

Abstract
Alphaherpesviruses, including pseudorabies virus (PRV), spread directionally within the nervous systems of their mammalian hosts. Three viral membrane proteins are required for efficient anterograde-directed spread of infection in neurons, including Us9 and a heterodimer composed of the glycoproteins gE and gI. We previously demonstrated that the kinesin-3 motor KIF1A mediates anterograde-directed transport of viral particles in axons of cultured peripheral nervous system (PNS) neurons. The PRV Us9 protein copurifies with KIF1A, recruiting the motor to transport vesicles, but at least one unidentified additional viral protein is necessary for this interaction. Here we show that gE/gI are required for efficient anterograde transport of viral particles in axons by mediating the interaction between Us9 and KIF1A. In the absence of gE/gI, viral particles containing green fluorescent protein (GFP)-tagged Us9 are assembled in the cell body but are not sorted efficiently into axons. Importantly, we found that gE/gI are necessary for efficient copurification of KIF1A with Us9, especially at early times after infection. We also constructed a PRV recombinant that expresses a functional gE-GFP fusion protein and used affinity purification coupled with mass spectrometry to identify gE-interacting proteins. Several viral and host proteins were found to associate with gE-GFP. Importantly, both gI and Us9, but not KIF1A, copurified with gE-GFP. We propose that gE/gI are required for efficient KIF1A-mediated anterograde transport of viral particles because they indirectly facilitate or stabilize the interaction between Us9 and KIF1A.
INTRODUCTION
Alphaherpesviruses are common pathogens that replicate and spread within the nervous systems of their mammalian hosts. Well-studied alphaherpesviruses include the human pathogens herpes simplex virus types 1 and 2 (HSV-1 and HSV-2) and varicella zoster virus (VZV), as well as the veterinary pathogen pseudorabies virus (PRV) (2, 3). Dissemination of viral particles within an infected host requires long-distance axonal transport of viral particles in neurons (5). Alphaherpesvirus infection of the peripheral nervous system (PNS) is initiated when virions enter axon termini and use retrograde axonal transport to travel toward neuron cell bodies (7, 8). Newly replicated progeny virions are assembled in the cell body and may be sorted into axons and transported back out toward the periphery (anterograde-directed spread) (10, 11). Three highly conserved viral membrane proteins are important for anterograde-directed spread of infection in vivo and in vitro, including Us9 and the glycoproteins gE and gI (13–21).
PRV strains with Us9 deleted are completely defective for axonal sorting of viral particles and exhibit no anterograde spread of infection (23, 24). Us9 is a small, type II, tail-anchored membrane protein that copurifies with the kinesin-3 motor KIF1A (6, 12). KIF1A is necessary for anterograde axonal movement of viral particles, but at least one additional viral protein is required to facilitate or stabilize its interaction with Us9; this protein (or proteins) is not expressed by the attenuated PRV strain Bartha (6). The PRV Bartha genome contains multiple mutations compared to virulent PRV, including a deletion in the coding region for gI, gE, Us9, and Us2 (28). Since deletion of gE or gI severely reduces but does not completely disrupt the anterograde spread capacity of PRV in vitro (24), we hypothesized that these two proteins may be involved in the Us9-KIF1A interaction to promote virion transport.
Both gE and gI are type I transmembrane proteins that form a heterodimer (referred to here as gE/gI) in the endoplasmic reticulum (ER) via noncovalent interactions between their ectodomains (30). Because gE and gI can be found in a complex and mutants with either gene deleted display a similar phenotype in animals, gE/gI may act as a single functional unit. Importantly, unlike Us9 null mutants, gE and gI single or double null mutants display a small-plaque phenotype in cultured epithelial cells (9), suggesting that this protein complex has multiple functions. Indeed, the gE/gI complex is involved in virion assembly, cell-to-cell spread, species-specific binding of immunoglobulin G as an Fc receptor, and mediating full virulence in animal infections (32, 34–38).
Reconciling the different functions of gE/gI with a common mechanism has been difficult. Previous experiments indicate that targeting of viral structural components and membrane proteins to axons requires both Us9 and gE/gI but that Us9 null mutants are more defective than single or double gE and gI mutants (15). Furthermore, the interactions of gE/gI with Us9 and with cellular proteins are not well understood. Here, we sought to elucidate the molecular mechanism by which gE/gI mediate anterograde spread of infection. We demonstrate that gE/gI but not Us2 are required for efficient axonal sorting and anterograde-directed transport of viral particles in axons. Importantly, we found that expression of gE/gI is required for efficient interaction between Us9 and KIF1A. We then used a direct biochemical approach to identify the host and viral proteins that interact with gE. PRV recombinants that express a functional gE-green fluorescent protein (GFP) fusion protein were constructed, and immunoaffinity purification methods with GFP antibodies were employed to isolate proteins that copurify with gE-GFP by mass spectrometry analysis. We identified 46 host and viral proteins that are in the complex with gE-GFP. Interestingly, the gE-GFP fusion protein copurified with Us9 but not with KIF1A during PRV infection. These results suggest that gE/gI directly or indirectly stabilize or facilitate Us9/KIF1A complex formation and in turn promote efficient axonal sorting and transport of viral particles in axons.
MATERIALS AND METHODS
Viruses and plasmids.
We used the wild-type (WT) PRV Becker strain as well as the previously described derivatives PRV 99 (gE and gI null) (39), and PRV 174 (Us2 null, expresses enhanced GFP [EGFP] from the Us2 locus) (41). For construction of PRV expressing gE-GFP (PRV 187), the gE open reading frame was ligated into the multiple-cloning site of pEGFP-N1 (Clontech) at the BamHI/EcoRI sites. This plasmid construct was designated pTK001. Next, pTK001 was cut with NheI/MfeI, and the fragment containing gE-GFP was ligated into the multiple-cloning site of pII1 (1). This plasmid was designated pTK013. To recombine gE-GFP into the Us4 locus of the PRV genome, linearized pTK013 was cotransfected with nucleocapsid DNA from PRV 758 (15) into PK15 cells. Virus produced after cotransfection was plated on PK15 cells, and plaques expressing gE-GFP were identified by epifluorescence microscopy, isolated, and subjected to three rounds of purification. To construct PRV 199 (gE-GFP, monomeric red fluorescent protein [mRFP]-VP26), PK15 cells were coinfected with PRV 187 and PRV 180 (WT mRFP-VP26) (4). Virus produced after coinfection was plated on PK15 cells, and plaques expressing both gE-GFP and mRFP-VP26 were identified by epifluorescence microscopy, isolated, and subjected to three rounds of purification.
We derived a PRV recombinant expressing GFP-Us9 in the PRV BaBe background, which carries the Bartha deletion within the unique short region of the genome including the coding regions for Us7 (gI), Us8 (gE), Us9, and Us2 (39). The open reading frame encoding the GFP-Us9 fusion sequence (44) was cloned into the pII1 shuttle vector (1), which contains flanking sequence homologous to the Us4 genome region, at the NheI/MfeI restriction sites; the resultant plasmid was designated pTK037. PK15 cells were then cotransfected with linearized pTK037 and nucleocapsid DNA from PRV BaBe. GFP-positive recombinant plaques showing a membrane-bound GFP localization pattern were picked and further subjected to three rounds of plaque purification. The resulting strain was designated PRV 444. We also derived a PRV BaBe recombinant expressing mRFP-VP26 (designated PRV 446). Briefly, PK15 cells were coinfected with PRV 180 (mRFP-VP26, Becker background) (4) and PRV 432 (PRV BaBe expressing GFP), and dual-fluorescent GFP/mRFP-positive plaques were picked and further subjected to three rounds of plaque purification. Three recombinants were isolated and then analyzed by Western blotting to confirm appropriate expression patterns. The resultant strain was designated PRV 438 and then used to coinfect PK15 cells along with PRV BaBe to derive the PRV BaBe/mRFP-VP26 recombinant (PRV 446), identified through loss of GFP fluorescence. The newly derived PRV 444 and PRV 446 recombinants were then coinfected to isolate a two-color recombinant (GFP-Us9, mRFP-VP26, null for gI, gE, Us9, and Us2), designated PRV 448.
For adenovirus (Ad) transduction experiments where GFP-Us9 is expressed, we used the previously described nonreplicating adenoviral vector Ad TK101 (here referred to as Ad GFP-Us9) (6).
Antibodies.
Rabbit polyclonal antisera specific for gE cytoplasmic tail (used at 1:1,000) (9), rabbit polyclonal antiserum specific for Us9 (used at 1:1,000) (12), the phospho-specific mouse monoclonal Us9 antibody 2D5E6 (used at 1:1,000) (22), and the rabbit polyclonal anti-GFP antibodies (25) have all been previously described. The mouse monoclonal antibody for cellular actin (AC-40, used at 1:5,000; Sigma-Aldrich, St. Louis, MO) was also employed.
Cell lines and primary neuron cultures.
Porcine kidney epithelial cells (PK15 cells) were maintained in Dulbecco modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin (HyClone, Logan, UT). The transformed neuronal cell line PC12 (26) has been used extensively to model primary neurons for infection with PRV and reproduces Us9-associated viral transport phenotypes (23, 27). PC12 cells were cultured on dishes coated with type 1 rat tail collagen (BD Biosciences, Bedford, MA) in 85% RPMI 1640 with 10% horse serum and 5% FBS (HyClone). PC12 cells were differentiated in RPMI 1640 supplemented with 1% horse serum and nerve growth factor (NGF) (Invitrogen, Carlsbad, CA) at 100 ng/ml. Differentiation medium was replaced every third day for 11 days before infection.
Dissociated cultures of primary rat superior cervical ganglion (SCG) neurons were prepared as previously detailed (27). Prior to plating, MatTek glass-bottom dishes (Ashland, MA) were coated with poly-dl-ornithine (Sigma-Aldrich) and murine laminin (Invitrogen). SCGs dissected from embryonic day 15.5 (E15.5) to E16.5 pregnant Sprague-Dawley rats (Hilltop Labs Inc., Pittsburgh, PA) were then plated and maintained in neuronal medium consisting of Neurobasal medium (Invitrogen) supplemented with 1% penicillin-streptomycin-glutamine (Invitrogen), B27 (Invitrogen), and 50 ng/ml NGF. Cultures were allowed to differentiate for at least 14 days prior to infection.
Chambered neuronal cultures.
SCG neurons cultured in modified Campenot chambers have been employed extensively in our laboratory to assay directional spread of viral infection (29). For chambered cultures, plastic tissue culture dishes were coated as described above. A series of parallel grooves were then etched across the surface and covered with 1% methylcellulose in DMEM. A CAMP320 three-chambered Teflon ring (Tyler Research; Edmonton, Alberta, Canada) was then coated with vacuum grease on one side and placed on top of the tissue culture surface, oriented such that the grooves extended across all three compartments. SCG neurons were then plated in one compartment (S compartment) and maintained in neuronal medium as for dissociated cultures. Following ∼17 days of culture, robust axonal extensions develop across the center (M compartment) into the far side (N compartment). A detector layer of PK15 cells (∼5 × 105 cells in neuronal medium supplemented with 1% FBS) was plated in the N compartment 24 h before any infections to amplify virus spread into this compartment.
Western blotting.
Cell lysates that were analyzed by Western blotting were prepared in RSB–NP-40 (10 mM Tris-HCl [pH 7.5], 10 mM NaCl, 1.5 mM MgCl2, 1% NP-40) with protease inhibitor cocktail at 1:100 (Sigma-Aldrich). For coimmunoprecipitation experiments, samples were prepared as previously described (6). All samples were heated to 65°C for 10 min, mixed with Laemmli sample buffer (Invitrogen), and run on one-dimensional SDS-polyacrylamide gels. Primary antibodies were diluted in 5% milk in Tris-buffered saline with Tween 20 (TBS-T) (50 mM Tris [pH 7.4], 200 mM NaCl, 0.1% Tween 20) and applied to the membrane for 1 h at room temperature. Membranes were then washed 3 times with TBS-T, and secondary horseradish peroxidase (HRP)-conjugated antibody (used at 1:20,000; KPL, Gaithersburg, MD) was applied for 1 h at room temperature. Signals were detected using the SuperSignal West Pico chemiluminescent substrate kit (Thermo-Fisher Scientific, Rockford, IL), according to the manufacturer's instructors.
Immunoaffinity purifications.
To isolate protein complexes, we employed high-affinity anti-GFP antibodies for immunoaffinity purification of viral protein complexes, as previously described (31, 33). Differentiated PC12 cells were infected with PRV strains expressing GFP-Us9 or gE-GFP fusion proteins, and cells were then lysed at the indicated time points (6). Cells were washed twice with phosphate-buffered saline (PBS) and then resuspended in a previously optimized immunoaffinity purification lysis buffer (20 mM HEPES-KOH [pH 7.4], 110 mM potassium phosphate, 2 mM MgCl2, 1 μM ZnCl2, 1 μM CaCl2, 0.1% Tween 20, 1% Triton X-100, and 150 mM NaCl) supplemented with protease inhibitor cocktail (Sigma-Aldrich) at 1:100. Samples were first homogenized by at least 15 passages though an 18-gauge needle and subsequently incubated for 30 min at 4°C. Following the lysis period, samples were rehomogenized and centrifuged at 20,800 × g for 10 min to pellet insoluble cellular debris. Dynal M-270 epoxy magnetic beads (Invitrogen) were coated with custom-made rabbit polyclonal anti-GFP antibodies as previously described (31) (5 μg antibodies per 1 mg of beads). Three milligrams of beads was added to each sample and then incubated for 1 h at 4°C. The beads were then washed six times with ice-cold lysis buffer, and protein complexes bound to the beads were eluted by incubation at 65°C for 10 min in 1× SDS-PAGE loading buffer (Invitrogen) supplemented with 60 mM dithiothreitol (DTT). For mass spectrometry (MS) experiments, samples were alkylated with 100 mM iodoacetamide (Sigma-Aldrich) at room temperature for 1 h in the dark.
Mass spectrometry and data analysis.
Reduced and alkylated GFP and gE-GFP affinity purification samples were resolved (approximately 3 cm) by SDS-PAGE, and proteins were stained with Coomassie blue. Each gel lane was cut into 1-mm slices, pooled into six fractions, and then digested in gel with trypsin as previously described (40). Nano-liquid chromatography-tandem MS (nLC-MS/MS) analysis of tryptic peptide fractions was performed on a Dionex Ultimate 3000 rapid-separation LC (RSLC) (Dionex, Amsterdam, the Netherlands) coupled online to an LTQ Orbitrap Velos mass spectrometer (Thermo-Fisher Scientific), as previously described (40). Proteome Discoverer (v1.3) (Thermo-Fisher Scientific) and Scaffold (v3.4) (Proteome Software Inc., Portland, OR) were used to perform database searches and peptide and protein validation, as previously described (42). Label-free spectral counting analyses were performed as previously described (40), with slight modification. To be considered for analysis, identified proteins had a minimum of 5 spectral counts. Spectral counts within the gE-GFP samples were normalized based on the average number of total identified spectra in the GFP control samples. Next, spectral counts between biological replicates of the GFP samples were averaged. Proteins unique to gE-GFP with at least 3.0-fold spectral count enrichment in gE-GFP versus the GFP control were considered putative gE interactions. The relative abundance of proteins that copurified with gE-GFP was calculated using the normalized spectral abundance factor (NSAF) method (43). Spectral counts of proteins detected in the gE-GFP sample were normalized by protein length, divided by the sum of all normalized spectral counts, and expressed as a percentage of the total abundance (Table 1).
Table 1.
Proteomic analysis of viral and host proteins that copurify with gE-GFP by mass spectrometry
| Protein category and name | Gene | Accession no. | Mol size (kDa) | Protein length (amino acids) | Assigned spectrum | Relative abundance (% NSAF) | Copurifies with GFP-Us9a |
|---|---|---|---|---|---|---|---|
| Viral | |||||||
| Glycoprotein E | Us8 | P08354 | 62 | 577 | 1,149 | 38.5 | + |
| Glycoprotein I | Us7 | Q6UK12 | 39 | 366 | 424 | 22.4 | |
| Glycoprotein H | UL22 | Q00660 | 72 | 686 | 265 | 7.5 | |
| Us9 | Us9 | Q5PP80 | 11 | 98 | 13 | 2.6 | + |
| UL43 | UL43 | Q9QQM8 | 38 | 373 | 22 | 1.1 | + |
| UL52 helicase/primase | UL52 | Q85228 | 103 | 962 | 36 | 0.7 | + |
| Glycoprotein C | UL44 | P06024 | 51 | 479 | 16 | 0.6 | + |
| Glycoprotein M | UL10 | Q85041 | 42 | 393 | 12 | 0.6 | + |
| Endoplasmic reticulum | |||||||
| Tecr | TECR | Q64232 | 36 | 308 | 22 | 1.4 | + |
| Smpd4 | SMPD4 | Q6ZPR5 | 93 | 823 | 42 | 1.0 | |
| Atp2a2 | ATP2A2 | P11507 | 115 | 1,043 | 55 | 1.0 | + |
| Sec61α1 | SEC61A1 | P61620 | 52 | 476 | 23 | 0.9 | + |
| Erlin-2 | ERLIN2 | B5DEH2 | 38 | 339 | 11 | 0.6 | |
| Ribophorin-1 | RPN1 | P07153 | 68 | 605 | 17 | 0.5 | |
| Sel1l | SEL1L | Q80Z70 | 89 | 794 | 18 | 0.4 | |
| Neuropathy target esterase | PNPLA6 | Q3TRM4 | 150 | 1,355 | 21 | 0.3 | |
| Chaperones | |||||||
| CCT-α | TCP1 | P28480 | 60 | 556 | 32 | 1.1 | |
| CCT-θ | CCT8 | P42932 | 60 | 548 | 30 | 1.1 | |
| CCT-λ | CCT3 | Q6P502 | 61 | 545 | 28 | 1.0 | |
| CCT-ζ | CCT6A | P80317 | 58 | 531 | 22 | 0.8 | |
| CCT-β | CCT2 | Q5XIM9 | 57 | 535 | 22 | 0.8 | |
| CCT-η | CCT7 | P80313 | 60 | 544 | 23 | 0.8 | |
| CCT-δ | CCT4 | Q7TPB1 | 58 | 539 | 21 | 0.8 | |
| Bat3/Bag6 | BAT3 | Q6MG49 | 115 | 1,146 | 33 | 0.7 | |
| CCT-ε | CCT5 | Q68FQ0 | 60 | 541 | 16 | 0.6 | |
| Immune | |||||||
| Complement C3 | C3 | P01027 | 186 | 1663 | 42 | 0.5 | |
| NF-κ-B-activating protein | NKAP | Q4V7C9 | 47 | 415 | 10 | 0.5 | |
| Secretory and cytoskeletal | |||||||
| Itm2b | ITM2B | Q5XIE8 | 30 | 266 | 19 | 1.4 | |
| Plectin | PLEC | Q9QXS1 | 534 | 4,691 | 179 | 0.7 | |
| Atp9a | ATP9A | O70228 | 119 | 1,047 | 14 | 0.3 | |
| Nuclear import/export | |||||||
| 14-3-3 β/α | YWHAB | P35213 | 28 | 246 | 20 | 1.6 | |
| Importin β | KPNB1 | P70168 | 97 | 876 | 28 | 0.6 | + |
| Transportin-1 | TNPO1 | Q8BFY9 | 102 | 898 | 18 | 0.4 | |
| Exportin-1 | XPO1 | Q6P5F9 | 123 | 1,071 | 13 | 0.2 | |
| Mitochondrial | |||||||
| Slc25a1 | SLC25A1 | P32089 | 34 | 311 | 11 | 0.7 | |
| Cds2 | CDS2 | Q91XU8 | 51 | 443 | 10 | 0.4 | |
| Miscellaneous | |||||||
| Scn3b | SCN3B | Q9JK00 | 25 | 215 | 12 | 1.1 | |
| Tho4 | THOC4 | O08583 | 27 | 255 | 11 | 0.8 | |
| Hnrnpa1 | HNRNPA1 | P04256 | 34 | 320 | 11 | 0.7 | |
| Slc2a3 | SLC2A3 | Q07647 | 54 | 493 | 16 | 0.6 | + |
| Poly(rC)-binding protein 3 | PCBP3 | P57722 | 39 | 371 | 11 | 0.6 | |
| HOS | FBXW11 | Q5SRY7 | 62 | 542 | 10 | 0.4 | |
| Myb-binding protein 1A | MYBBP1A | O35821 | 152 | 1,344 | 22 | 0.3 | |
| Ikbkap | IKBKAP | Q8VHU4 | 149 | 1,331 | 11 | 0.2 | |
| Caspr1 | CNTNAP1 | P97846 | 156 | 1,381 | 12 | 0.2 | |
| Ube3c | UBE3C | Q80U95 | 124 | 1,083 | 14 | 0.2 |
From reference 20.
Fluorescence microscopy imaging.
To visualize the subcellular localization of GFP-Us9 or gE-GFP and mRFP-VP26 in neuronal cell bodies, we utilized a Perkin-Elmer spinning-disc confocal microscope (Perkin-Elmer, Waltham, MA). Dissociated SCG cultures were infected with PRV 341, PRV 448, or PRV 199 and fixed at 8 h postinfection (hpi) with 4% paraformaldehyde prior to fluorescence microscopy visualization.
Live-cell imaging of the recombinant fluorescent strains PRV 341, PRV 448, and PRV 199 was performed in dissociated SCG cultures as previously described for the wild-type GFP-Us9 fusion protein (44). All imaging was performed with a Nikon Ti-Eclipse epifluorescence inverted microscope designed for rapid, serial acquisition of multiple fluorescent channels under a heated cell culture chamber (Live Cell Instrument, Seoul, South Korea). Cultures were imaged between 6 and 16 h postinfection. Manual quantification of mobile/stalled capsids in 10 movies from 2 biological replicates, each 3 min in length, for PRV 341 and PRV 448 was performed. Statistical analyses of capsids quantified from movies were performed using the Prism software package (Prism GraphPad Software, Inc.). For quantification of colocalization of GFP and mRFP signals on anterograde-directed puncta during infection with PRV 199, a total of 65 puncta were manually scored from 5 movies from 2 biological replicates.
RESULTS
Both gE and gI, but not Us2, are required for anterograde spread of infection in neurons.
We recently demonstrated that the kinesin-3 motor KIF1A copurifies with the viral membrane protein Us9 (6). While Us9 is necessary for directly or indirectly interacting with KIF1A and recruiting the motor complex to transport vesicles, at least one additional viral protein that is missing or mutated in PRV Bartha is required for this interaction (6). The genome sequences of wild-type PRV Becker and Bartha have been completed (28). Compared to the 67 wild-type protein-coding regions, PRV Bartha has missense or silent mutations in at least 46 proteins as well as a deletion in the unique short region of the PRV genome that removes the coding sequences for gI (Us7), gE (Us8), Us9, and Us2. Since both gE and gI have a well-established role in anterograde spread in neurons (13–15, 17–19), the function of the heterodimer formed by these two proteins may be required for mediating the interaction between Us9 and KIF1A. However, since the Us2 protein is also mutated in Bartha and, like gE, copurifies with Us9 (6) we also assessed whether it is required for anterograde spread of infection using a previously described compartmentalized neuronal culture system (Fig. 1) (24). The PRV Us2 null mutant (PRV 174) spread with an efficiency similar to that of the wild-type strain PRV Becker, while the gE/gI null strain (PRV 99) showed a 2.5-log reduction in anterograde spread. These findings confirm that gE/gI, but not Us2, are required for efficient anterograde spread in neurons. We then assessed the anterograde spread capacity of the newly derived PRV 444, which expresses GFP-Us9 but not gI, gE, or Us2 and also does not have the point mutations of PRV Bartha. Compared with the wild-type strain expressing GFP-Us9 (PRV 340), PRV 444 displayed a 2.5-log anterograde spread defect. This defect was comparable to the 2.5-log spread defect for the gE/gI null mutant (PRV 99) compared with PRV Becker. Taken together, these results indicate that Us2 is not required for anterograde transport and that PRV 444 recapitulates the spread phenotype associated with deletion of gE and gI.
Fig 1.

gE and gI, but not Us2, are required for efficient anterograde spread in neurons. Quantification of the efficiency of anterograde axonal spread using a chambered neuronal culture system is shown. Cell bodies in the soma (S) compartment were infected at a high multiplicity of infection (MOI) with the indicated PRV strains. At 24 hpi, the contents of the soma (S) and neurite (N) compartments were collected separately, and titers were determined on PK15 cells using a standard plaque assay. The following PRV strains were used: Becker (wild type), PRV 174 (Us2 null), PRV 99 (null for gE and gI), PRV 340 (GFP-Us9), and PRV 444 (expresses GFP-Us9 and null for gE, gI, and Us2). Point estimates reflect viral titers in each compartment. Data are from two biological replicates, each performed in triplicate. Lines indicate medians.
Expression of gE/gI is required for efficient Us9-dependent axonal sorting and anterograde transport of viral particles.
Since gE copurifies with Us9 and is required for efficient axonal targeting of viral structural components (6, 15), we first tested whether gE/gI are required for proper localization of GFP-Us9 in cell bodies and axons. In primary rat superior cervical ganglion (SCG) neurons infected with PRV expressing GFP-Us9 and mRFP-VP26 (red capsid), GFP-Us9 localizes predominantly to intracellular puncta within the cell body (6) (Fig. 2A). Similarly, in the absence of gE, gI, and Us2, infected cell bodies contained puncta that were dually labeled for GFP-Us9 and mRFP-tagged capsids (Fig. 2A). This is consistent with previous work showing that gE/gI are not required for incorporation of Us9 into secondary envelopment membranes and mature virions (46). Furthermore, in both wild-type- and gE/gI null-infected neurons, we observed puncta that were singly labeled with GFP or mRFP (Fig. 2A). These represent GFP-Us9-containing vesicles or unenveloped capsids, respectively.
Fig 2.

gE/gI are required for efficient axonal sorting and anterograde transport of viral particles. (A) Dissociated SCG cultures were infected with PRV 341 (expresses GFP-Us9 and the capsid protein mRFP-VP26) or PRV 448 (expresses GFP-Us9 and mRFP-VP26 and is null for gE, gI, and Us2). Infected cells were fixed at 8 hpi and visualized by confocal microscopy. Each image represents a maximum-intensity projection. (B) Live-cell imaging of anterograde transport of virions at 8 hpi with PRV 448, showing GFP-Us9 and mRFP-VP26 channels over time. The arrowhead indicates the position of a dually labeled viral particle at different time points. In each frame, the arrow above the axon indicates the direction of anterograde movement. (C) Quantification of anterograde transport of capsids in axons infected with PRV 341 or PRV 448. Ten axons from two biological replicates were imaged for 3 min each, and the numbers of anterograde, retrograde, and stalled capsids were manually quantified. The number of viral particles counted in each axon was normalized to axon length. Error bars indicate standard errors of the means (SEM). *, P < 0.001; N.S., not significant. (D) Differentiated PC12 cells were infected with PRV Becker or PRV 99 (null for gE and gI), lysed at 12 hpi, and subjected to Western blot analysis using the indicated antibodies.
We next determined whether gE/gI affect the efficiency of axonal transport by visualizing fluorescently labeled viral particles in axons. Neurons were infected with wild-type or gE/gI null PRV strains expressing GFP-Us9 and mRFP-tagged capsids and imaged at 8 h postinfection (hpi). In the absence of gI, gE, and Us2, we observed that GFP-Us9 and mRFP-VP26 dually labeled puncta underwent anterograde-directed axonal transport (Fig. 2B). This is consistent with previous work showing that PRV particles undergo secondary envelopment in cell bodies prior to Us9-dependent axonal sorting and transport (10, 23, 44). Quantification of these transport events revealed that the number of anterograde-directed viral particles in the gI, gE, and Us2 null PRV mutant was reduced by approximately 4-fold compared to that the wild-type strain (Fig. 2C). Retrograde motion of capsids, as well as the number of stalled capsids, did not differ significantly between the two recombinants. These results suggest that gE/gI facilitate efficient axonal transport of Us9-containing viral particles.
Previously, we demonstrated that efficient anterograde transport in axons required phosphorylation of Us9 at S51 and S53 (22). We therefore used phospho-specific antibodies raised against these residues to determine if Us9 is inefficiently phosphorylated in the absence of gE/gI. Us9 was phosphorylated at wild-type levels at these key residues in the presence (PRV Becker infection) or absence (PRV 99 infection) of gE/gI (Fig. 2D). These results indicate that gE/gI expression does not impact Us9 posttranslation modification.
Efficient interaction between Us9 and KIF1A requires expression of gE and gI.
Since anterograde-directed spread of PRV is dependent upon the recruitment of the kinesin-3 motor KIF1A by Us9 and gE/gI are also required for efficient sorting into axons, we assessed whether expression of gE/gI is necessary to mediate the Us9-KIF1A interaction. We compared the efficiency of KIF1A copurification with GFP-Us9 during infection of differentiated PC12 cells with PRV expressing GFP-Us9 in a wild-type background (PRV 340), GFP-Us9 in a gE, gI, and Us2 null background (PRV 444), and GFP-Us9 transduced by a nonreplicating adenoviral transduction vector (Ad TK101). Immunoaffinity purification experiments were performed using custom-made high-affinity anti-GFP antibodies, which have been previously used to isolate GFP-tagged PRV membrane protein complexes (6, 33). Lysis and immunoaffinity purification conditions were optimized to preserve lipid raft membrane microdomains; incorporation of Us9 within these membrane domains is necessary for Us9 to mediate anterograde spread and for interactions with KIF1A (6, 45). At 16 hpi, KIF1A copurified with GFP-Us9 during wild-type infection, but this interaction was not seen in the absence of the other PRV proteins (Fig. 3A). Furthermore, at this time postinfection, GFP-Us9 does not copurify with KIF1A in the absence of gE, gI, and Us2 expression. Given that Us2 is not required for anterograde-directed spread, these findings suggested that expression of gE/gI is required for Us9-KIF1A interactions. Since the Us9-KIF1A interaction decreases at late time points during infection (6), we performed this coimmunoaffinity purification analysis at three time points (8, 16, and 24 hpi) (Fig. 3B). For PRV 444, Us9-KIF1A complex formation was markedly reduced at all three time points, although a small quantity of interactions were detected at 8 hpi. The reduced Us9-KIF1A complex formation is consistent with the severe reduction, but not complete absence, of anterograde-directed spread observed in vitro during gE/gI null infections (Fig. 1).
Fig 3.
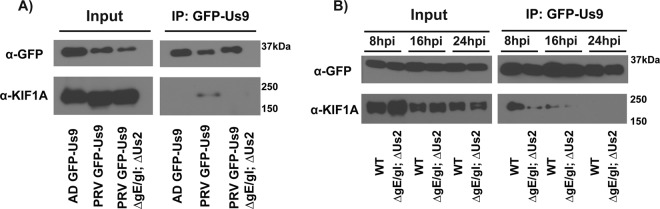
Expression of gE/gI is necessary for efficient Us9-KIF1A interactions. (A) Differentiated PC12 cells were infected with the indicated PRV strains, lysed at 16 hpi, and subject to immunoaffinity purification (IP) using anti-GFP antibodies. Western blot analysis was used to assess the efficiency of KIF1A copurification with GFP-Us9 following infection with the indicated viral strains. (B) Western blot analysis of immunoaffinity purifications of differentiated PC12 cells infected with the indicated viral strains over the course of infection.
Subcellular localization and transport of functional GFP-tagged gE fusion proteins.
To visualize the subcellular localization of gE in neurons and then assay potential interacting partners, we constructed PRV recombinants that express functional full-length gE-GFP fusion proteins. To verify that this fusion protein was functional for anterograde spread, we first infected chambered neuronal cultures with wild-type PRV expressing GFP (PRV 151), PRV gE null (PRV 758), or PRV gE-GFP (PRV 187) (Fig. 4). At 24 hpi, viral titers in the S compartment were similar for all three PRV recombinants, indicating that these viruses replicated efficiently in cell bodies. Spread of the PRV gE null mutant to the N compartment was markedly reduced compared to that of the wild-type control (mean of 6.4 × 104 PFU/ml for PRV 758 and 2.9 × 107 PFU/ml for PRV 151), as previously reported (24). In contrast, anterograde spread of PRV expressing gE-GFP was essentially identical to that of wild-type PRV expressing soluble GFP (mean of 1.3 × 107 PFU/ml for PRV 187). These experiments indicate that the gE-GFP fusion protein is functional for promoting anterograde spread of infection in vitro.
Fig 4.
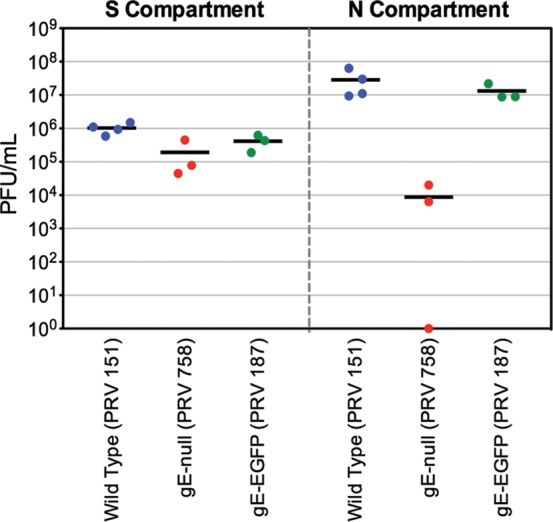
PRV expressing gE-GFP is competent for efficient anterograde spread in vitro. Viral titers in the S (left panel) and N (right panel) compartments of chambered SCG cultures at 24 hpi with PRV 151 (GFP), PRV 758 (gE null), or PRV 187 (gE-GFP) are shown. Point estimates reflect viral titers in each compartment. Lines indicate the medians.
To further establish functionality of the gE-GFP fusion protein in infected neurons, we utilized confocal microscopy to visualize fluorescent protein localization and then performed live imaging experiments to characterize particle dynamics. We infected SCG neurons with PRV 199 (expresses both mRFP-VP26 and gE-GFP) (Fig. 5A). At 8 hpi, gE-GFP localized predominantly to intracellular vesicular compartments, with fluorescence barely detectable at the plasma membrane. In live-cell imaging studies with SCG neurons infected with PRV 199 (Fig. 5B) we observed significant colocalization of gE and capsid protein fluorescent signals on punctate structures moving in the anterograde direction. Approximately 95% of anterograde-directed capsid puncta labeled with mRFP-VP26 also had detectable gE-GFP signal (Fig. 5C). These data are similar to previous reports where over 95% of anterograde-moving capsids colabeled with GFP-Us9 and mCitrine-KIF1A (6, 44). We conclude that nearly all mRFP-VP26-positive capsids undergo anterograde transport in complex with detectable levels of gE-GFP.
Fig 5.

Visualization of gE-GFP in PRV-infected SCG neurons. (A) Confocal microscopy of an SCG cell body infected with PRV 199 (expresses gE-GFP and mRFP-VP26) at 8 hpi. (B) Live-cell epifluorescence microscopy imaging of SCG axons was performed at 8 hpi for 3 min per axon. GFP and mRFP channels are shown sequentially. Two anterograde-directed puncta, each representing enveloped viral particles, are highlighted with black and green triangles, respectively. (C) Anterograde-directed mRFP-VP26-positive punctate structures were scored for colocalization with gE-GFP. A total of 65 puncta were counted across 5 movies, representative of 2 biological replicates.
Identification of gE-GFP interactions with viral and host proteins.
Since gE and gI are required for efficient complex formation between Us9 and KIF1A and we previously identified the host and viral proteins in the Us9/KIF1A complex (6), we sought to determine the proteins that interact with gE under similar conditions. We performed coimmunoaffinity purifications from PC12 cells infected with PRV 187 (gE-GFP) or PRV 151 (diffusible GFP) as a control. At 20 hpi, cells were harvested and extracted with lysis buffer that preserves the incorporation of gE within lipid rafts (45). Whole-cell lysates were subjected to immunoaffinity purification using the same high-affinity polyclonal anti-GFP antibodies described above for isolating GFP-Us9 complexes. Immunoisolates of gE-GFP and GFP were resolved by SDS-PAGE, digested in-gel with trypsin, and analyzed by nLC-tandem MS (MS/MS) using an LTQ-Orbitrap Velos mass spectrometer. Immunoaffinity purification and mass spectrometry analysis of GFP-Us9 (from PRV 340-infected cells at 20 hpi) were performed in parallel using identical extraction and purification conditions. Data from GFP-Us9 purifications have been previously published (6). A minimum of three unique peptides in at least one replicate was required for protein identification. The specificity of protein interactions in the gE-GFP sample compared to the GFP controls was assessed by a previously described label-free spectral counting approach (40) to assess the relative enrichment of each protein in the gE-GFP sample compared to the GFP controls. Proteins with an average spectral count enrichment of at least 3-fold compared to the GFP controls and a minimum of five spectra were considered putative gE-associated proteins.
Overall, 46 proteins that met these criteria were identified. These proteins were manually categorized based on their subcellular localization and function (Table 1). The relative abundance of each protein was calculated using the normalized spectral abundance factor (NSAF) method (43). For each putative gE-associated protein, the number of spectral counts was normalized by the protein's length and then divided by the sum of all normalized spectral counts and expressed as a percentage (Table 1). As expected, gE was the most abundant protein, followed by its well-characterized binding partner gI (30). Six additional viral proteins were enriched in the gE-GFP sample, five of which represent structural proteins belonging to the envelope layer of PRV extracellular virions (Us9, UL43, and glycoproteins gC, gH, and gM) (42). With the exception of gH, these viral proteins also copurified with GFP-Us9 at 20 hpi (6).
Additionally, 38 cellular proteins copurified with gE-GFP. Among the host proteins that copurified with gE-GFP were all eight subunits of the TRiC/CCT chaperonin complex, a 1-MDa hetero-oligomer that assists the folding of proteins in eukaryotes (47). Additionally, we identified two isoforms of Bat3/Bag6 (homologues of yeast Get4). Bat3/Bag6 functions as a cytosolic chaperone that facilitates targeting of tail-anchored membrane proteins to the ER (48). Of the cellular proteins we detected, five proteins also copurified with GFP-Us9 (6). These include the ER proteins Sec61a1, very-long-chain enoyl coenzyme A (enoyl-CoA) reductase (Tecr), and ATP2a2 (an ER ATPase subunit), as well as importin β and the glucose transporter Slc2a3. Importantly, KIF1A did not copurify with gE-GFP under these conditions, though Us9 was detectable in the complex. This finding indicates that gE/gI may facilitate or stabilize the interaction between Us9 and KIF1A indirectly. It also remains possible that our biochemical purification of gE-interacting proteins under these conditions may not preserve weak interaction between gE and KIF1A.
DISCUSSION
Directional spread of alphaherpesvirus infection requires long-distance transport of viral particles in axons. We previously demonstrated that KIF1A mediates Us9-dependent axonal sorting and anterograde transport of viral particles (6). Here, we utilized fluorescent fusion proteins and biochemical purifications to identify a role for the viral proteins gE and gI in mediating efficient anterograde spread of infection. We extended previous observations that gE/gI are required for efficient axonal sorting of Us9-containing viral particles through live-cell imaging and infections of chambered neuronal cultures. Furthermore, we confirmed that the Us2 protein, which copurifies with Us9, is not required for anterograde spread in vitro (Fig. 1) and in vivo (T. del Rio and L. W. Enquist, unpublished data). By immunoaffinity purification, we demonstrated that gE/gI are required for facilitating or stabilizing the interaction between Us9 and KIF1A. Since KIF1A does not copurify with gE-GFP, we propose that gE/gI indirectly mediate or stabilize the Us9-KIF1A interaction.
The gE/gI proteins are important for cell-to-cell spread in nonneuronal cells and anterograde spread in neurons, but it is not clear if these two phenotypes reflect common or distinct mechanisms. The gE protein contains conserved endocytosis motifs in its cytoplasmic tail that are required for efficient cell-to-cell spread (49, 50). Since gE and other viral membrane proteins copurify with lipid raft-associated GFP-Us9 (6), the endocytosis function of gE/gI may be important for recovery of these proteins from the plasma membrane. These endocytic vesicles may serve as substrates for secondary envelopment (51) or for fusion with other secondary envelopment membranes. This process would ensure that newly made viral particles contain all the necessary viral and host membrane proteins for carrying out the sequential molecular steps leading to egress and reentry. For example, an axon-targeted viral particle must contain Us9 in the transport vesicle membrane to recruit KIF1A for axonal sorting. The virion envelope inside the transport vesicle must carry the viral fusion glycoprotein complex gB/gH/gL to enable entry into a new cell after release from the axon (52, 53). However, other data are not compatible with this model. First, gE mutant proteins with truncated cytoplasmic tails are incorporated into extracellular virions, although with reduced efficiency compared to full-length gE (49). This finding indicates that the endocytosis motifs of gE/gI are not required for virion assembly or release from nonneuronal cells. Second, expression of gE is not required for incorporation of Us9 into virus envelopes (Fig. 2A) (46). Finally, gE and gI null mutants show reduced but measureable anterograde neuronal spread in vitro, even though expression of these proteins is required for efficient Us9-KIF1A complex formation (Fig. 3). Previously, Ch'ng and Enquist found in Campenot chamber experiments that gE null mutants produced minimally detectable anterograde-directed spread at early times after infection though viral titers in the axonal compartment did increase at later times (15, 24). In this study, we found that the Us9-KIF1A interaction in the absence of gE/gI is detected by Western blot analysis only at early times postinfection (Fig. 4B). Therefore, the increased anterograde directed spread by gE/gI null mutants late in infection likely reflects a limited number of early spread events that then amplify in the detector cell layer. We recently demonstrated that the interaction between Us9 and KIF1A is more robust at early times postinfection (6). Reduced copurification of KIF1A with GFP-Us9 is correlated with depletion of KIF1A over the course of infection and is dependent on Us9 expression. This is consistent with previous measurements showing that the overall number of stalled capsids in axons increases over the course of infection (5).
One hypothesis for how gE/gI function to promote anterograde spread via Us9-KIF1A binding is by selective membrane partitioning. Since the secondary envelopment membrane is ultimately divided into two functionally distinct membranes, i.e., the virion envelope and the transport vesicle, gE/gI may be involved in selective targeting or partitioning of viral proteins between these two topologically distinct compartments. This would ensure, for example, that gB/gH/gL is localized to the virion envelope in order to facilitate membrane fusion while Us9 is localized to the transport vesicle to facilitate axonal sorting. Taylor et al. previously hypothesized that GFP-Us9 could be selectively enriched in the transport vesicle membrane compared to the virion envelope membrane (44). In agreement with this, a recent report from Bohannon et al. provides evidence that the secondary envelopment process results in the asymmetric distribution of membrane and tegument proteins within mature virions (54). This work provides evidence that secondary envelopment and virion assembly produce asymmetric viral particles and suggests a framework for how gE/gI (and, potentially, other proteins) may function to establish this asymmetry. In this model, viral particles that are within transport vesicles containing Us9-rich membranes would be more likely to recruit KIF1A and undergo axonal sorting.
gE/gI may also facilitate the formation of KIF1A-Us9 complexes indirectly by targeting Us9 to the correct membrane microdomains. Proteomic analysis of gE-GFP immunoaffinity purifications revealed that gE-GFP copurifies with several host proteins involved in a variety of cellular functions (Table 1). Of particular interest is Bat3/Bag6, which targets tail-anchored membrane proteins to the ER (48). Us9 is a tail-anchored membrane protein that must be incorporated within lipid rafts in order to interact with KIF1A and facilitate anterograde spread (6, 45). However, gE expression is not required for Us9 lipid raft localization, such that the copurification of Bat3/Bag6 with gE may simply be a marker of the subcellular localization of these proteins. Further work is necessary to determine whether the interaction between Bat3/Bag6 and gE is important for efficient membrane localization of Us9.
The limited overlap between host proteins that copurify with GFP-Us9 and gE-GFP, respectively, may reflect their localization to different membrane microdomains during recruitment of the KIF1A motor complex. Under this model, gE would function indirectly to stabilize Us9-KIF1A complexes. Alternatively, gE/gI may reside in the same membrane microdomain as Us9 but not directly interact with Us9-KIF1A protein complexes. Thus, gE/gI interactions with KIF1A may be more indirect or transient than interactions between Us9 and KIF1A. An additional explanation for the differences in proteins that copurify with gE-GFP and GFP-Us9 relates to the stability of these complexes during the infection time course. The Us9-KIF1A complex is highly enriched at early times postinfection (6). Over the course of infection, we demonstrated that KIF1A levels are diminished in the cell (6), such that at later times postinfection, Us9 proteins are likely in great excess to KIF1A. This is consistent with our finding that gE-GFP copurifies with Us9 but not KIF1A at 20 hpi. However, based on our previous proteomic analysis, the reduced copurification of KIF1A with GFP-Us9 at late time points is the result of a regulated process. The remaining proteins that consistently copurified with GFP-Us9 over the course of infection, including gE, did not follow this trend (6). Furthermore, immunoaffinity purification of gE-GFP was performed in parallel with purification of GFP and GFP-Us9 using identical lysis buffer and purification conditions. Mass spectrometry analysis of these samples showed that KIF1A was detected robustly and reproducibly only in the GFP-Us9 sample (6). Thus, the majority of the host proteins that copurify with gE-GFP but not GFP-Us9 at 20 hpi may reflect gE's other functions besides axonal sorting. These proteins may copurify with gE proteins that are localized to membranes that are not involved in KIF1A-dependent transport.
Overall, our findings suggest that the glycoproteins gE and gI mediate anterograde spread of infection indirectly by facilitating efficient interactions between Us9 and KIF1A, which are directly required for axonal sorting and transport. Further work is needed to establish the mechanisms by which gE/gI mediate Us9-KIF1A interactions as well as the molecular function of this heterodimer during infection of nonneuronal cells.
ACKNOWLEDGMENTS
We thank Jens-Bernhard Bosse (Enquist lab) for assistance with spinning-disc confocal microscopy experiments.
This work was supported by National Institutes of Health grants to L.W.E. (R37 NS33506, R01 NS060699, and P40 RR18604) and I.M.C. (DP1 DA026192), a Human Frontiers Science Program Organization award to I.M.C (RGY0079/2009-C), a National Science Foundation graduate research fellowship to T.K. (DGE-0646086), and an American Cancer Society postdoctoral research fellowship to M.P.T. (PF-1005701-MPC).
Footnotes
Published ahead of print 26 June 2013
REFERENCES
- 1.Smith BN, Banfield BW, Smeraski CA, Wilcox CL, Dudek FE, Enquist LW, Pickard GE. 2000. Pseudorabies virus expressing enhanced green fluorescent protein: a tool for in vitro electrophysiological analysis of transsynaptically labeled neurons in identified central nervous system circuits. Proc. Natl. Acad. Sci. U. S. A. 97:9264–9269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Smith G. 2012. Herpesvirus transport to the nervous system and back again. Annu. Rev. Microbiol. 66:153–176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mettenleiter TC, Ehlers B, Muller T, Yoon K-J, Teifke JP. 2012. Herpesviruses, p 421–446 In Zimmerman J, Karriker L, Ramirez A, Schwartz K, Stevenson G. (ed), Diseases of swine, 10th ed. John Wiley & Sons, Inc., New York, NY [Google Scholar]
- 4.Rio TD, Chng TH, Flood EA, Gross SP, Enquist LW. 2005. Heterogeneity of a fluorescent tegument component in single pseudorabies virus virions and enveloped axonal assemblies. J. Virol. 79:3903–3919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Smith GA, Gross SP, Enquist LW. 2001. Herpesviruses use bidirectional fast-axonal transport to spread in sensory neurons. Proc. Natl. Acad. Sci. U. S. A. 98:3466–3470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kramer T, Greco TM, Taylor MP, Ambrosini AE, Cristea IM, Enquist LW. 2012. Kinesin-3 mediates axonal sorting and directional transport of alphaherpesvirus particles in neurons. Cell Host Microbe 12:806–814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zaichick SV, Bohannon KP, Hughes A, Sollars PJ, Pickard GE, Smith GA. 2013. The herpesvirus VP1/2 protein is an effector of dynein-mediated capsid transport and neuroinvasion. Cell Host Microbe 13:193–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Koyuncu OO, Perlman DH, Enquist LW. 2013. Efficient retrograde transport of pseudorabies virus within neurons requires local protein synthesis in axons. Cell Host Microbe 13:54–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tirabassi RS, Enquist LW. 2000. Role of the pseudorabies virus gI cytoplasmic domain in neuroinvasion, virulence, and posttranslational N-linked glycosylation. J. Virol. 74:3505–3516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kratchmarov R, Taylor MP, Enquist LW. 2012. Making the case: married versus separate models of alphaherpes virus anterograde transport in axons. Rev. Med. Virol. 22:378–391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kramer T, Enquist LW. 2013. Directional spread of alphaherpesviruses in the nervous system. Viruses 5:678–707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brideau AD, Banfield BW, Enquist LW. 1998. The Us9 gene product of pseudorabies virus, an alphaherpesvirus, is a phosphorylated, tail-anchored type II membrane protein. J. Virol. 72:4560–4570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Husak PJ, Kuo T, Enquist LW. 2000. Pseudorabies virus membrane proteins gI and gE facilitate anterograde spread of infection in projection-specific neurons in the rat. J. Virol. 74:10975–10983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tirabassi RS, Townley RA, Eldridge MG, Enquist LW. 1997. Characterization of pseudorabies virus mutants expressing carboxy-terminal truncations of gE: evidence for envelope incorporation, virulence, and neurotropism domains. J. Virol. 71:6455–6464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ch'ng TH, Enquist LW. 2005. Efficient axonal localization of alphaherpesvirus structural proteins in cultured sympathetic neurons requires viral glycoprotein E. J. Virol. 79:8835–8846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brideau AD, Card JP, Enquist LW. 2000. Role of pseudorabies virus Us9, a type II membrane protein, in infection of tissue culture cells and the rat nervous system. J. Virol. 74:834–845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McGraw HM, Awasthi S, Wojcechowskyj JA, Friedman HM. 2009. Anterograde spread of herpes simplex virus type 1 requires glycoprotein E and glycoprotein I but not Us9. J. Virol. 83:8315–8326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Snyder A, Polcicova K, Johnson DC. 2008. Herpes simplex virus gE/gI and US9 proteins promote transport of both capsids and virion glycoproteins in neuronal axons. J. Virol. 82:10613–10624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Al-Mubarak A, Simon J, Coats C, Okemba JD, Burton MD, Chowdhury SI. 2007. Glycoprotein E (gE) specified by bovine herpesvirus type 5 (BHV-5) enables trans-neuronal virus spread and neurovirulence without being a structural component of enveloped virions. Virology 365:398–409 [DOI] [PubMed] [Google Scholar]
- 20.Butchi NB, Jones C, Perez S, Doster A, Chowdhury SI. 2007. Envelope protein Us9 is required for the anterograde transport of bovine herpesvirus type 1 from trigeminal ganglia to nose and eye upon reactivation. J. Neurovirol. 13:384–388 [DOI] [PubMed] [Google Scholar]
- 21.Brum MCS, Coats C, Sangena RB, Doster A, Jones C, Chowdhury SI. 2009. Bovine herpesvirus type 1 (BoHV-1) anterograde neuronal transport from trigeminal ganglia to nose and eye requires glycoprotein E. J. Neurovirol. 15:196–201 [DOI] [PubMed] [Google Scholar]
- 22.Kratchmarov R, Taylor MP, Enquist LW. 2013. Role of us9 phosphorylation in axonal sorting and anterograde transport of pseudorabies virus. PLoS One 8:e58776. 10.1371/journal.pone.0058776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lyman MG, Feierbach B, Curanovic D, Bisher M, Enquist LW. 2007. Pseudorabies virus Us9 directs axonal sorting of viral capsids. J. Virol. 81:11363–11371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ch'ng TH, Enquist LW. 2005. Neuron-to-cell spread of pseudorabies virus in a compartmented neuronal culture system. J. Virol. 79:10875–10889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cristea IM, Williams R, Chait BT, Rout MP. 2005. Fluorescent proteins as proteomic probes. Mol. Cell Proteomics 4:1933–1941 [DOI] [PubMed] [Google Scholar]
- 26.Greene LA, Tischler AS. 1976. Establishment of a noradrenergic clonal line of rat adrenal pheochromocytoma cells which respond to nerve growth factor. Proc. Natl. Acad. Sci. U. S. A. 73:2424–2428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ch'ng TH, Flood EA, Enquist LW. 2005. Culturing primary and transformed neuronal cells for studying pseudorabies virus infection. Methods Mol. Biol. 292:299–316 [DOI] [PubMed] [Google Scholar]
- 28.Szpara ML, Tafuri YR, Parsons L, Shamim SR, Verstrepen KJ, Legendre M, Enquist LW. 2011. A wide extent of inter-strain diversity in virulent and vaccine strains of alphaherpesviruses. PLoS Pathog. 7:e1002282. 10.1371/journal.ppat.1002282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Curanovic D, Ch'ng TH, Szpara M, Enquist L. 2009. Compartmented neuron cultures for directional infection by alpha herpesviruses. Curr. Protoc. Cell Biol. Chapter 26:Unit 26.4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Whealy ME, Card JP, Robbins AK, Dubin JR, Rziha HJ, Enquist LW. 1993. Specific pseudorabies virus infection of the rat visual system requires both gI and gp63 glycoproteins. J. Virol. 67:3786–3797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cristea IM, Chait BT. 2011. Conjugation of magnetic beads for immunopurification of protein complexes. Cold Spring Harb. Protoc. 2011:pdb.prot5610. 10.1101/pdb.prot5610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pomeranz LE, Reynolds AE, Hengartner CJ. 2005. Molecular biology of pseudorabies virus: impact on neurovirology and veterinary medicine. Microbiol. Mol. Biol. Rev. 69:462–500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cristea IM, Chait BT. 2011. Affinity purification of protein complexes. Cold Spring Harb. Protoc. 2011:pdb.prot5611. 10.1101/pdb.prot5611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Johnson DC, Webb M, Wisner TW, Brunetti C. 2001. Herpes simplex virus gE/gI sorts nascent virions to epithelial cell junctions, promoting virus spread. J. Virol. 75:821–833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dingwell KS, Johnson DC. 1998. The herpes simplex virus gE-gI complex facilitates cell-to-cell spread and binds to components of cell junctions. J. Virol. 72:8933–8942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Farnsworth A, Wisner TW, Johnson DC. 2007. Cytoplasmic residues of herpes simplex virus glycoprotein gE required for secondary envelopment and binding of tegument proteins VP22 and UL11 to gE and gD. J. Virol. 81:319–331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Han J, Chadha P, Starkey JL, Wills JW. 2012. Function of glycoprotein E of herpes simplex virus requires coordinated assembly of three tegument proteins on its cytoplasmic tail. Proc. Natl. Acad. Sci. U. S. A. 109:19798–19803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mettenleiter TC. 2003. Pathogenesis of neurotropic herpesviruses: role of viral glycoproteins in neuroinvasion and transneuronal spread. Virus Res. 92:197–206 [DOI] [PubMed] [Google Scholar]
- 39.Card JP, Whealy ME, Robbins AK, Enquist LW. 1992. Pseudorabies virus envelope glycoprotein gI influences both neurotropism and virulence during infection of the rat visual system. J. Virol. 66:3032–3041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tsai Y-C, Greco TM, Boonmee A, Miteva Y, Cristea IM. 2012. Functional proteomics establishes the interaction of SIRT7 with chromatin remodeling complexes and expands its role in regulation of RNA polymerase I transcription. Mol. Cell Proteomics 11:60–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Clase AC, Lyman MG, del Rio T, Randall JA, Calton CM, Enquist LW, Banfield BW. 2003. The pseudorabies virus Us2 protein, a virion tegument component, is prenylated in infected cells. J. Virol. 77:12285–12298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kramer T, Greco TM, Enquist LW, Cristea IM. 2011. Proteomic characterization of pseudorabies virus extracellular virions. J. Virol. 85:6427–6441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zybailov B, Mosley AL, Sardiu ME, Coleman MK, Florens L, Washburn MP. 2006. Statistical analysis of membrane proteome expression changes in Saccharomyces cerevisiae. J. Proteome Res. 5:2339–2347 [DOI] [PubMed] [Google Scholar]
- 44.Taylor MP, Kramer T, Lyman MG, Kratchmarov R, Enquist LW. 2012. Visualization of an alphaherpesvirus membrane protein that is essential for anterograde axonal spread of infection in neurons. mBio 3:e00063–12. 10.1128/mBio.00063-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lyman MG, Curanovic D, Enquist LW. 2008. Targeting of pseudorabies virus structural proteins to axons requires association of the viral Us9 protein with lipid rafts. PLoS Pathog. 4:e1000065. 10.1371/journal.ppat.1000065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brack AR, Klupp BG, Granzow H, Tirabassi R, Enquist LW, Mettenleiter TC. 2000. Role of the cytoplasmic tail of pseudorabies virus glycoprotein E in virion formation. J. Virol. 74:4004–4016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kalisman N, Adams CM, Levitt M. 2012. Subunit order of eukaryotic TRiC/CCT chaperonin by cross-linking, mass spectrometry, and combinatorial homology modeling. Proc. Natl. Acad. Sci. U. S. A. 109:2884–2889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Leznicki P, Clancy A, Schwappach B, High S. 2010. Bat3 promotes the membrane integration of tail-anchored proteins. J. Cell Sci. 123:2170–2178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tirabassi RS, Enquist LW. 1999. Mutation of the YXXL endocytosis motif in the cytoplasmic tail of pseudorabies virus gE. J. Virol. 73:2717–2728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brideau AD, Enquist LW, Tirabassi RS. 2000. The role of virion membrane protein endocytosis in the herpesvirus life cycle. J. Clin. Virol. 17:69–82 [DOI] [PubMed] [Google Scholar]
- 51.Hollinshead M, Johns HL, Sayers CL, Gonzalez-Lopez C, Smith GL, Elliott G. 2012. Endocytic tubules regulated by Rab GTPases 5 and 11 are used for envelopment of herpes simplex virus. EMBO J. 31:4204–4220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Curanovic D, Enquist LW. 2009. Virion-incorporated glycoprotein B mediates transneuronal spread of pseudorabies virus. J. Virol. 83:7796–7804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Curanovic D, Enquist L. 2009. Directional transneuronal spread of α-herpesvirus infection. Future Virol. 4:591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bohannon KP, Jun Y, Gross SP, Smith GA. 2013. Differential protein partitioning within the herpesvirus tegument and envelope underlies a complex and variable virion architecture. Proc. Natl. Acad. Sci. U. S. A. 110:E1613–E1620 [DOI] [PMC free article] [PubMed] [Google Scholar]