. have previously reported (32, 33) that resting CD4+ T cells can be infected by HIV-1 after being cocultured with EC+ but remain in a resting state. We examined cell activation markers CD25, CD69, and HLA-DR using flow cytometry various days postinfection as well as GFP levels. In the experiment whose results are shown in Fig. 2A, samples were taken 5 days postinfection. Even though there were higher levels of infection in T cells cocultured with EC than in CD3/28-activated T cells, few T cells cocultured with EC expressed activation markers. In T cells cocultured with EC+, about 3 to 8% of T cells typically expressed some activation markers. These cells may recognize allogeneic MHC class II on EC and become activated. However, the proportion of T cells that were infected (an average of about 29% in this experiment) was typically much higher than the proportion of cells that were activated (an average of about 4% in this experiment). This means that most of the infected cells were not activated. In T cells cocultured with EC−, which do not express MHC class II, the proportion of activated T cells was typically lower than for EC+ cocultures and either similar to or slightly higher than that observed for resting T cells alone. In each of the cultures, Ki67 levels were also measured as a marker for cell proliferation, and there was very little Ki67 expression, except for in CD3/CD28-activated T cells (data not shown).
Fig 2.

Infected resting T cells cocultured with EC remained in a resting state. (A) Resting T cells were cultured alone, with EC treated with IFN-γ, with untreated EC, or with CD3/28 activation and infected with a reporter virus expressing GFP. Expressions of GFP as well as activation markers (combined CD69, HLA-DR, or CD25) were measured on day 5 postinfection. (B) In a separate experiment, activation markers CD69, HLA-DR, and CD25 were measured individually on day 10 postinfection. (C) Flow cytometry plots showing both GFP expression and activation markers for EC+ cultures from the same experiment as in panel B. The percentage of cells in each quadrant is indicated. (D) Analysis of infection rates in activation marker-positive cells versus activation marker-negative cells from the same experiment as in panel B. Samples were taken in duplicate (A) or quadruplicate (B and D), and means ± standard errors are plotted, representative of four separate experiments. #, not enough HLA-DR+ cells for analysis. *, P < 0.05, Student t test.
We further analyzed among the small proportion of EC-stimulated T cells that expressed any activation markers which activation marker or markers (CD69, HLA-DR, or CD25) were upregulated most strongly. In a separate experiment, T cells cultured alone or with EC were stained with PE-conjugated antibodies against CD69, HLA-DR, or CD25. We found that CD69 was upregulated on more cells among EC-cocultured cells than HLA-DR and CD25 (Fig. 2B).
Interestingly, significant proportions (about 30%) of GFP+ cells in EC+ cultures were CD69+, while in the same cultures, very few GFP+ cells were HLA-DR+ and almost none were CD25+ (Fig. 2C). Moreover, even though cells bearing activation markers from EC cultures were preferentially infected, the preferences were not equal among populations defined by these three activation markers (Fig. 2D). CD69-positive cells had a much higher infection rate (defined as the GFP+ proportion in total CD69+ cells) than CD69-negative cells (defined as the GFP+ proportion in total CD69− cells), and infection rates in HLA-DR-positive cells were also significantly higher than in HLA-DR-negative cells, though the difference was smaller than that of the samples with CD69 markers. However, CD25-positive cells had just slightly higher infection rates than CD25-negative cells. In EC− cultures, the difference was more pronounced, as almost all (95%) CD69+ cells were infected, whereas infection rates among marker-negative cells were similar. Thus, the virus seemed to preferentially infect CD69+ cells in both EC+ and EC− cultures.
EC-stimulated resting CD4+ T cells that become infected are mainly in cell cycle stage G0/G1a.
Korin and Zack suggested that cell cycle G1b was required for HIV infection in CD4+ T cells (13), as they found much higher infection rates in cells arrested in G1b than in those arrested in G1a. Therefore, we examined cell cycle status of resting CD4+ T cells stimulated by EC, particularly to determine whether they were in G1b. We followed the general scheme of infection in Fig. 1A and stained T cells with 7-AAD (DNA) and pyronin Y (RNA) on day 6 postinfection. We used N-butyrate to arrest cells in G1a to serve as a control. As shown in Fig. 3A, a very small percentage of T cells cocultured with EC+ were in G1b (or in G2/M), and almost none of the T cells cocultured with EC− were in G1b or G2/M. To further examine whether infected T cells from the EC cocultures were in G1b, we sorted GFP+ cells at day 5 postinfection and immediately analyzed them for cell cycle stage. As shown in Fig. 3B, while a high proportion of CD3/28-activated T cells were in G1b or G2/M, there were very few cells in G1b or G2/M for resting T cells alone (R) or T cells in EC cocultures. Even though infected (GFP+) cells contained a higher proportion of cells in G1b or G2/M, the vast majority of the infected cells were not in G1b or G2/M. Thus, we concluded that infected resting CD4+ T cells stimulated by EC were mostly in the G0/G1a phase of the cell cycle.
Fig 3.

Cell cycle analysis of CD4+ T cells cultured alone or with EC. (A) Resting T cells were cultured alone, with EC treated with IFN-γ, with untreated EC, or with CD3/28 activation, and on day 6 after coculture, cell cycle analyses were performed by staining for DNA (7-AAD) and RNA (PY). Populations for various cell cycle status and their percentages are indicated on the plots. (B) Resting T cells were cultured alone, with EC treated with IFN-γ, with untreated EC, or with CD3/28 activation and infected with a reporter virus expressing GFP. Five days postinfection, T cells from these various cultures were each sorted into GFP+ and GFP− populations. Cell cycle analyses were then performed on each population from various cultures by staining DNA (7-AAD) and RNA (PY). Panel B shows the means ± standard errors of duplicated samples and is representative of three similar experiments.
Resting memory T cells are preferentially infected compared to naive T cells when cocultured with EC.
Choi et al. showed that when cocultured with EC+, memory T cells were infected, while naive T cells were not infected. In order to examine viral infection in these two subsets of resting T cells in our system, we sorted naive (CD45RAhi and CD45RO−) and memory (CD45RAlo and CD45RO+) populations from freshly purified resting T cells using flow cytometry, cultured them alone or with EC, and infected them with a pseudotyped reporter virus. As seen in Fig. 4A, memory T cells were infected at much higher rates than naive T cells in EC-stimulated cultures (both EC+ and EC−). EC-stimulated memory T cells were preferentially infected compared to EC-stimulated naive T cells, though EC-stimulated naive T cells still were infected more than naive T cells cultured alone.
Fig 4.
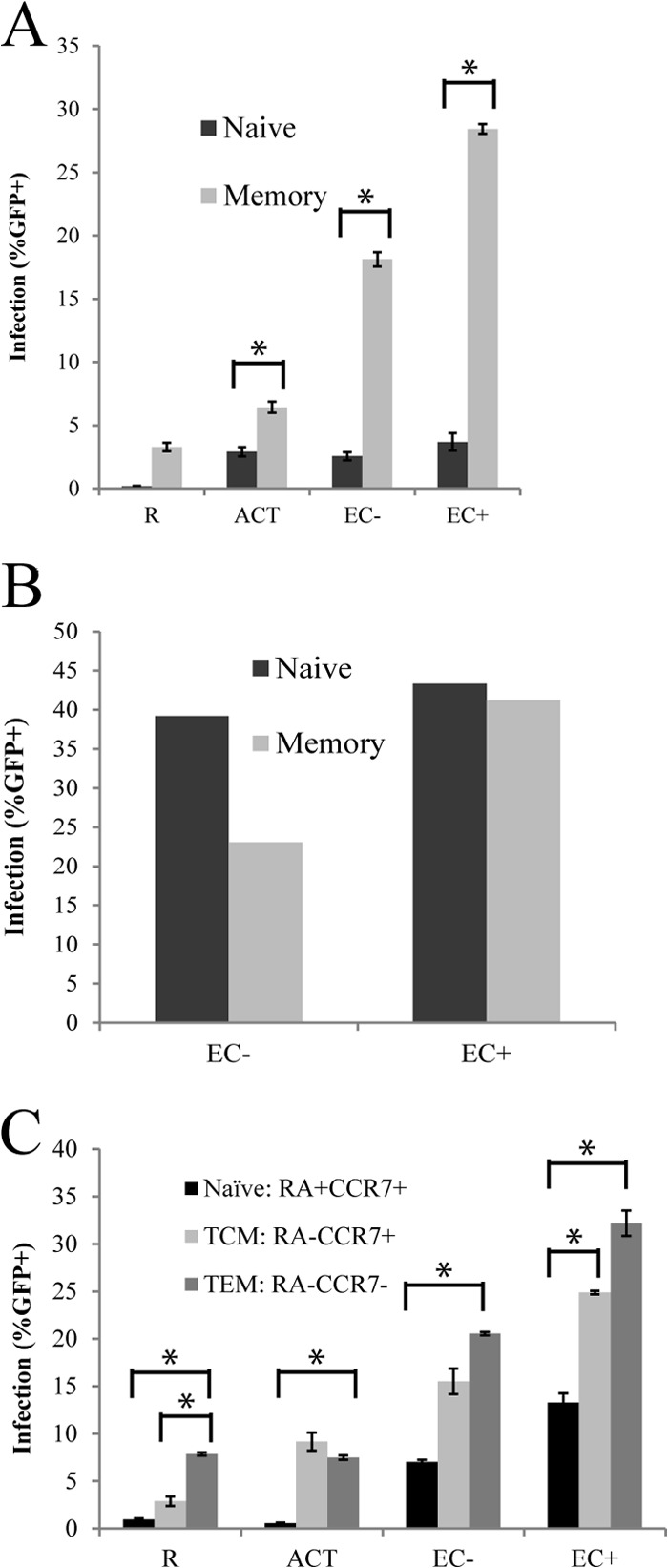
Memory T cells were preferentially infected compared to naive T cells when cocultured with EC. (A) Resting CD4+ T cells were sorted for naive (CD45RAhi and CD45RO−) and memory (CD45RAlo and CD45RO+) populations, cocultured separately with EC+ and EC−, and infected with a reporter virus expressing GFP overnight in culture. GFP expression was measured day 6 postinfection. (B) Resting CD4+ T cells were sorted and cocultured similarly as for panel A and then infected with spinoculation (2 h at 1,200 × g). GFP expression was also measured on day 6 postinfection. (C) Resting CD4+ T cells were sorted into naive, TCM, and TEM cell populations and cocultured and infected in a manner similar to that for panel A. Panels A and C show the means ± standard errors of duplicated samples and are representative of four and two similar experiments, respectively. Panel B shows a representative of two experiments with nonreplicated samples. *, P < 0.05, Student t test.
This suggested that signals provided by EC to memory T cells were able to overcome the restrictions to a much greater extent than in naive cells. This is consistent with the fact that EC express CD58 but not the costimulatory molecules CD80/86 and thus are better at stimulating memory T cells than naive T cells. Naive T cells generally require a stronger costimulatory signal (e.g., through CD80/86) for activation than memory T cells (reviewed in reference 38).
In some experiments, we used spinoculation (2 h at 1,200 × g) instead of overnight culture infection. The results using this method were quite different than with the overnight infection method. As shown in Fig. 4B, naive and memory cells were infected equally well, and in EC− cocultures, naive cells even seemed more susceptible to infection than memory cells. Since spinoculation may introduce additional variables not relevant to the in vivo situation, subsequent experiments were performed using overnight infection.
Effector memory T cells are preferentially infected compared to central memory T cells in EC cocultures.
We wished to determine within the memory population whether there was preferential infection of effector or central memory T cells (TEM or TCM cells, respectively). In order to determine viral infection rates in these two groups of memory T cells, we sorted TCM (CCR7+ and CD45RAlo), TEM (CCR7− and CD45RAlo), and naive (CD45RAhi and CD45RO−) populations from freshly purified resting T cells using flow cytometry, cocultured them with EC, and infected them with a pseudotyped reporter virus. As shown in Fig. 4C, in EC cocultures, the effector memory subset had the highest infection rate, followed by central memory T cells, while naive T cells had the lowest infection rate. This suggested that TEM cells may have a lower threshold for stimulation than TCM cells, which is consistent with current understanding about TEM and TCM cell activation signals (reviewed in reference 39).
Latent viral infection in resting T cells cocultured with EC.
So far, we have found that resting CD4+ T cells cocultured with EC can be productively infected to significantly higher levels than resting T cells cultured alone. In order to determine whether in vitro infection of resting T cells cocultured with EC could result in postintegration latent infection, we followed the scheme of infection in Fig. 1A. On day 9 postinfection, after most unintegrated viral DNA had decayed (19), GFP− cells were sorted and activated with PMA and ionomycin (PMA-I) for 2 days (Fig. 1A). PMA is known to reactivate latent HIV. If we detected a significant increase of GFP expression cells after T cells were stimulated with PMA-I, compared with unstimulated cells, the proportion of GFP+ cells in excess would be considered to have been latently infected cells. PMA-I stimulation also promotes viral integration. Therefore, if there are unintegrated proviruses in the cells, upon PMA-I stimulation, they will become integrated and subsequently express GFP. In order to prevent de novo viral integration (thus to ensure detection of postintegration latency), the integrase inhibitor raltegravir was also included in the cultures. As shown in Fig. 5B, the increase in GFP expression in PMA-I-stimulated cultures compared with unstimulated cultures was most dramatic for the EC− culture but was also statistically significant for the resting-alone (R) and EC+ cultures (Student t test, P < 0.05). Thus, in resting T cells cultured alone or with EC, there was postintegration latent infection. No increase in GFP expression was seen in activated T cells upon PMA-I stimulation, indicating that there were no latently infected cells.
Fig 5.

Latency in resting CD4+ T cells cocultured with EC. (A) Resting CD4+ T cells with or without EC coculture were infected, and on day 9 postinfection, GFP− cells were sorted and stimulated with PMA-ionomycin or cultured alone for 2 days. Raltegravir was added to prevent de novo viral integration. GFP expression in resting T cells from a EC− coculture are shown, either unstimulated or stimulated with PMA-I. (B) Experiment similar to that in panel A. GFP expressions were compared in unstimulated cultures versus PMA-I-stimulated cultures. Samples were taken in duplicate (ACT) or quadruplicate (all others) depending on the number of cells available. (C) Resting CD4+ T cells were isolated from four suppressed patients and cultured alone, with CD3/28 activation, or with EC cocultures in limiting dilutions. Frequencies of latently infected cells (IUPM) were determined by Poisson statistics. (D) Latently infected long-lived resting T cells (B2T) were cultured alone (B2T alone), with activation (CD3/28 or PMA), or with EC cocultures for 6 days. Samples were taken in quadruplicates, and means ± standard errors are plotted in panels B and C, representative of three and two experiments, respectively. *, P < 0.05, Student t test.
EC do not reactivate latent HIV.
Because we found that coculturing T cells with EC resulted in both productive and latent infection of resting CD4+ T cells, we next asked whether EC can reactivate latent HIV already formed in resting CD4+ T cells. First, we used resting T cells from suppressed patients on suppressive HAART regimens. Resting CD4+ T cells were purified from HIV+ donors whose viral loads were suppressed by therapy to <50 copies/ml for at least 6 months. The cells were activated by anti-CD3/CD28 antibodies or cocultured with EC treated or not with IFN-γ. Virus production was measured on day14 by detecting capsid protein p24 in the supernatants. Resting T cells were cultured in 5-fold limiting dilutions starting at 5 million cells per well, and the frequencies of latently infected cells (infectious units per million cells [IUPM]) were determined by Poisson statistics. For 4 patients, even though CD3/28 activation resulted in detection of latently infected cells (up to 14 IUPM), no virus production was detected in EC cocultures (up to 5 million cells) (Fig. 5C). Based on input cell number, resting T cells alone and EC cocultures had <0.32 IUPM (shown as 0 in Fig. 5C). This result suggests that EC cannot reactivate latent virus in resting T cells from patients.
In the standard viral outgrowth assay (40), after activation of resting CD4+ T cells, allogeneic CD4+ T lymphoblasts are added to amplify the infection in vitro. With the EC cocultures, we did not add allogeneic lymphoblasts because such addition would activate resting T cells through mixed-lymphocyte reactions and cytokine production. Therefore, low levels of viral activation or viral production stimulated by EC might not be captured. Thus, we used an in vitro primary cell model of latency (latently infected B2Tcells [35]) to evaluate whether EC could activate latent virus. B2T cells were generated by transducing primary CD4+ T cells with Bcl-2 to promote longevity in vitro. The B2T cells were infected with a modified reporter virus that expresses GFP. GFP+ cells were removed at day 3 postinfection by flow cytometry, and the remaining GFP− cells contained latently infected cells (and uninfected cells). In this system, reactivation of latent virus was indicated by expression of GFP upon stimulation.
Latently infected B2T cells were cultured alone, with EC (with or without IFN-γ), or with CD3/28 or PMA activation. GFP expression, as an indication of reactivation from latency, was measured after 6 days. As shown in Fig. 5D, EC did not induce more GFP expression in B2T compared to the GFP level in B2T alone, while CD3/28 and PMA activation induced significantly more GFP expression. Taken together, these results demonstrated that EC do not reactivate latent virus.
Cytokine CCL19, CCL21, IL-7, or IL-15 levels do not correlate with infection of resting T cells in cocultures.
Choi et al. (32, 33) showed that the interactions between endothelial cells and T cells required cell-cell contact and were dependent upon MHC class II and CD58 on the endothelial cells. However, signals through T cell receptor (TCR)-MHC and CD2-CD58 were not sufficient in inducing HIV infection of resting T cells, and soluble factors were implicated. They excluded the following cytokines using antibody blocking: IL-2, IL-6, tumor necrosis factor (TNF), and BMP-6. We then tested cytokines IL-7 and IL-15 and chemokines CCL19 and CCL21, as they were found to render resting T cells permissive for HIV replication (30, 31). We collected supernatants from resting T cells, CD3/CD28-activated T cells, EC+ and EC−, and MCF7, a human breast cancer cell line that when cocultured with resting T cells did not induce significant HIV infection (Fig. 6A). We also cocultured resting T cells with EC+, EC−, and MCF7 cells for 6 days and then collected supernatants from the cocultures. We then measured levels of CCL19, CCL21, IL-7, and IL-15 using ELISA. As shown in Fig. 6B to E, EC cocultures did not induce high levels of any of the cytokines or chemokines, especially compared with MCF7 cell cocultures. Therefore, none of these cytokines or chemokines correlated with infection of resting T cells in EC coculture.
Fig 6.

Cytokine secretions do not correlate with infection of resting T cells in cocultures. (A) Resting T cells were cultured alone, with MCF7 cells, with EC treated with IFN-γ, with untreated EC, or with CD3/28 activation and infected with a reporter virus expressing GFP. Expression of GFP was measured on day 6 postinfection. Supernatants from resting T cells, activated T cells, and MCF7 cells or EC only or cocultured with T cells were collected and measured for expression of CCL19 (B), CCL21 (C), IL-7 (D), and IL-15 (E) using ELISA. Samples were taken in duplicates, and means ± standard errors are plotted, representative of two or three experiments. *, P < 0.05, Student t test.
DISCUSSION
In this study, we demonstrated that EC stimulation can render resting CD4+ T cells permissive for HIV infection. The resulting cells remained largely unactivated and showed a lower death rate than activated T cells. Among resting CD4+ T cells stimulated by EC, memory T cells, particularly effector memory T cells, were preferentially infected, even though naive T cells also had increased infection rates compared with unstimulated T cells. Both productive and latent infections were observed in endothelial-cell-stimulated resting CD4+ T cells infected in vitro, though EC stimulation did not result in reactivation of latent virus ex vivo.
This study confirms and extends initial studies by Choi et al. (32, 33). We confirmed that (i) EC stimulation dramatically enhances productive HIV infection of resting CD4+ T cells, (ii) such infected T cells remained in a resting phenotype, and (iii) among these infected resting T cells, memory T cells were preferentially infected rather than naive T cells.
There were several differences as well. For instance, Choi et al. found measurable infection only in EC+ (treated with IFN-γ) cocultures and not in EC− (untreated) cocultures or T cells cultured alone, but we were able to measure infection in both EC− coculture and T cells cultured alone. We also found that both EC+ and EC− stimulation of T cells resulted in increased infection rates compared with resting T cells alone, though EC− cocultures had lower rates than EC+ cocultures. We hypothesize that the difference is largely due to the increased sensitivity in our system, since we used a pseudotyped reporter virus and thus were able to examine infection at the single-cell level. Choi et al. used a replication-competent virus in their in vitro infections and measured viral infection by the accumulation of p24 antigen in the culture supernatants. Measuring the accumulation of p24 is not as sensitive, and this method measures the accumulated effect of multiple rounds of infection, as p24 was typically detected at day 9 to 12 postinfection. Therefore, even if EC+ cocultures had a small increase in infection rate (as was observed in our system), the compounding effects of multiple rounds of infection in vitro may have resulted in much higher difference in infection rates between EC+ and EC− cocultures. In our system, we used a reporter virus capable of only single-round infection, and the infection was measured by GFP expression by individual cells. This gave us the advantage of detecting exactly what proportions of the resting T cells were infected and following the detailed kinetics of viral gene expression. Choi et al. found the stimulating effects of endothelial cells only in EC+ cocultures, but in our system, generally speaking, any effect that was observed for EC+ cocultures was also observed in EC− cocultures in a lower magnitude. In addition, Choi found increased infection rates in memory T cells stimulated with EC+ but not in naive T cells. However, even though in our system memory T cells stimulated by EC had a much higher infection rate than naive T cells stimulated by EC, we were able to detect a difference in infection rates between naive T cells stimulated with EC and these cells without EC stimulation. This again was due to the increased sensitivity of our system.
Consistent with the results of Choi et al., we observed slower kinetics of viral infection in resting T cells stimulated by EC than in CD3/28-activated T cells (Fig. 1C). For EC-stimulated T cells, viral gene expression peaked after day 5 postinfection, whereas for CD3/28-activated T cells, the peak was at day 2 to 3. The slower kinetics probably reflect the fact that EC-stimulated T cells were largely still in a resting state in which key events such as reverse transcription occur slowly (18). More interestingly, in EC-stimulated cells, the proportion of GFP+ T cells was stable after day 5 or 6 postinfection, whereas in CD3/28-activated T cells, there was a decline of GFP+ cells after day 2 to 3. The decline was probably due to cell death, as infected activated T cells seemed to die more rapidly than uninfected activated T cells (data not shown). Generally speaking, after infection, there was substantially more cell death in CD3/28-activated cultures than in EC-stimulated cultures, particularly in T cells stimulated by EC− (Fig. 1D). This is probably due to the fact that nonproliferating T cells are less susceptible to viral cytopathic effects, such as Vpr-induced arrest of the cell cycle at G2 and the subsequent induction of apoptosis (41, 42). Since EC-stimulated T cells mostly remained in a resting state and in cell cycle stage G0/G1a (Fig. 2 and 3), they probably survived better than activated T cells. If this proves to be true in vivo, then EC would be a significant contributor to viral propagation in the host because EC stimulation allows resting T cells to be infected while still maintaining their viability.
Our data also demonstrated that resting CD4+ T cells did not have to be fully activated, nor did they have to proliferate or even be in cell cycle, to support productive HIV infection. In our study, resting T cells stimulated by EC displayed a resting phenotype, with only a small percentage of cells expressing any activation makers (Fig. 2), and they were mostly in cell cycle stage G0/G1a (Fig. 3). The barrier to proviral integration in resting T cells may be overcome by some degree of activation that is short of full immune activation through the T cell receptor, as long as some threshold is reached. Memory T cells, particularly effector memory T cells, are likely closer to the threshold than naive T cells, as the highest level of productive infection was achieved in effector memory populations (Fig. 4). Soluble factors have been in several studies to provide such partial activation. These include the proinflammatory cytokines IL-2, IL-4, IL-7, and IL-15 (30) and chemokines CCL19 and CCL21 (31). In our study, we found no correlation between infection rates of EC stimulated resting T cells and levels of cytokines IL-7 and IL-15 or chemokines CCL19 and CCL21 in the supernatants of the cultures (Fig. 6). This suggests that these cytokines and chemokines are not involved in EC stimulation of resting T cells. Choi et al. (32, 33) found that the interaction between EC and T cells involved CD2-CD58 interactions. We have previously shown that CD2-CD58 interactions are involved in activation of macaque resting CD4+ T cells by Epstein-Barr virus (EBV)-transformed human B cells (43). In future studies, we will explore other soluble factors and cell signaling molecules as potential mechanisms for subactivation stimulation by endothelial cells.
The most significant and interesting result from this study was the finding that EC stimulation of resting CD4+ T cells induced latent infection, most prominently in EC− cocultures (Fig. 5A and B). EC+ coculture also induced a statistically significant level of latency, though it was lower than with EC− coculture. This was most likely because there were more activated T cells (5 to 8%) in EC+ cocultures, and activated T cells were more prone to productive infection and infection-induced cell death. In fact, we often observed a moderate decline of GFP+ cells in EC+ cocultures from day 6 to day 8 postinfection (Fig. 1C). Preferential death of infected cells was probably also the reason why activated T cells did not have significant levels of latency. Endothelial cells line the interior of blood and lymphatic vessels in the lymphoid tissues and have frequent interactions with T cells. While activated T cells would most likely die after viral infection, EC-stimulated resting T cells could be readily infected (up to 40%, Fig. 6A), and in some cases (up to 1.5% [Fig. 5B]), latent infection could be established, allowing the cells to escape the cytopathic effect of the virus and killing by CD8+ T cells. Thus, our results suggest that EC may be involved in the establishment of a latent reservoir for HIV in CD4+ resting T cells in vivo.
Finally, even though EC stimulation induced both productive and latent infections in resting CD4+ T cells, it failed to reactivate latent virus. In other words, EC stimulation seemed to facilitate viral integration and viral expression in resting CD4+ T cells during de novo infection, but not expression of virus from already integrated provirus in resting T cells (Fig. 5C and D). This indicates that the mechanisms involved in the restriction of HIV infection of resting T cells (such as SAMHD1 [44, 45]) are different than those involved in the maintenance of latency. Since EC stimulation must have provided for the activation of essential transcription factors needed for productive infection of resting T cells, latency may be maintained through mechanisms other than unavailability of these factors. Similar arguments could be made regarding nucleotide levels. Mechanisms such as transcriptional interference, DNA methylation, or repressive histone modifications were implicated in maintenance of latency (46–48), and they may not be overcome by EC stimulation.
In summary, we found that EC had a significant impact on direct infection of HIV-1 in resting CD4+ T cells, facilitating both productive and latent infection. Direct infection of resting T cells stimulated by EC has significant implications in vivo because there are many more resting T cells in vivo than activated T cells, and if HIV infection does not require T cell activation, there would be many more potential target cells. Moreover, endothelial cells line the blood and lymphatic vessels in the lymphoid tissues and have frequent interactions with T cells in vivo. EC interactions with T cells in vivo may explain how resting CD4+ T cells are productively infected in lymphoid tissues. We also showed for the first time that EC-stimulated resting T cells can harbor latent virus and that infected resting T cells stimulated by EC had higher viability than infected activated T cells. These results suggest a new mechanism for latency formation in resting CD4+ T cells in vivo. Although latency may be established when activated CD4+ T cells revert back to a resting state, infection and survival of EC-stimulated resting CD4+ T cells may provide an additional route. Further understanding of the interaction between EC and T cells will be highly beneficial to our efforts to understand and eliminate latent HIV.
ACKNOWLEDGMENTS
We thank Mary Dekker, John Morris, and Jonas Lopez for excellent technical assistance, Hao Zhang and Louis King for flow cytometric sorting, Joel Blankson for patient contact, Lin Shen, Shan Liang, Jason Dinoso, and Sifei Xing for providing plasmids, virus, and cells, Jun Lai and Lori Keen for managerial assistance, and Jaehyuk Choi and Jordan S. Pober for helpful discussions on endothelial cells. We also thank the HIV− donors and HIV+ patients.
This study was supported by NIH grants 43222 to R.F.S. and AI096991 to A.S., the Howard Hughes Medical Institute, and Calvin College.
Footnotes
Published ahead of print 3 July 2013
REFERENCES
- 1.Gulick RM, Mellors JW, Havlir D, Eron JJ, Gonzalez C, McMahon D, Richman DD, Valentine FT, Jonas L, Meibohm A, Emini EA, Chodakewitz JA. 1997. Treatment with indinavir, zidovudine, and lamivudine in adults with human immunodeficiency virus infection and prior antiretroviral therapy. N. Engl. J. Med. 337:734–739 [DOI] [PubMed] [Google Scholar]
- 2.Hammer SM, Squires KE, Hughes MD, Grimes JM, Demeter LM, Currier JS, Eron JJ, Jr, Feinberg JE, Balfour HH, Jr, Deyton LR, Chodakewitz JA, Fischl MA. 1997. A controlled trial of two nucleoside analogues plus indinavir in persons with human immunodeficiency virus infection and CD4 cell counts of 200 per cubic millimeter or less. AIDS Clinical Trials Group 320 Study Team. N. Engl. J. Med. 337:725–733 [DOI] [PubMed] [Google Scholar]
- 3.Perelson AS, Essunger P, Cao Y, Vesanen M, Hurley A, Saksela K, Markowitz M, Ho DD. 1997. Decay characteristics of HIV-1-infected compartments during combination therapy. Nature 387:188–191 [DOI] [PubMed] [Google Scholar]
- 4.Davey RT, Jr, Bhat N, Yoder C, Chun TW, Metcalf JA, Dewar R, Natarajan V, Lempicki RA, Adelsberger JW, Miller KD, Kovacs JA, Polis MA, Walker RE, Falloon J, Masur H, Gee D, Baseler M, Dimitrov DS, Fauci AS, Lane HC. 1999. HIV-1 and T cell dynamics after interruption of highly active antiretroviral therapy (HAART) in patients with a history of sustained viral suppression. Proc. Natl. Acad. Sci. U. S. A. 96:15109–15114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alexaki A, Liu Y, Wigdahl B. 2008. Cellular reservoirs of HIV-1 and their role in viral persistence. Curr. HIV Res. 6:388–400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chun TW, Stuyver L, Mizell SB, Ehler LA, Mican JA, Baseler M, Lloyd AL, Nowak MA, Fauci AS. 1997. Presence of an inducible HIV-1 latent reservoir during highly active antiretroviral therapy. Proc. Natl. Acad. Sci. U. S. A. 94:13193–13197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Finzi D, Hermankova M, Pierson T, Carruth LM, Buck C, Chaisson RE, Quinn TC, Chadwick K, Margolick J, Brookmeyer R, Gallant J, Markowitz M, Ho DD, Richman DD, Siliciano RF. 1997. Identification of a reservoir for HIV-1 in patients on highly active antiretroviral therapy. Science 278:1295–1300 [DOI] [PubMed] [Google Scholar]
- 8.Wong JK, Hezareh M, Gunthard HF, Havlir DV, Ignacio CC, Spina CA, Richman DD. 1997. Recovery of replication-competent HIV despite prolonged suppression of plasma viremia. Science 278:1291–1295 [DOI] [PubMed] [Google Scholar]
- 9.Lassen KG, Ramyar KX, Bailey JR, Zhou Y, Siliciano RF. 2006. Nuclear retention of multiply spliced HIV-1 RNA in resting CD4+ T cells. PLoS Pathog. 2:e68. 10.1371/journal.ppat.0020068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Siliciano JD, Kajdas J, Finzi D, Quinn TC, Chadwick K, Margolick JB, Kovacs C, Gange SJ, Siliciano RF. 2003. Long-term follow-up studies confirm the stability of the latent reservoir for HIV-1 in resting CD4+ T cells. Nat. Med. 9:727–728 [DOI] [PubMed] [Google Scholar]
- 11.Williams SA, Greene WC. 2007. Regulation of HIV-1 latency by T-cell activation. Cytokine 39:63–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chun TW, Engel D, Mizell SB, Ehler LA, Fauci AS. 1998. Induction of HIV-1 replication in latently infected CD4+ T cells using a combination of cytokines. J. Exp. Med. 188:83–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Korin YD, Zack JA. 1998. Progression to the G1b phase of the cell cycle is required for completion of human immunodeficiency virus type 1 reverse transcription in T cells. J. Virol. 72:3161–3168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Spina CA, Guatelli JC, Richman DD. 1995. Establishment of a stable, inducible form of human immunodeficiency virus type 1 DNA in quiescent CD4 lymphocytes in vitro. J. Virol. 69:2977–2988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stevenson M, Stanwick TL, Dempsey MP, Lamonica CA. 1990. HIV-1 replication is controlled at the level of T cell activation and proviral integration. EMBO J. 9:1551–1560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zack JA, Arrigo SJ, Weitsman SR, Go AS, Haislip A, Chen IS. 1990. HIV-1 entry into quiescent primary lymphocytes: molecular analysis reveals a labile, latent viral structure. Cell 61:213–222 [DOI] [PubMed] [Google Scholar]
- 17.Zack JA, Haislip AM, Krogstad P, Chen IS. 1992. Incompletely reverse-transcribed human immunodeficiency virus type 1 genomes in quiescent cells can function as intermediates in the retroviral life cycle. J. Virol. 66:1717–1725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pierson TC, Zhou Y, Kieffer TL, Ruff CT, Buck C, Siliciano RF. 2002. Molecular characterization of preintegration latency in human immunodeficiency virus type 1 infection. J. Virol. 76:8518–8531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhou Y, Zhang H, Siliciano JD, Siliciano RF. 2005. Kinetics of human immunodeficiency virus type 1 decay following entry into resting CD4+ T cells. J. Virol. 79:2199–2210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chun TW, Carruth L, Finzi D, Shen X, DiGiuseppe JA, Taylor H, Hermankova M, Chadwick K, Margolick J, Quinn TC, Kuo YH, Brookmeyer R, Zeiger MA, Barditch-Crovo P, Siliciano RF. 1997. Quantification of latent tissue reservoirs and total body viral load in HIV-1 infection. Nature 387:183–188 [DOI] [PubMed] [Google Scholar]
- 21.Lassen K, Han Y, Zhou Y, Siliciano J, Siliciano RF. 2004. The multifactorial nature of HIV-1 latency. Trends Mol. Med. 10:525–531 [DOI] [PubMed] [Google Scholar]
- 22.Eckstein DA, Penn ML, Korin YD, Scripture-Adams DD, Zack JA, Kreisberg JF, Roederer M, Sherman MP, Chin PS, Goldsmith MA. 2001. HIV-1 actively replicates in naive CD4(+) T cells residing within human lymphoid tissues. Immunity 15:671–682 [DOI] [PubMed] [Google Scholar]
- 23.Li Q, Duan L, Estes JD, Ma ZM, Rourke T, Wang Y, Reilly C, Carlis J, Miller CJ, Haase AT. 2005. Peak SIV replication in resting memory CD4+ T cells depletes gut lamina propria CD4+ T cells. Nature 434:1148–1152 [DOI] [PubMed] [Google Scholar]
- 24.Nishimura Y, Brown CR, Mattapallil JJ, Igarashi T, Buckler-White A, Lafont BA, Hirsch VM, Roederer M, Martin MA. 2005. Resting naive CD4+ T cells are massively infected and eliminated by X4-tropic simian-human immunodeficiency viruses in macaques. Proc. Natl. Acad. Sci. U. S. A. 102:8000–8005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ostrowski MA, Chun TW, Justement SJ, Motola I, Spinelli MA, Adelsberger J, Ehler LA, Mizell SB, Hallahan CW, Fauci AS. 1999. Both memory and CD45RA+/CD62L+ naive CD4(+) T cells are infected in human immunodeficiency virus type 1-infected individuals. J. Virol. 73:6430–6435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wightman F, Solomon A, Khoury G, Green JA, Gray L, Gorry PR, Ho YS, Saksena NK, Hoy J, Crowe SM, Cameron PU, Lewin SR. 2010. Both CD31(+) and CD31 naive CD4(+) T cells are persistent HIV type 1-infected reservoirs in individuals receiving antiretroviral therapy. J. Infect. Dis. 202:1738–1748 [DOI] [PubMed] [Google Scholar]
- 27.Zhang Z, Schuler T, Zupancic M, Wietgrefe S, Staskus KA, Reimann KA, Reinhart TA, Rogan M, Cavert W, Miller CJ, Veazey RS, Notermans D, Little S, Danner SA, Richman DD, Havlir D, Wong J, Jordan HL, Schacker TW, Racz P, Tenner-Racz K, Letvin NL, Wolinsky S, Haase AT. 1999. Sexual transmission and propagation of SIV and HIV in resting and activated CD4+ T cells. Science 286:1353–1357 [DOI] [PubMed] [Google Scholar]
- 28.Kreisberg JF, Yonemoto W, Greene WC. 2006. Endogenous factors enhance HIV infection of tissue naive CD4 T cells by stimulating high molecular mass APOBEC3G complex formation. J. Exp. Med. 203:865–870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kinter A, Moorthy A, Jackson R, Fauci AS. 2003. Productive HIV infection of resting CD4+ T cells: role of lymphoid tissue microenvironment and effect of immunomodulating agents. AIDS Res. Hum. Retroviruses 19:847–856 [DOI] [PubMed] [Google Scholar]
- 30.Unutmaz D, KewalRamani VN, Marmon S, Littman DR. 1999. Cytokine signals are sufficient for HIV-1 infection of resting human T lymphocytes. J. Exp. Med. 189:1735–1746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Saleh S, Solomon A, Wightman F, Xhilaga M, Cameron PU, Lewin SR. 2007. CCR7 ligands CCL19 and CCL21 increase permissiveness of resting memory CD4+ T cells to HIV-1 infection: a novel model of HIV-1 latency. Blood 110:4161–4164 [DOI] [PubMed] [Google Scholar]
- 32.Choi J, Walker J, Boichuk S, Kirkiles-Smith N, Torpey N, Pober JS, Alexander L. 2005. Human endothelial cells enhance human immunodeficiency virus type 1 replication in CD4+ T cells in a Nef-dependent manner in vitro and in vivo. J. Virol. 79:264–276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Choi J, Walker J, Talbert-Slagle K, Wright P, Pober JS, Alexander L. 2005. Endothelial cells promote human immunodeficiency virus replication in nondividing memory T cells via Nef-, Vpr-, and T-cell receptor-dependent activation of NFAT. J. Virol. 79:11194–11204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mochizuki H, Schwartz JP, Tanaka K, Brady RO, Reiser J. 1998. High-titer human immunodeficiency virus type 1-based vector systems for gene delivery into nondividing cells. J. Virol. 72:8873–8883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang HC, Xing S, Shan L, O'Connell K, Dinoso J, Shen A, Zhou Y, Shrum CK, Han Y, Liu JO, Zhang H, Margolick JB, Siliciano RF. 2009. Small-molecule screening using a human primary cell model of HIV latency identifies compounds that reverse latency without cellular activation. J. Clin. Invest. 119:3473–3486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schmid I, Cole SW, Korin YD, Zack JA, Giorgi JV. 2000. Detection of cell cycle subcompartments by flow cytometric estimation of DNA-RNA content in combination with dual-color immunofluorescence. Cytometry 39:108–116 [DOI] [PubMed] [Google Scholar]
- 37.Zhang H, Zhou Y, Alcock C, Kiefer T, Monie D, Siliciano J, Li Q, Pham P, Cofrancesco J, Persaud D, Siliciano RF. 2004. Novel single-cell-level phenotypic assay for residual drug susceptibility and reduced replication capacity of drug-resistant human immunodeficiency virus type 1. J. Virol. 78:1718–1729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Choi J, Enis DR, Koh KP, Shiao SL, Pober JS. 2004. T lymphocyte-endothelial cell interactions. Annu. Rev. Immunol. 22:683–709 [DOI] [PubMed] [Google Scholar]
- 39.Sallusto F, Geginat J, Lanzavecchia A. 2004. Central memory and effector memory T cell subsets: function, generation, and maintenance. Annu. Rev. Immunol. 22:745–763 [DOI] [PubMed] [Google Scholar]
- 40.Siliciano JD, Siliciano RF. 2005. Enhanced culture assay for detection and quantitation of latently infected, resting CD4+ T-cells carrying replication-competent virus in HIV-1-infected individuals. Methods Mol. Biol. 304:3–15 [DOI] [PubMed] [Google Scholar]
- 41.Jowett JB, Planelles V, Poon B, Shah NP, Chen ML, Chen IS. 1995. The human immunodeficiency virus type 1 vpr gene arrests infected T cells in the G2 + M phase of the cell cycle. J. Virol. 69:6304–6313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stewart SA, Poon B, Jowett JB, Chen IS. 1997. Human immunodeficiency virus type 1 Vpr induces apoptosis following cell cycle arrest. J. Virol. 71:5579–5592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shen A, Yang HC, Zhou Y, Chase AJ, Boyer JD, Zhang H, Margolick JB, Zink MC, Clements JE, Siliciano RF. 2007. Novel pathway for induction of latent virus from resting CD4(+) T cells in the simian immunodeficiency virus/macaque model of human immunodeficiency virus type 1 latency. J. Virol. 81:1660–1670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Baldauf HM, Pan X, Erikson E, Schmidt S, Daddacha W, Burggraf M, Schenkova K, Ambiel I, Wabnitz G, Gramberg T, Panitz S, Flory E, Landau NR, Sertel S, Rutsch F, Lasitschka F, Kim B, Konig R, Fackler OT, Keppler OT. 2012. SAMHD1 restricts HIV-1 infection in resting CD4(+) T cells. Nat. Med. 18:1682–1687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Descours B, Cribier A, Chable-Bessia C, Ayinde D, Rice G, Crow Y, Yatim A, Schwartz O, Laguette N, Benkirane M. 2012. SAMHD1 restricts HIV-1 reverse transcription in quiescent CD4(+) T-cells. Retrovirology 9:87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Blazkova J, Trejbalova K, Gondois-Rey F, Halfon P, Philibert P, Guiguen A, Verdin E, Olive D, Van Lint C, Hejnar J, Hirsch I. 2009. CpG methylation controls reactivation of HIV from latency. PLoS Pathog. 5:e1000554. 10.1371/journal.ppat.1000554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kauder SE, Bosque A, Lindqvist A, Planelles V, Verdin E. 2009. Epigenetic regulation of HIV-1 latency by cytosine methylation. PLoS Pathog. 5:e1000495. 10.1371/journal.ppat.1000495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shan L, Yang HC, Rabi SA, Bravo HC, Shroff NS, Irizarry RA, Zhang H, Margolick JB, Siliciano JD, Siliciano RF. 2011. Influence of host gene transcription level and orientation on HIV-1 latency in a primary-cell model. J. Virol. 85:5384–5393 [DOI] [PMC free article] [PubMed] [Google Scholar]