

Abstract
Hepatitis C virus (HCV) infection is a major cause of severe liver disease. Interferon (IFN)/ribavirin treatment remains the standard therapeutic regimen for HCV infection in most countries. IFN-stimulated genes are believed to contribute to antiviral effects. However, emerging evidence suggests that microRNAs (miRNAs), a class of noncoding small RNAs, are involved in the control of viral infection. Here, we systematically profiled the hepatocyte expression of a set of 750 miRNAs in response to alpha interferon (IFN-α) and interleukin-28B (IL-28B) treatments. The anti-HCV activity of differentially expressed miRNAs was evaluated using cell culture-derived HCV in vitro. The results demonstrate that let-7b had a significant anti-HCV effect by inhibiting HCV replication and viral protein translation in human hepatoma cells. In particular, we show that the inhibition of let-7b attenuated the anti-HCV effects of IFN-α and IL-28B. Furthermore, we show that the host factor insulin-like growth factor 2 mRNA-binding protein 1 (IGF2BP1) is a target of let-7b. IGF2BP1 was required for HCV replication, and its expression was downregulated by IFN-α and IL-28B. Deletion of the wild-type seed region of let-7b abolished its antiviral activity. Finally, we demonstrate that other let-7 family miRNAs were able to inhibit HCV and to suppress IGF2BP1 expression. In conclusion, we provide an example of a host miRNA regulated by type I and type III IFNs that inhibits HCV replication and infectivity by targeting host targets. These results highlight the important role of miRNAs in the host antiviral immune response and provide a novel candidate for anti-HCV therapy.
INTRODUCTION
The hepatitis C virus (HCV), which affects more than 2.35% of the world's population (1), is a major pathogen in liver disease. The current standard antiviral treatment for HCV infection is a combination of peginterferon alfa, ribavirin, and one of protease inhibitors (2). Recently, an increasing number of directly acting antivirals (DAAs) have been identified that target HCV NS3/4A protease (3, 4), NS5B polymerase (5), NS4B (6), or NS5A (7). The utilization of DAAs marks a milestone in anti-HCV therapy.
Interferons (IFNs) are integral to innate mammalian antiviral immunity. IFNs bind to their receptors on the cell membrane, activate the JAK/STAT signaling pathway, and induce IFN-stimulated genes (ISGs). Several type I IFN-regulated gene products, such as protein kinase R, the 2′-5′ oligoadenylate synthetase/RNase L system, the adenosine deaminase ADAR1, and the Mx GTPases, are important effectors of these cytokines' antiviral properties (8). Adding to what is known about protein-encoding genes, an emerging theme is that noncoding genes play an important role in IFN-dependent antiviral activity (9).
MicroRNAs (miRNAs) are endogenous, noncoding RNA molecules ∼22 nucleotides in length that are involved in the posttranscriptional regulation of gene expression by a process termed RNA interference (RNAi) (10). Since the discovery of the first miRNA, lin-4, in 1993 (11), more than 2,000 putative miRNAs encoded by the human genome have been catalogued (miRBase v19). miRNAs play key roles in diverse regulatory pathways, with the potential for one miRNA to regulate numerous mRNAs and for one mRNA to be targeted by multiple miRNAs (12). Of particular interest, the 6- to 8-bp “seed region” at the 5′ end of the miRNA-mRNA heteroduplex usually mediates translational repression of the target mRNA (13).
miRNA-mediated gene silencing has been shown to play an important role in viral pathogenesis by modulating viral replication through the targeting of viral sequences or the regulation of host genes. During HIV-1 infection, a cluster of cellular miRNAs were shown to directly target the 3′-untranslated regions (3′UTRs) of HIV-1 mRNAs, leading to their translational suppression (14). Three other human miRNAs—miR-323, miR-491, and miR-654—inhibit replication of the H1N1 influenza A virus through binding to the viral PB1 gene (15). A hepatocyte-specific miRNA, miR-122, was identified as a positive regulatory factor for HCV replication based on its ability to bind the HCV genome (16). The miRNA miR-141 was shown to promote efficient HCV replication by suppressing DLC-1 (17). The IFN-β-induced miRNAs—miR-196, miR-296, miR-351, miR-431, and miR-448—have sequences partially complementary to HCV genome and result in the suppression of HCV replication (9).
In addition to the role played by type I IFNs, the recently discovered type III IFNs have been strongly associated with HCV infection and treatment (18). Type III IFNs include interleukin-29 (IL-29), IL-28A, and IL-28B (also known as IFN-λ1, IFN-λ2, and IFN-λ3, respectively). Generally, type III IFNs upregulate ISGs with a different kinetic profile than type I IFNs and induce a distinct set of genes, which might account for the functional differences between type I and III IFNs (19). Our previous study showed that IL-28B exhibits efficient antiviral activity against HCV in vitro with fewer adverse effects than IFN-α due to its restricted cell tropism, thus representing a novel antiviral candidate for improved treatment of HCV-infected patients (20). However, the miRNAs regulated by type III IFN are largely unidentified.
In the present study, we systematically analyzed and compared host miRNA expression profiles under IFN-α or IL-28B treatment. The anti-HCV activities of differentially expressed miRNAs were measured in cells transfected with miRNA mimics or inhibitors. Interestingly, let-7 family miRNAs demonstrated strong anti-HCV activity, and the detailed mechanisms of that activity were characterized. Our findings suggest that miRNAs such as let-7 are exceptionally promising candidates for therapeutic approaches to treat HCV infection.
MATERIALS AND METHODS
Cells and reagents.
The human hepatoma cell line Huh7 was from Apath, Inc. (Brooklyn, NY), and Huh7.5.1 was provided by Francis Chisari (Scripps Research Institute, La Jolla, CA). The 2−3+ cell line harboring the HCV genotype 1b replicon genome was provided by Stanley Lemon (University of Texas Medical Branch, Galveston, TX). The cell culture was performed as described previously (20). The primary fetal liver cells (PFLCs) were prepared as described previously (21). Recombinant human IL-28B was produced in Pichia pastoris, and its activity was evaluated (20). Cyclosporine, 2′-C-methylcytidine, and BMS-790052 were purchased from Sigma (St. Louis, MO) or synthesized. Antibodies were obtained from Sigma (β-actin), Thermo Scientific (HCV Core), and Cell Signaling Technology (IGF2BP1). Horseradish peroxidase-conjugated and DyLight-conjugated secondary antibodies are purchased from Jackson ImmunoResearch Laboratories (West Grove, PA).
TaqMan MicroRNA profiling.
The human hepatoma cell line Huh7 was cultured in 100-mm plates and treated with 1 ng of IFNa2b/ml, 12 ng of IL-28B/ml, or phosphate-buffered saline (PBS) for 12 h. The PBS treatment served as a negative control. The concentrations of the IFNs were higher than their 90% inhibitory concentrations, which had been measured in our laboratory (20). The IFN-stimulated cells and the PBS-treated cells were harvested in TRIzol reagent, and the total RNA was isolated according to the manufacturer's instructions. The purification of total RNA was necessary and was performed using the mirVana miRNA isolation kit (Ambion, Austin, TX). The miRNA expression profiling was analyzed with TaqMan Array Human MicroRNA A+B Cards Set v3.0 on an Applied Biosystems 7900HT Fast Real-Time PCR system by CapitalBio (Beijing, China). The miRNA nomenclature used here is based on miRBase v14. The miRNAs that were differentially expressed in IFN-treated cells compared to PBS-treated cells were identified using DataAssist 2.0 software with the relevant fold change set at >2-fold.
Bioinformatics.
The miRNAs differentially expressed (>10.0-fold) after IFN treatment were subjected to target prediction and pathway enrichment analyses. The potential miRNA-mRNA target interactions were predicted using miRanda (http://www.microrna.org/), miRDB (http://mirdb.org/miRDB/), PITA (http://genie.weizmann.ac.il/pubs/mir07/mir07_prediction.html), and Targetscan (http://www.targetscan.org/). The predicted target mRNAs simultaneously identified by all four algorithms were selected for analysis in a pathway enrichment assay with Molecule Annotation System 3.0 (MAS: http://bioinfo.capitalbio.com/mas3/) and the KEGG database (http://www.genome.jp/kegg/pathway.html). Hierarchical clustering was performed with Gene Cluster 3.0 and Eisen's TreeView (Stanford University, Palo Alto, CA). Clustering data were generated in the form of the negative log of the P values. A P value cutoff of 0.005 was used to identify significantly enriched pathways.
RNAs and RNA transfection.
The mimics and inhibitors for Homo sapiens miRNAs, the negative control mimics and inhibitors (cel-miR-67-3p), and the mimic for customized mutant has-let-7b were obtained from RiboBio (Guangzhou, China). All of the small interfering RNAs (siRNA), including the scrambled negative control siRNA, were purchased from RiboBio. The sequences for the CD81 siRNA (siCD81) and the IGF2BP1 siRNAs (siIGF2BP1) were as follows: siCD81, 5′-GGACCAGAUCGCCAAGGAU-dTdT-3′; siIGF2BP1-#1, 5′-CGGGAAAGUAGAAUUACAA-dTdT-3′; siIGF2BP1-#2, 5′-CGAAACACCUGACUCCAAA-dTdT-3′; and siIGF2BP1-#3, 5′-CCUGAAGAAGGUAGAGCAA-dTdT-3′. Transfection of miRNA or siRNA was performed using Lipofectamine RNAiMAX (Invitrogen) according to the manufacturer's protocol. Transfection of in vitro-synthesized RNA was performed using DMIRE-C (Invitrogen) according to the manufacturer's instructions.
In vitro transcription.
The linear pFK-Luc-Jc1 plasmid with a T7 promoter was used for in vitro transcription of Jc1-Luc HCV RNA. The plasmid containing the HCV 5′UTR-directed luciferase gene used for HCV internal ribosome entry site (IRES)-directed luciferase reporter assay was derived from the plasmid pFK-Luc-Jc1. RNAs were transcribed with a RiboMAX large-scale RNA production system (Promega, Madison, WI) according to the manufacturer's instructions.
Reporter constructs.
The pmirGLO plasmid vector obtained from Promega contains cDNA sequences encoding the firefly luciferase (F-luc) reporter gene and the Renilla luciferase (R-luc) gene, which acts as an internal control reporter. For the luciferase reporter assay of let-7b targeting insulin-like growth factor 2 mRNA binding protein 1 (IGF2BP1), the pmirGLO-IGF2BP1-3′UTR-Wild and pmirGLO-IGF2BP1-3′UTR-Mutation luciferase reporter constructs contained, respectively, fragments with bp 3233 to 5933 from the wild-type IGF2BP1-3′UTR and the same 2,701-bp fragment mutated to generate nucleotide mismatches in the “seed region” matching let-7b. The wild-type 2,701-bp fragment contains three putative let-7b seed match sites (two exact matches to positions 1 to 8 of let-7b and one exact match to positions 2 to 8 of let-7b). Site-directed mutagenesis was performed using overlap-extension by PCR. The pmirGLO-3′UTR-Mutation#1 construct contains a mutation at match site 1, as depicted in Fig. 5A (right). pmirGLO-3′UTR-Mutation#1+2 contains mutations at sites 1 and 2, and pmirGLO-3′UTR-Mutation#1+2+3 harbors mutations at sites 1, 2, and 3. The successful subcloning of each construct was confirmed by restriction enzyme digestion and sequencing.
Fig 5.

Inhibition of HCV by let-7b via targeting cellular IGF2BP1. (A) Schematic of the seed region match between let-7b and the putative IGF2BP1 3′UTR. The mutation of five nucleotides in the seed match is shown (left). The positions of three seed match sites for let-7b and the IGF2BP1 3′UTR in the luciferase reporter construct pmirGLO-IGF2BP1-3′UTR are indicated (right). (B) Huh7.5.1 cells were transfected with 50 nM let-7b mimics or NC mimics and incubated for 1 to 3 days as indicated. The levels of endogenous IGF2BP1 mRNA were then quantified using real-time qRT-PCR. (C) Huh7.5.1 cells were transfected with differing concentration of let-7b mimics or NC mimics, incubated for 2 days, and harvested for Western blotting of IGF2BP1 protein levels. (D and E) Huh7.5.1 cells in 48-well plates were cotransfected with 50 nM the indicated miRNA mimics and 0.1 μg of the indicated reporter constructs, respectively (the pmirGLO-IGF2BP1-3′UTR/empty construct contains or does not contain a 2,701-bp fragment from the wild-type IGF2BP1 3′UTR in the downstream of Firefly luciferase gene [D]; the pmirGLO-3′UTR-Wild and pmirGLO-3′UTR-Mutation constructs contain wild-type and mutated IGF2BP1 3′UTR fragments as described in Materials and Methods and supplemental Materials and Methods [E]). Firefly and Renilla luciferase activities were measured at 24 h posttransfection. (F) Huh7.5.1 cells were transfected with 50 nM siRNAs as indicated, incubated for 2 days, and then infected with Jc1-Luc HCVcc (MOI = 0.1). Luciferase activity was measured 2 days after infection (upper panel). In parallel, the transfected cells were harvested to evaluate their IGF2BP1 protein levels by Western blotting (lower panel). The data represent means ± the standard deviations of at least three independent experiments (*, P < 0.05, **, P < 0.01; ***, P < 0.001).
Luciferase reporter assays.
Huh7.5.1 cells were seeded into 48-well plates 1 day before transfection. The next day, 0.1 μg of construct was transfected into the cells in each well using Lipofectamine LTX (Invitrogen). At 6 h after DNA transfection, the cells were transfected with 50 nM miRNA mimics. The luciferase reporter activities were measured at 24 h after RNA transfection using a dual luciferase reporter assay system (Promega). The F-luc activity was normalized to the R-luc activity.
Real-time quantitative reverse transcription-PCR (qRT-PCR).
Total intracellular RNA was extracted using TRIzol reagent (Invitrogen) according to the manufacturer's instructions. The absolute quantification of HCV RNA replication has been described previously (20). For the relative quantification of IGF2BP1 mRNA, the primer pair 5′-CCTGCTGGCTCAGTATGGT-3′ and 5′-GACATTCACCACTGCCGTCTC-3′ was used as previously described (22). The expression of β-actin was measured using the primer set 5′-CCAACCGCGAGAAGATGA-3′ and 5′-CCAGAGGCGTACAGGGATAG-3′ and served as the endogenous control. The real-time quantifications of mRNAs were performed by using a QuantiFast SYBR green RT-PCR kit (Qiagen, Düsseldorf, Germany). For the relative quantification of miRNA, a mirVana miRNA isolation kit (Ambion) was used to isolate total RNA according to the manufacturer's protocol. The reverse transcription reaction and quantitative PCR were then carried out using miRNA analysis kits specific for each individual miRNA (Applied Biosystems, Foster City, CA) according to the manufacturer's protocol. The expression level of the U6 gene was used as the endogenous control. All of the quantifications were performed with an ABI Prism 7500 system (Applied Biosystems).
Western blotting.
Western blotting was performed according to the standard protocols of our laboratory, as reported elsewhere (20).
Production and infection of HCVcc and HCVpp.
The Jc1-Luc HCVcc production and infection have been described in detail previously (20). The production and infection procedures of HCV pseudoviral particles (HCVpp) and VSV-pseudotyped lentivirus (VSVpp) were described elsewhere (23, 24).
Indirect immunofluorescent staining.
Indirect immunofluorescent staining of HCVcc-infected Huh7.5.1 cells was performed with antibodies against the HCV Core protein as previously described (23). Nuclei were stained with DAPI (4′,6′-diamidino-2-phenylindole). Images were captured using a Leica TCS-SL confocal microscope.
Statistical analyses.
The values shown in graphs are presented as means ± the standard deviations of at least three independent experiments. A Student t test was used to analyze the difference between the means for two independent samples, and P values of <0.05 were considered statistically significant.
RESULTS
IFN-α and IL-28B regulate distinct miRNA expression profiles in hepatocytes.
To systematically elucidate the changes in host miRNA expression stimulated by type I or type III IFNs, we used high-throughput qRT-PCR technology to analyze the expression of 750 miRNAs in IFN-α-treated or IL-28B-treated Huh7 cells. The relative expression data for the analyzed miRNAs are summarized in Table S1 in the supplemental material. Hierarchical clustering analysis of the differentially expressed miRNAs suggested that treatment of cells with type I or type III IFN resulted in overlapping and distinct patterns of miRNA expression (Fig. 1A). Of the 160 miRNAs found to be differentially expressed (fold change > 2.0) in response to IFN-α or IL-28B treatment, 46 were similarly affected by the two treatments (Fig. 1B). Interestingly, IFN-α and IL-28B treatments demonstrated opposing effects on the expression of several miRNAs (has-miR-215, miR-503, miR-616, miR-641, and miR-1282), as shown in bold in Table S1 in the supplemental material. miRNAs function by modulating their mRNA targets. By target prediction and pathways enrichment analyses, potential pathways modulated by up- or downregulated miRNAs (fold change > 10.0) may mediate the antiviral and immunomodulatory effects of IFNs (Fig. 1C and D). The majority of these pathways were associated with cancer, cell growth and death, immunity, signal transduction, endocrine function, and cell-cell communication (Fig. 1C and D). Taking these findings together, the characterization of the IFN-α- and IL-28B-regulated host miRNA expression signatures provides a powerful tool for dissecting the novel mechanisms of IFN actions.
Fig 1.

miRNA expression profiles and pathways in IFN-α- and IL-28B-stimulated hepatocytes. (A) Unsupervised hierarchical clustering of the changes in miRNA expression correlates with IFN-α and IL-28B treatment in Huh7 cells. A pseudocolor scale outlines the expression levels represented in the heat map. Data with a fold change threshold of at least 2.0 are depicted in the heat map. The fold change data for differentially expressed miRNAs were transformed to a log scale before clustering. (B) Venn diagram of differentially expressed miRNAs (>2.0-fold). Significantly expressed miRNAs (>10.0-fold) during IFN treatment were subjected to target prediction and pathway enrichment analyses as described in Materials and Methods. All pathways were selected by using a P value cutoff of 0.005 and clustered into categories using pathway ontology in the KEGG pathway database. The pathways corresponding to upregulated (C) and downregulated (D) miRNAs are shown. Bars below the zero line depict the pathways that the implicated mRNAs were upregulated by downregulated miRNAs with IFN treatment, while bars above the zero line show pathways represented by downregulated mRNAs. The y axis indicates the significance of enrichment (in the form of the number of target mRNAs).
let-7b inhibits HCV infection.
To determine whether the differentially expressed miRNAs can regulate HCV infection, we performed a functional anti-HCV screen by transfecting Huh7.5.1 cells with the synthetic mimics of upregulated miRNAs or the inhibitors of downregulated miRNAs and then infected those cells with Jc1-Luc reporter virus. The miRNAs that were expressed differentially (fold change > 2.0) in response to either or both of IFN-α and IL-28B treatments were selected to perform the functional analysis for antivirus. The results for a total of 18 miRNAs with significant anti-HCV activity are summarized in Table 1. Notably, let-7b had the most potent anti-HCV activity. let-7b inhibited HCV infection in dose- and time-dependent manners (Fig. 2A and B). Here, the potential cytotoxicity at 75 nM in Fig. 2A was caused by miRNA transfection reagent (data not shown). A similar inhibitory effect on JFH-1 HCVcc infection was observed using immunofluorescent staining with an antibody against the HCV core protein on Huh7.5.1 cells transfected with let-7b mimics (Fig. 2C). In accordance with previous reports (20, 25), the suppression of endogenous miR-122 or the addition of IFN-α resulted in a marked decrease in HCV infection, and these conditions served as positive controls (Fig. 2C). The overexpression of let-7b and the suppression of miR-122 in the cells were confirmed by qRT-PCR (Fig. 2D). The effects of miRNA transfection on cell growth and viability were also determined. let-7b did not affect cell growth and viability (data not shown). When used in combination with the known HCV inhibitors cyclosporine A, 2′-C-methylcytidine, IFN-α, or IL-28B, let-7b overexpression produced additive effects on HCV inhibition (Fig. 2E). These findings indicated that let-7b is a negative regulator of HCV infection.
Table 1.
miRNAs with anti-HCVcc ability and their different expression in response to IFNs treatment
| Type of regulation and miRNAa | Fold change |
Mean antiviral abilityb (% of control) ± SD | Genome location(s) | Potential targets | |
|---|---|---|---|---|---|
| IFN-α2b | IL-28B | ||||
| Upregulation (mimics) | |||||
| has-miR-122-5p (PC) | –72.81 ± 21.3 | 18q21.31 | |||
| cel-miR-67-3p (NC) | –1.64 ± 12.18 | ||||
| hsa-let-7b | 2.1513 | 2.2533 | 82.38 ± 1.38 | 22q13.31 | HMGA2, IGF2BP1 |
| hsa-miR-143-3p | 2.0304 | 8.1919 | 65.81 ± 3.35 | 5q32 | SLC30A8, ABL2 |
| hsa-miR-301b | 4.1337 | 4.0949 | 52.41 ± 4.55 | 22q11.21 | MIER1, SLAIN1 |
| hsa-miR-181a-3p | 4.0368 | 4.0325 | 49.76 ± 7.86 | 1q32.1 | PIAS2, CFL2 |
| hsa-let-7f | 2.2799 | 1.7353 | 49.51 ± 18.24 | 9q22.32, Xp11.22 | HMGA2, IGF2BP1 |
| hsa-miR-499a-5p | 16.4561 | 1.0188 | 46.72 ± 9.26 | 20q11.22 | SOX6, SLC30A4 |
| hsa-miR-1267 | 0.9692 | 261561.2641 | 46.64 ± 6.89 | 13 | VGLL3, SHROOM2 |
| hsa-miR-582-3p | 2.0655 | 2.1033 | 45.42 ± 0.12 | 5q12.1 | EIF2S1, RREB1 |
| hsa-miR-145-5p | 1.037 | 2.0252 | 35.93 ± 15.72 | 5q32 | FAM108C1, FSCN1 |
| hsa-miR-181a-2-3p | 2.0333 | 2.0405 | 32.9 ± 16.2 | 9q33.3 | ARID4A, CUTC |
| hsa-miR-181c-3p | 1.0163 | 16.9214 | 32.74 ± 16.92 | 19p13.13 | FAM122B, CFL2 |
| Downregulation (inhibitors) | |||||
| has-miR-122-5p (PC) | 56.56 ± 4.11 | 18q21.31 | |||
| cel-miR-67-3p (NC) | –0.33 ± 8.53 | ||||
| hsa-miR-1225-3P | 0.0655 | 0.0657 | 80.1 ± 1.83 | 16p13.3 | AFF4, GSG1L |
| hsa-miR-518b | 0.1258 | 0.1261 | 79.01 ± 0.1 | 19q13.42 | EGR1, WDR1 |
| hsa-miR-449a | 0.4947 | 0.5015 | 77.63 ± 5.83 | 5q11.2 | HCN3, FAM76A |
| hsa-miR-1296 | 0.0156 | 0.9807 | 75.68 ± 8.07 | 10 | TBC1D25, HYOU1 |
| hsa-miR-184 | 0.4786 | 0.4798 | 63.44 ± 12.13 | 15q25.1 | EPB41L5, SF1 |
| hsa-miR-190b | 0.2525 | 0.2475 | 61.89 ± 14.48 | 1q21.3 | OTUD4, NEUROD1 |
| hsa-miR-212-3p | 0.1258 | 0.2532 | 56.26 ± 16.15 | 17p13.3 | TMEM106B, ZNF516 |
PC, positive control; NC, negative control.
Huh7.5.1 cells transfected with miRNA mimics (upregulation, 50 nM) or inhibitors (downregulation, 100 nM) were infected with Jc1-Luc HCVcc. The cells were cultured for 2 days to measure the intracellular luciferase activity. Reductions in HCVcc luciferase activity are given represented as the percentage of NC (n = 3). NC would be set to 0%.
Fig 2.

Characterization of the anti-HCV activity of let-7b. Huh7.5.1 cells were transfected with various concentrations of let-7b mimics and incubated for 2 days (A) or transfected with 50 nM let-7b mimics and incubated for different times (B), followed by infection with Jc1-Luc HCVcc (MOI = 0.1). The infected cells were cultured for an additional 2 days, and the luciferase activity was used to measure HCV propagation. NC, negative control mimics. Statistical analysis was performed to compare the HCV levels in let-7b-transfected and NC cells (*, P < 0.05; **, P < 0.01). (C) Huh7.5.1 cells were transfected with the indicated miRNAs (mimics, 50 nM; inhibitors, 100 nM) and cultured for 2 days, followed by infection with JFH1 HCVcc (MOI = 0.4). Huh7.5.1 cells were challenged with JFH1 HCVcc (MOI = 0.4) 1 day before IFN-α (0.5 ng/ml) treatments. After miRNA transfection or the addition of IFN-α, the cells were cultured for 2 additional days. An miR-122 inhibitor and IFN-α served as the positive controls. Immunofluorescent staining and confocal microscopy were performed using an anti-Core antibody and DAPI. (D) Detection of let-7b or miR-122 expressions in the indicated samples by real-time qRT-PCR was used to confirm the mimic and inhibitor transfections. (E) Huh7.5.1 cells were transfected with 50 nM let-7b mimics or NC mimics. After 2 days, the cells were infected with Jc1-Luc HCVcc (MOI = 0.1) 1 day before administering the indicated drug treatments. The cells were cultured 2 days longer in the presence of drugs, and luciferase activity was measured to assess HCV propagation. Error bars denote the standard deviation from the mean of three independent experiments. Statistical significance was analyzed using Student t test (*, P < 0.05; **, P < 0.01). For ease of comparison, all of the bar graph values were normalized to the mock-transfected control.
let-7b suppresses HCV translation and replication in hepatocytes.
To further delineate the individual steps in the HCV life cycle affected by let-7b, the effects of let-7b on viral entry, replication, translation, and release were evaluated. As shown in Fig. 3A, the overexpression of let-7b showed no effect on HCVpp entry into the hepatocytes compared to the positive control (siRNA targeting CD81 [siCD81]). The overexpression of let-7b in genotype 1b HCV replicon cells (2−3+), however, reduced both HCV RNA abundance and viral protein expression, similar to the effects of knocking down miR-122 (Fig. 3B and C). By using an HCV IRES-directed luciferase reporter assay, the effect of let-7b on viral translation was evaluated. As shown in Fig. 3D, transfection of let-7b decreased HCV IRES activity by ∼3-fold. In addition, after blocking HCV replication step by chemicals 2′C-Mec or BMS-790052, let-7b showed inhibitory effects on HCV early translation (Fig. 3E). Finally, transfection of let-7b in HCV-infected Huh7.5.1 cells suppressed the progeny virus infectivity in culture supernatants (Fig. 3F).
Fig 3.

let-7b inhibits HCV replication and translation. (A) Huh7.5.1 cells were transfected with the indicated miRNA mimics or siRNA (50 nM) and incubated for 2 days prior to infection with VSVpp (VSV-pseudotyped lentivirus) and HCVpp. A siRNA for CD81 (siCD81) was used as the positive control for HCV entry. (B) The 2−3+ cells were transfected with the indicated mimics and inhibitors and incubated for 3 days. The miR-122 inhibitor served as the positive control. Total cellular RNA was isolated using TRIzol reagent for real-time qRT-PCR analyses of HCV genomes. (C) Cell lysates from the samples described above were analyzed by Western blotting to determine HCV core protein levels. The detection of β-actin was used to control for sample loading. (D) HCV IRES-directed luciferase reporter assay. Huh7.5.1 cells were transfected with indicated miRNA mimics (50 nM) and incubated for 2 days. The cells were then transfected with in vitro-synthesized HCV IRES-directed luciferase mRNA. The luciferase activity was measured 1 day after mRNA transfection. (E) Huh7.5.1 cells transfected with miRNA mimics (50 nM) were transfected with in vitro-synthesized Jc1-Luc genomic RNA and treated with 2′C-Mec (10 μM) or BMS-790052 (1 nM) simultaneously. Luciferase activity was measured to assess HCV mRNA translation at the indicated time points. (F) Huh7.5.1 cells infected with Jc1-Luc HCVcc were transfected with mimics (50 nM) or inhibitors (100 nM) and incubated for 2 days. Supernatants from the transfected cells were used to inoculate naive Huh7.5.1 cells. After a 2-day incubation, luciferase activity in the inoculated cells was measured. A miR-122 inhibitor served as the positive control. The data represented means ± the SD (n = 3; *, P < 0.05; **, P < 0.01).
Downregulation of let-7b attenuates the antiviral activity of IFNs.
It was reported that miR-122, a liver-specific miRNA, was essential for HCV replication and that the upregulation of miR-122 reduced the IFN-β-dependent inhibition of HCV (9). To determine whether let-7b participates in the IFN-mediated inhibition of HCV, we transfected a let-7b inhibitor into Huh7.5.1 cells infected with Jc1-Luc HCVcc and then treated the cells with either IFN-α or IL-28B. As shown in Fig. 4A, IFN-α or IL-28B treatment of mock-transfected cells led to a significant reduction in HCV propagation. This reduction was not affected by transfection of the cells with a negative control inhibitor. However, transfecting the cells with a let-7b inhibitor rescued infection from the suppressive effect of IFN-α, altering it from ∼90% inhibition to ∼75% inhibition. Comparable results were observed in assays using IL-28B in place of IFN-α (Fig. 4A). It has been reported that IFN-β treatment resulted in a transient but pronounced downmodulation of miR-122 (9). As a positive control, transfection of cells with a miR-122 mimic also antagonized the anti-HCV effects of IFN-α or IL-28B in a pattern similar to that observed with let-7b inhibition (Fig. 4B). These data suggest that the induction of let-7b might contribute to the antiviral effects of IFNs.
Fig 4.

Inhibition of let-7b attenuates IFN-α- and IL-28B-mediated anti-HCV activity. Huh7.5.1 cells infected with Jc1-Luc HCVcc were transfected with either a let-7b inhibitor (A) or an miR-122 mimic (B) 2 days prior to treatment with IFN-α (5 ng/ml) or IL-28B (50 ng/ml). The luciferase activity was measured 1 day after IFN treatment (means ± the standard deviations of three independent experiments; P values are from Student t tests; NS, no significance).
let-7b inhibits HCV by targeting host factor IGF2BP1.
During the course of our study, Cheng et al. reported that let-7b inhibited HCV infectivity by interacting with the HCV genome (26). Here, we investigated whether there was another mechanism contributing to let-7b's anti-HCV activity. An online search of the TargetScan 5.2, Diana-MicroT, MicroRNA.org, TargetMiner, and miRDB databases revealed at least five putative let-7b seed match sites present in the 3′UTR of IGF2BP1 mRNA (Fig. 5A, left). IGF2BP1 is one of the predicted targets of let-7b. IGF2BP1 was identified in a HCV IRES-mediated translation complex and enhanced HCV translation (27). To experimentally verify that IGF2BP1 is an endogenous target of let-7b, we transfected Huh7.5.1 cells with the synthetic let-7b mimic and measured the IGF2BP1 mRNA and protein levels by qRT-PCR and Western blotting, respectively. The overexpression of let-7b in Huh7.5.1 cells resulted in significant reductions in the endogenous levels of IGF2BP1 mRNA (Fig. 5B) and protein (Fig. 5C).
To further demonstrate that let-7b targets the 3′UTR of IGF2BP1 mRNA, a reporter plasmid (pmirGLO-IGF2BP1-3′UTR) was constructed containing the IGF2BP1 3′-UTR downstream of the firefly luciferase open reading frame (Fig. 5A, right). As shown in Fig. 5D, the cotransfection of cells with pmirGLO-IGF2BP1-3′UTR and the let-7b mimic significantly decreased firefly luciferase reporter activity compared to cotransfection with the reporter construct and a negative control mimic or the mu7b mimic in which the seed match sites for the 3′UTR of IGF2BP1 mRNA were abolished.
Next, three independent mutant luciferase reporter plasmids were constructed. These plasmids contained one, two, or three 5-nucleotide mutations at the let-7b seed match sites in the IGF2BP1 mRNA 3′-UTR and were designated pmirGLO-3′UTR-Mutation#1, pmirGLO-3′UTR-Mutation#1+2, and pmirGLO-3′UTR-Mutation#1+2+3, respectively (Fig. 5E). The luciferase reporter assay demonstrated that transfection of cells with the let-7b mimic significantly decreased the luciferase activity of the cotransfected wild-type IGF2BP1 mRNA reporter construct, whereas the stepwise addition of binding-site mutations to the IGF2BP1 mRNA 3′UTR progressively restored luciferase activity, with a complete reversal of the inhibition of luciferase activity achieved when all binding sites were mutated (Fig. 5E). Next, to investigate the role of IGF2BP1 in HCV infection, endogenous IGF2BP1 was knocked down in HCV-infected Huh7.5.1 cells using synthetic siRNAs. As shown in Fig. 5F, endogenous IGF2BP1 was required for HCV propagation in hepatocytes, which is consistent with a previous report (27). These results suggest that the mechanism through which let-7b inhibits HCV involves the targeting of host IGF2BP1 mRNA.
IFN-α and IL-28B downregulate IGF2BP1.
Based on our confirmation of IGF2BP1 as a let-7b target and on the observation that let-7b could be induced by IFNs, we hypothesized that IFNs might regulate IGF2BP1 expression. To test this hypothesis, Huh7.5.1 cells were treated with 1 ng of IFN-α/ml or 15 ng of IL-28B/ml, and the IGF2BP1 mRNA and protein levels were measured by qRT-PCR and Western blotting, respectively. As expected, IFN-α and IL-28B were able to suppress IGF2BP1 expression (Fig. 6).
Fig 6.
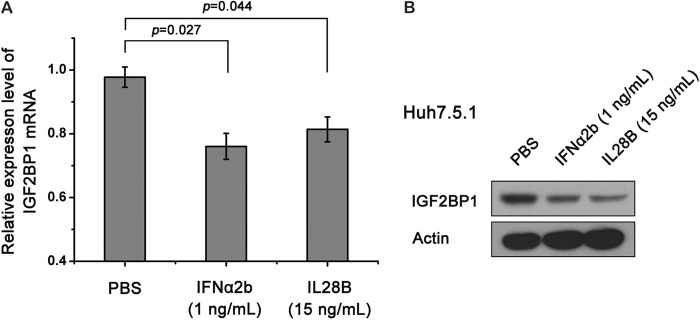
IFN-α and IL-28B treatments suppress IGF2BP1 expression. Huh7.5.1 cells were treated with the indicated concentrations of IL-28B or IFN-α for 3 days. IGF2BP1 mRNA (A) and protein (B) levels were evaluated by real-time qRT-PCR and Western blotting, respectively. The data represent means ± the standard deviations (n = 4). P values were determined using a Student t test.
let-7 family members show anti-HCV activity and inhibit IGF2BP1 expression.
Using sequence alignment analysis, we found that the eight members of the let-7 family possess the consensus seed sequence and have different endogenous expression patterns in Huh7.5.1 cells (Fig. 7A and B). To investigate the effects of these let-7 family members on HCV infection, we transfected cells with the miRNA mimic for each of the eight members, and the results showed that all of them significantly inhibited the replication of HCV (Fig. 7D and E). All let-7 family members were able to reduce the expression of IGF2BP1 (Fig. 7C). Each miRNA's 2- to 8-nucleotide seed sequence was important in determining its specific mRNA target. The antiviral activity of let-7b was abolished when the let-7b seed sequence specific for the 3′UTR of IGF2BP1 mRNA was mutated (Fig. 7F).
Fig 7.

let-7 family members inhibit HCVcc replication and IGF2BP1 expression. (A) Multiple alignments of eight let-7 family members. (B) The expression levels of let-7 family members in Huh7.5.1 cells were detected by real-time qRT-PCR. (C) Huh7.5.1 cells were transfected with mimics for individual let-7 family members (50 nM) and incubated for 2 days, and the IGF2BP1 protein levels in the cell lysates were then determined by Western blotting. (D) Mimics for individual let-7 family members were transfected into 2−3+ cells. After the cells were cultured for 3 days, the HCV Core protein levels in the cell lysates were evaluated by Western blotting. (E) Huh7.5.1 cells were transfected with mimics for individual let-7 family members (50 nM), incubated for 2 days, and then infected with Jc1-Luc HCVcc. The HCVcc-infected cells were cultured 2 days further, and the luciferase activity in the cells was measured. (F) Huh7.5.1 cells were transfected with 50 nM let-7b, mu7b, or NC mimics and then infected with Jc1-Luc HCVcc. After the HCVcc-infected cells were cultured for 2 additional days, their luciferase activities were measured. Error bars denote the standard deviations from the mean of three independent experiments. Statistical analyses were performed between let-7b or mu7b and negative control using the Student t test.
let-7b inhibits HCV infection and IGF2BP1 expression in PFLCs.
To better understand the regulation of let-7b on HCV infection and IGF2BP1 expression, we investigated the effects of let-7b in cultured PFLCs. In accordance with the results observed in hepatoma cells, overexpression of let-7b in PFLCs reduced Jc1-Luc HCVcc replication (Fig. 8A) and decreased the endogenous IGF2BP1 expression in both mRNA and protein levels (Fig. 8B and C).
Fig 8.

let-7b suppresses HCV infection and IGF2BP1 expression in PFLCs. (A) PFLCs were seeded in 48-well plates and transfected with the indicated miRNA mimics (50 nM) or inhibitors (100 nM). The cells were cultured for 2 days, followed by transfection with 1.2 μg of in vitro-transcribed Jc1-Luc RNA. The transfected cells were cultured 1 days further, and the luciferase activity was measured. The IGF2BP1 mRNA (B) and protein (C) levels were evaluated, respectively, by real-time qRT-PCR and Western blotting in PFLCs that were transfected with 50 nM miRNA mimics for 2 days. Transfection of siIGF2BP1 served as a positive control.
DISCUSSION
IFN was discovered more than a half century ago. Since then, IFNs have been used broadly in medical therapies and scientific studies addressing various viral infections and cancers. For more than 2 decades, IFN-α treatment in combination with ribavirin has formed the cornerstone of therapy for HCV infection. In 2009, an inspiring discovery was made that showed that single nucleotide polymorphisms (SNPs) linked to the type III IFN IL-28B are major host determinants at the population level for determining IFN treatment outcomes (18). Several miRNAs have been reported to respond to the type I IFN stimulation and to contribute to IFN-dependent antiviral immunity (9). Aside from the fact that type I and type III IFNs are both known to facilitate JAK-STAT signal transduction and the antiviral innate immune response, there are no currently available data comparing how stimulation with the different types of IFNs affects cellular miRNA profiles (20). Therefore, we described and compared the changes in miRNA expression after IFN-α and IL-28B treatments. There were differences between the IFN-α- and IL-28B-dependent miRNA expression patterns both in terms of the direction in which miRNA expression was modulated and the extent to which expression was modulated (Fig. 1A and see Table S1 in the supplemental material). Notably, five miRNAs were upregulated in response to one IFN treatment but downregulated in response to the other IFN treatment, as indicated in boldface in Table S1 in the supplemental material, which strongly indicates that there are distinct regulatory consequences for type I and III IFN stimulation. Indeed, both our study and previous reports have shown that type I IFN treatment of Huh7 results in a temporary 20 to 50% attenuation of miR-122 expression (see Table S1 in the supplemental material), which is evidence that IFN-responsive miRNAs contribute to IFN-mediated antiviral defense (9, 28, 29). Here, the miRNA expression patterns were not exactly consistent with previous reports probably because the miRNAs expression analysis were performed in different types of cells (IFN-α- or IL-28B-stimulated hepatocytes versus IFN-α/β- or IFN-γ-stimulated lymphocytes) by different methods (real-time qRT-PCR versus oligonucleotide probe microchip) (9).
We previously reported that IL-28B has a more restricted tissue distribution than IFN-α (20), and the pathway enrichment assay performed in the present study demonstrated that IL-28B has more limited signaling effects than IFN-α in Huh7 (Fig. 1C and D). The major pathways associated with the IFN-regulated miRNAs were cancer, cell growth and death, immunity, and signal transduction, a finding consistent with the major biological functions of IFNs, which are cell proliferation, antiviral and antitumor activity, and immune regulation. Together with the reported observation that type III IFN regulated signal transduction and ISG expression in a manner distinct from that of IFN-α (30), our cumulative findings support the idea that IL-28B is not simply redundant with type I IFN.
A number of cellular miRNAs have been found to regulate HCV infection (9, 25, 31, 32). In response to IFN treatment, host cells initiate an antiviral defense mechanism that involves the induction of certain miRNAs that suppress viral mRNA replication and the inhibition of other cellular miRNAs that promote the viral life cycle. The present study shows that HCV replication was affected by the upregulation of 11 miRNAs and the downregulation of 7 miRNAs (Table 1). The correlation between antiviral activity and the fold change in the expression of these IFN-regulated miRNAs was not linear. It may be that miRNAs with a high fold change in expression but limited anti-HCV activity participate in other IFN-related cellular processes, such as immune regulation. For example, several miRNAs can directly regulate IFN-β protein expression in mammalian cells (33).
The inhibition of viral infection by miRNA might occur either via the targeting of viral sequences or by the regulation of host genes that participate in the viral life cycle. Although let-7b has been suggested to bind to NS5B coding sequences and to the 5′UTR of the HCV genome, the possibility that it suppresses HCV by regulating host targets had not been previously excluded (26). Our extensive in silico prediction identified human IGF2BP1 as a possible target of let-7b. The attenuation of IGF2BP1 expression can be achieved using a let-7b mimic and IFN treatments and inhibits HCV infection (Fig. 5 and 6). These results support the theory that let-7b inhibits HCV infection via both targeting HCV genome and host gene. It is notable that the IFN-dependent inhibition of IGF2BP1 expression may be mediated through the activation of let-7b or the let-7 family members let-7f, let-7g, and let-7g* (see Table S1 in the supplemental material). The ability of one miRNA to regulate numerous mRNAs and the potential for one mRNA to be targeted by multiple miRNAs suggests that there may be other host genes regulated by let-7b that limit HCV proliferation; further research is needed to explore this possibility. The results presented here support the theory that, in addition to the innate immune signaling pathways, the steps linking IFN treatment to changes in host protein expression may involve the participation of noncoding genes.
The let-7 family consists of 12 genes encoding nine distinct miRNAs. Notably, although the members share the seed region, anti-HCV activity, and the ability to suppress IGF2BP1 expression (Fig. 7), they are not functionally redundant with one another. That the let-7 family members are each unique was supported by their differing expression levels after IFN treatment, the differing expression levels in hepatocytes, and the differing degrees to which they inhibited HCV replication and suppressed target protein expression (Fig. 7). An explanation for this phenomenon is that differences in the expression and regulation of the miRNAs were cell specific, localization dependent, and time specific.
Our finding that the transfection of Huh7.5.1 cells with a let-7b inhibitor did not enhance HCV proliferation to a statistically significant extent (Fig. 4) is inconsistent with a previous publication (26). The earlier study, which found that increased HCV infection corresponded to reduced let-7b expression, used a different cell line (26); the Huh7.5.1 cells we used already express very low endogenous levels of let-7b, such that reducing let-7b expression further may not permit significant improvement in HCV replication.
Given that cellular miRNAs play important roles in modulating host-pathogen interactions and that the IFN system represents a vital arm of innate immunity, it is not surprising to find that IFN-responsive miRNAs contribute to antiviral defense through the targeting of host cell factors and the viral genome. Our results provide the first comparative analysis of miRNA expression profiles modulated in response to type I and type III IFNs. Furthermore, our findings provide an example of a single miRNA influencing infection through two mechanisms: suppressing the expression of specific host genes and inhibiting viral mRNA expression. The in vivo overexpression of let-7b in a nude mouse model did not affect body weight (34). The ever-increasing number of IFN-related miRNAs provides novel candidates for the development of therapeutic antiviral agents. The mechanisms through which IFNs regulate miRNA expression need to be explored in more detail. As a class of molecules, miRNAs, through their posttranscriptional regulation of the immune system, have the potential to become yet another weapon against viral infection.
Supplementary Material
ACKNOWLEDGMENTS
We thank T. Wakita, C. Rice, F. Chisari, and S. Lemon for kindly providing the cell lines and reagents.
This study was supported by grants from the National Science and Technology Major Project of China (2012ZX10002007-003 and 2013ZX10004-601), the National Basic Research Program of China (2011CB504800), the National Natural Science Foundation of China (grant 81271831), and the Fundamental Research Funds for the Central Universities (grant 2012Y01) and by an intramural grant from the Institute of Pathogen Biology (2013IPB103).
Footnotes
Published ahead of print 3 July 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.00802-13.
REFERENCES
- 1.Lavanchy D. 2011. Evolving epidemiology of hepatitis C virus. Clin. Microbiol. Infect. 17:107–115 [DOI] [PubMed] [Google Scholar]
- 2.Hotho DM, de Bruijne J, Spaan M, Treitel MA, Boonstra A, de Knegt RJ, Janssen HL, Reesink HW. 2013. Sustained virologic response after therapy with the HCV protease inhibitor narlaprevir in combination with peginterferon and ribavirin is durable through long-term follow-up. J. Viral. Hepat. 20:e78–e81 [DOI] [PubMed] [Google Scholar]
- 3.McHutchison JG, Everson GT, Gordon SC, Jacobson IM, Sulkowski M, Kauffman R, McNair L, Alam J, Muir AJ. 2009. Telaprevir with peginterferon and ribavirin for chronic HCV genotype 1 infection. N. Engl. J. Med. 360:1827–1838 [DOI] [PubMed] [Google Scholar]
- 4.Poordad F, McCone J, Jr, Bacon BR, Bruno S, Manns MP, Sulkowski MS, Jacobson IM, Reddy KR, Goodman ZD, Boparai N, DiNubile MJ, Sniukiene V, Brass CA, Albrecht JK, Bronowicki JP. 2011. Boceprevir for untreated chronic HCV genotype 1 infection. N. Engl. J. Med. 364:1195–1206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Membreno FE, Lawitz EJ. 2011. The HCV NS5B nucleoside and non-nucleoside inhibitors. Clin. Liver Dis. 15:611–626 [DOI] [PubMed] [Google Scholar]
- 6.Cho NJ, Dvory-Sobol H, Lee C, Cho SJ, Bryson P, Masek M, Elazar M, Frank CW, Glenn JS. 2010. Identification of a class of HCV inhibitors directed against the nonstructural protein NS4B. Sci. Transl. Med. 2:15ra16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gao M, Nettles RE, Belema M, Snyder LB, Nguyen VN, Fridell RA, Serrano-Wu MH, Langley DR, Sun JH, O'Boyle DR, 2nd, Lemm JA, Wang C, Knipe JO, Chien C, Colonno RJ, Grasela DM, Meanwell NA, Hamann LG. 2010. Chemical genetics strategy identifies an HCV NS5A inhibitor with a potent clinical effect. Nature 465:96–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Katze MG, He Y, Gale M., Jr 2002. Viruses and interferon: a fight for supremacy. Nat. Rev. Immunol. 2:675–687 [DOI] [PubMed] [Google Scholar]
- 9.Pedersen IM, Cheng G, Wieland S, Volinia S, Croce CM, Chisari FV, David M. 2007. Interferon modulation of cellular microRNAs as an antiviral mechanism. Nature 449:919–922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bartel DP. 2004. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116:281–297 [DOI] [PubMed] [Google Scholar]
- 11.Lee RC, Feinbaum RL, Ambros V. 1993. The Caenorhabditis elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 75:843–854 [DOI] [PubMed] [Google Scholar]
- 12.Lim LP, Lau NC, Garrett-Engele P, Grimson A, Schelter JM, Castle J, Bartel DP, Linsley PS, Johnson JM. 2005. Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs. Nature 433:769–773 [DOI] [PubMed] [Google Scholar]
- 13.Lewis BP, Burge CB, Bartel DP. 2005. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 120:15–20 [DOI] [PubMed] [Google Scholar]
- 14.Huang J, Wang F, Argyris E, Chen K, Liang Z, Tian H, Huang W, Squires K, Verlinghieri G, Zhang H. 2007. Cellular microRNAs contribute to HIV-1 latency in resting primary CD4+ T lymphocytes. Nat. Med. 13:1241–1247 [DOI] [PubMed] [Google Scholar]
- 15.Song L, Liu H, Gao S, Jiang W, Huang W. 2010. Cellular microRNAs inhibit replication of the H1N1 influenza A virus in infected cells. J. Virol. 84:8849–8860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Randall G, Panis M, Cooper JD, Tellinghuisen TL, Sukhodolets KE, Pfeffer S, Landthaler M, Landgraf P, Kan S, Lindenbach BD, Chien M, Weir DB, Russo JJ, Ju J, Brownstein MJ, Sheridan R, Sander C, Zavolan M, Tuschl T, Rice CM. 2007. Cellular cofactors affecting hepatitis C virus infection and replication. Proc. Natl. Acad. Sci. U. S. A. 104:12884–12889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Banaudha K, Kaliszewski M, Korolnek T, Florea L, Yeung ML, Jeang KT, Kumar A. 2011. MicroRNA silencing of tumor suppressor DLC-1 promotes efficient hepatitis C virus replication in primary human hepatocytes. Hepatology 53:53–61 [DOI] [PubMed] [Google Scholar]
- 18.Tanaka Y, Nishida N, Sugiyama M, Kurosaki M, Matsuura K, Sakamoto N, Nakagawa M, Korenaga M, Hino K, Hige S, Ito Y, Mita E, Tanaka E, Mochida S, Murawaki Y, Honda M, Sakai A, Hiasa Y, Nishiguchi S, Koike A, Sakaida I, Imamura M, Ito K, Yano K, Masaki N, Sugauchi F, Izumi N, Tokunaga K, Mizokami M. 2009. Genome-wide association of IL-28B with response to pegylated interferon-alpha and ribavirin therapy for chronic hepatitis C. Nat. Genet. 41:1105–1109 [DOI] [PubMed] [Google Scholar]
- 19.Thomas E, Gonzalez VD, Li Q, Modi AA, Chen W, Noureddin M, Rotman Y, Liang TJ. 2012. HCV infection induces a unique hepatic innate immune response associated with robust production of type III interferons. Gastroenterology 142:978–988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cheng M, Si Y, Yang Y, Liu X, Gong Q, Zhao J, Niu Y, Li X, Jin Q, Yang W. 2012. Recombinant human interleukin 28B: anti-HCV potency, receptor usage and restricted cell-type responsiveness. J. Antimicrob. Chemother. 67:1080–1087 [DOI] [PubMed] [Google Scholar]
- 21.Andrus L, Marukian S, Jones CT, Catanese MT, Sheahan TP, Schoggins JW, Barry WT, Dustin LB, Trehan K, Ploss A, Bhatia SN, Rice CM. 2011. Expression of paramyxovirus V proteins promotes replication and spread of hepatitis C virus in cultures of primary human fetal liver cells. Hepatology 54:1901–1912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stoskus M, Gineikiene E, Valceckiene V, Valatkaite B, Pileckyte R, Griskevicius L. 2011. Identification of characteristic IGF2BP expression patterns in distinct B-ALL entities. Blood Cells Mol. Dis. 46:321–326 [DOI] [PubMed] [Google Scholar]
- 23.Si Y, Liu S, Liu X, Jacobs JL, Cheng M, Niu Y, Jin Q, Wang T, Yang W. 2012. A human claudin-1-derived peptide inhibits hepatitis C virus entry. Hepatology 56:507–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang W, Qiu C, Biswas N, Jin J, Watkins SC, Montelaro RC, Coyne CB, Wang T. 2008. Correlation of the tight junction-like distribution of Claudin-1 to the cellular tropism of hepatitis C virus. J. Biol. Chem. 283:8643–8653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jopling CL, Yi M, Lancaster AM, Lemon SM, Sarnow P. 2005. Modulation of hepatitis C virus RNA abundance by a liver-specific MicroRNA. Science 309:1577–1581 [DOI] [PubMed] [Google Scholar]
- 26.Cheng JC, Yeh YJ, Tseng CP, Hsu SD, Chang YL, Sakamoto N, Huang HD. 2012. let-7b is a novel regulator of hepatitis C virus replication. Cell. Mol. Life Sci. 69:2621–2633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Weinlich S, Huttelmaier S, Schierhorn A, Behrens SE, Ostareck-Lederer A, Ostareck DH. 2009. IGF2BP1 enhances HCV IRES-mediated translation initiation via the 3′UTR. RNA 15:1528–1542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sarasin-Filipowicz M, Krol J, Markiewicz I, Heim MH, Filipowicz W. 2009. Decreased levels of microRNA miR-122 in individuals with hepatitis C responding poorly to interferon therapy. Nat. Med. 15:31–33 [DOI] [PubMed] [Google Scholar]
- 29.Gong BD, Xie Q, Xiang XG, Wang L, Zhao GD, An FM, Wang H, Lin LY, Yu H, Bao SS. 2010. Effect of ribavirin and interferon beta on miRNA profile in the hepatitis C virus subgenomic replicon-bearing Huh7 cells. Int. J. Mol. Med. 25:853–859 [DOI] [PubMed] [Google Scholar]
- 30.Marcello T, Grakoui A, Barba-Spaeth G, Machlin ES, Kotenko SV, MacDonald MR, Rice CM. 2006. Interferons alpha and lambda inhibit hepatitis C virus replication with distinct signal transduction and gene regulation kinetics. Gastroenterology 131:1887–1898 [DOI] [PubMed] [Google Scholar]
- 31.Murakami Y, Aly HH, Tajima A, Inoue I, Shimotohno K. 2009. Regulation of the hepatitis C virus genome replication by miR-199a. J. Hepatol. 50:453–460 [DOI] [PubMed] [Google Scholar]
- 32.Bandyopadhyay S, Friedman RC, Marquez RT, Keck K, Kong B, Icardi MS, Brown KE, Burge CB, Schmidt WN, Wang Y, McCaffrey AP. 2011. Hepatitis C virus infection and hepatic stellate cell activation downregulate miR-29: miR-29 overexpression reduces hepatitis C viral abundance in culture. J. Infect. Dis. 203:1753–1762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Witwer KW, Sisk JM, Gama L, Clements JE. 2010. MicroRNA regulation of IFN-β protein expression: rapid and sensitive modulation of the innate immune response. J. Immunol. 184:2369–2376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen F, Chen C, Yang S, Gong W, Wang Y, Cianflone K, Tang J, Wang DW. 2012. let-7b inhibits human cancer phenotype by targeting cytochrome P450 epoxygenase 2J2. PLoS One 7:e39197. 10.1371/journal.pone.0039197 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.