

Abstract
Passive immunoprophylaxis or immunotherapy with norovirus-neutralizing monoclonal antibodies (MAbs) could be a useful treatment for high-risk populations, including infants and young children, the elderly, and certain patients who are debilitated or immunocompromised. In order to obtain antinorovirus MAbs with therapeutic potential, we stimulated a strong adaptive immune response in chimpanzees to the prototype norovirus strain Norwalk virus (NV) (genogroup I.1). A combinatorial phage Fab display library derived from mRNA of the chimpanzees' bone marrow was prepared, and four distinct Fabs reactive with Norwalk recombinant virus-like particles (rVLPs) were recovered, with estimated binding affinities in the subnanomolar range. Mapping studies showed that the four Fabs recognized three different conformational epitopes in the protruding (P) domain of NV VP1, the major capsid protein. The epitope of one of the Fabs, G4, was further mapped to a specific site involving a key amino acid residue, Gly365. One additional specific Fab (F11) was recovered months later from immortalized memory B cells and partially characterized. The anti-NV Fabs were converted into full-length IgG (MAbs) with human γ1 heavy chain constant regions. The anti-NV MAbs were tested in the two available surrogate assays for Norwalk virus neutralization, which showed that the MAbs could block carbohydrate binding and inhibit hemagglutination by NV rVLP. By mixing a single MAb with live Norwalk virus prior to challenge, MAbs D8 and B7 neutralized the virus and prevented infection in a chimpanzee. Because chimpanzee immunoglobulins are virtually identical to human immunoglobulins, these chimpanzee anticapsid MAbs may have a clinical application.
INTRODUCTION
Noroviruses (NoVs) are a leading cause of epidemic gastroenteritis in both children and adults worldwide (1). Outbreaks commonly occur in settings such as hospitals, nursing homes, cruise ships, university dormitories, and military barracks. Although NoV illnesses are generally self-limiting, increased morbidity and mortality have been reported among vulnerable populations such as infants, the elderly, and immunocompromised individuals (2–6). It is estimated that NoV infection may account for up to 200,000 deaths per year in infants and young children in developing countries (7).
Currently, there are no vaccines or specific antiviral therapies available for the treatment of NoV infections, due largely to the unavailability of permissive cell culture systems and animal disease models. Most information regarding host immunity to NoV infection has originated from human challenge studies and epidemiological investigations (8–13). As a result, the immune correlates of protection are poorly understood. Successes in expression of recombinant virus-like particles (rVLPs) that mimic the antigenic structure of authentic virions (14–16) and identification of histo-blood group antigens (HBGAs) as cellular binding ligands for NoV infection (17–20) have facilitated efforts toward the development of prevention and treatment strategies (21–24).
Noroviruses are nonenveloped, ∼38-nm icosahedral viruses with an ∼7.5-kb, single-stranded, positive-sense RNA genome that encodes three open reading frames (ORFs). ORF1 encodes RNA-dependent RNA polymerase, while ORFs 2 and 3 encode the major (VP1) and minor (VP2) capsid proteins, respectively. VP1 is structurally divided into the shell (S) domain, which forms the internal structural core of the particle, and the protruding (P) domain, which is exposed on the outer surface (15). The P domain is further subdivided into the P1 subdomain (residues 226 to 278 and 406 to 520) and the P2 subdomain (residues 279 to 405) (15). P2 represents the most exposed surface of the viral particle and is involved in interactions with both neutralizing antibodies (Abs) and HBGA oligosaccharides (25–28).
Noroviruses are divided into five distinct genogroups (genogroup I [GI] to GV) based on VP1 sequence similarity. Virus strains from GI and GII are responsible for most human infections, and these genogroups are further subdivided into more than 25 different genotypes (29). Although human NoV GII.4 strains are now recognized as the predominant genotype, the GI.1 Norwalk virus (NV) is an established reference strain for the human noroviruses and has been studied extensively in human volunteers and chimpanzees (30). Early human challenge studies with NV provided evidence for short-term, but not long-term (>2 years), homotypic immunity following infection with NV (9, 10, 12) and also showed the absence of heterotypic immunity when cross-challenged with the GII.1 Hawaii virus (13). Later human challenge studies showed an association between secretor status and susceptibility to NV infection (17–20). Chimpanzees were found to be susceptible to NV infection (31) and proved useful as a model for studies of norovirus pathogenesis and vaccine development (31, 32). Finally, our understanding of human NoV virion structure is based largely on X-ray crystallographic and cryo-electron microscopy analyses of NV rVLPs (15, 16), and these rVLPs have been established recently as a promising norovirus vaccine candidate in a multicenter clinical efficacy trial (21).
The aim of the present study was to generate clinically useful chimpanzee-derived monoclonal antibodies (MAbs) against the well-studied NV. Since chimpanzee immunoglobulins (Igs) are virtually identical to human Igs (33) and a human MAb administered to a chimpanzee had an effective half-life comparable to the half-life of human IgG in humans (34), chimpanzee-derived MAbs may be administered to humans without modification. Antibodies to NV were induced in chimpanzees by an immunization strategy of infection and priming with live NV followed by boosting with NV rVLPs. A panel of NV-specific MAbs was then generated by phage display library technology from chimpanzee bone marrow B cells and immortalized memory B cells derived from peripheral blood mononuclear cells (PBMCs).
MATERIALS AND METHODS
Quantification of the NV challenge pool.
Data from 15 chimpanzees experimentally inoculated intravenously with various 10-fold serial dilutions (100 to 10−5 of a standard fecal suspension of NV [32; this study and our unpublished data]) were evaluated with the Reed-Muench equation (35) to yield a quantitative titer of the virus, expressed in 50% chimpanzee infectious doses (CID50).
Immunization of chimpanzees with Norwalk virus and construction of combinatorial Fab antibody library.
Chimpanzee 1618 was inoculated orally with 101 CID50 of prototype NV in a stool filtrate (preparation 8 FIIa F/T, 1976) and was boosted intravenously with NV rVLPs. Bone marrow was aspirated from the chimpanzee at days 60, 74, and 88 after the boost. Bone marrow-derived lymphocytes were prepared by centrifugation on a Ficoll-Hypaque gradient (GE Healthcare, Piscataway, NJ) and were used for construction of a combinatorial Fab phage display library, as described previously (36, 37). A Fab phage display library containing γ1 heavy chain and κ and λ light chains with a diversity of >108 was constructed. The chimpanzees were housed at Bioqual, Inc., in accordance with the Guide for the Care and Use of Laboratory Animals (38). The chimpanzee protocol (designated LID 15) was approved by the NIAID and Bioqual Institutional Animal Care and Use Committees. The facilities were fully accredited by the American Association for Accreditation of Laboratory Animal Care International.
Panning of the phage library and selection of NV-specific Fabs.
Phages were produced from the library, as described previously (37), and panned by affinity binding against NV rVLPs adsorbed to a 96-well enzyme-linked immunosorbent assay (ELISA) plate (Nunc, Rochester, NY). For each cycle of panning, 100 μl of NV rVLPs at 5 μg/ml in phosphate-buffered saline (PBS) was used to coat each well of an ELISA plate, and specific phages were selected by incubating 1012 phages with the coated VLP. The NV-specific Fab clones were enriched by three cycles of panning against the NV rVLP. After panning, 96 single phage-Fab clones were cultured in a 96-well plate for phage production. The resulting phages were screened for specific binding to NV rVLP by phage ELISA (37). Clones that bound to the NV rVLP but not to bovine serum albumin (BSA) were scored as NV-specific clones.
Isolation of NV-specific antibodies by immortalization of memory B cells.
Peripheral blood (50 ml) was collected from the chimpanzee immunized with NV rVLPs. The PBMCs were isolated by Ficoll-Hypaque density gradient centrifugation. B cells were first enriched from PBMCs by using the BD IMag human B-lymphocyte enrichment set (BD Biosciences, San Jose, CA) and then incubated with phycoerythrin (PE)-labeled anti-CD27 (Invitrogen Life Technologies, Grand Island, NY), PE-Cy5-labeled anti-CD22 (Invitrogen), Alexa 700-labeled anti-CD38 (eBioscience, San Diego, CA), and Atto 633-labeled anti-IgG (Jackson ImmunoResearch, West Grove, PA) Abs for 30 min at 4°C. The IgG-positive (IgG+) memory B cells were isolated by fluorescence-activated cell sorter (FACS) analysis based on positivity for CD27, CD22, and IgG markers and negativity for CD38. The FACS-sorted cells were used immediately for B-cell immortalization.
Memory B cells were cultured in U-bottom 96-well plates (Nunc) at 40 cells per well in RPMI 1640 medium supplemented with 10% fetal bovine serum, 50 U/ml penicillin, and 50 μg/ml streptomycin (all from Invitrogen Life Technologies). The cells were immortalized by maintenance for 3 weeks in the presence of 1 × 105 irradiated MRC-5 feeder cells (ATCC, Manassas, VA), 30% Epstein-Barr virus (EBV)-containing supernatant of the B95-8 cell line, and 1 μg/ml CpG2006 (39). The supernatant fluid of each well was screened by ELISA for binding to NV rVLPs, as described above. The total RNA was extracted from the cells producing Abs specific to NV rVLPs by using the ZR RNA MicroPrep kit (Zymo Research, Irvine, CA). cDNA synthesis and PCR amplification of VH and VL genes were carried out as described previously for phage library construction (37).
DNA sequence analysis of specific Fabs.
The genes coding for the variable regions of the heavy (VH) and light (VL) chains of NV-specific Fabs were sequenced. Fabs with distinct VH and VL sequences were regarded as separate Fab clones. The presumed immunoglobulin family usage, germ line origin, and somatic mutations were identified by comparison with data deposited in the IMGT sequence database (http://www.imgt.org/).
Production of Fabs and IgGs.
Constructs for Fab and full-length IgG1 expression were made as described previously (37). Histidine-tagged Fab was expressed in Escherichia coli and was affinity purified on a nickel column. IgG was expressed in transiently transfected 293T mammalian cells and purified on a protein A column. Both Fab and IgG were further purified through a cation-exchange SP column (GE Healthcare). Purities of the Fab and IgG were evaluated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), and the protein concentrations were determined by optical density (OD) measurements at 280 nm, with an A280 of 1.35 corresponding to 1.0 mg/ml.
ELISA.
Wells in a 96-well ELISA plate were coated with 100 μl of NV rVLPs or other NoV rVLPs at a concentration of 1.5 μg/ml, followed by incubation with 3% nonfat dry milk in PBS for blocking. After washing, the plate was incubated with 3-fold serially diluted anti-NV Fabs for 2 h at room temperature (RT). After washing, the plate was incubated for 1 h with horseradish peroxidase-conjugated antibodies against human IgG F(ab′)2. The color was developed by the addition of tetramethylbenzidine (TMB) reagent (KPL, Gaithersburg, MD), and color development was stopped with H2SO4 after 10 min. The OD at 450 nm was read in an ELISA plate reader. The data were plotted and the dose-response curves were generated with Prism software (Graphpad Software, Inc., San Diego, CA).
Affinity measurement by SPR.
Kinetics of binding of antibody Fab fragments to NV rVLPs were assessed by surface plasmon resonance (SPR) using a Biacore 1000 instrument (GE Healthcare). Running buffer contained 10 mM HEPES (pH 7.4), 150 mM NaCl, and 0.005% Tween 20 (HBS-EP buffer). NV rVLPs were coupled to the surface of a CM5 chip by using the manufacturer-recommended protocol for amine coupling. Briefly, a 7-min injection at 10 μl/min of a mixture of 0.4 M 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) and 0.1 M N-hydroxysulfosuccinimide (NHS) was used to activate the surface, and 25 μg/ml NV rVLPs in 10 mM acetate buffer (pH 4.5) was then injected across the surface to achieve 200 resonance units (RU), followed by deactivation of the surface by a 7-min injection of 1 M ethanolamine at 10 μl/min. Fabs at a concentration series of 1, 2.5, 5, 7.5, and 10 nM were passed over the NV rVLP surface for 2 min at 30 μl/min. Following dissociation for 6 min at 30 μl/min, the surface was regenerated by injecting a solution of Gentle Ag/Ab elution buffer (pH 6.6) (Thermo Scientific, Rockford, IL) supplemented with 0.01% (vol/vol) Triton X-100 (Sigma, St. Louis, MO) for 1 min at 50 μl/min. Data were analyzed with Biacore software (version 2.1.2), using the Langmuir model.
Competitive SPR.
Cross-competition between Fabs was carried out by SPR. NV rVLPs were coupled to the surface of a CM5 chip, as described above, to achieve approximately 200 RU. All Fabs were tested at a concentration of 100 nM. The first Fab was injected and observed for the binding of analyte to ligand via sensorgram. When the surface was saturated, the second Fab was injected immediately, and binding activity was observed. The surface was regenerated, and the order of injection of the Fabs was reversed to perform a two-way competition experiment. This procedure was repeated for all Fabs with all possible binary combinations, and the binding signal for each pair of Fabs was recorded and analyzed. An increase in the binding signal above 10% following injection of the second Fab was defined as an absence of competition between the two Fabs.
Epitope mapping by immunoprecipitation.
To express NV VP1 mutants with consecutive N-terminal deletions in vitro, the corresponding truncated ORF2 DNA fragments were PCR amplified by using the Elongase amplification system (Invitrogen) with plasmid pNV101 (40). The PCR-employed sense primers included a T7 RNA polymerase promoter sequence followed by a short noncoding sequence (GGGAACAGACCACC), an AUG codon, and the in-frame sequence of the NV ORF2 region of interest. The antisense primer contained a sequence complementary to the 3′ end of NV ORF2. To express the NV VP1 mutants with C-terminal deletions, the DNA fragments were amplified by using a sense primer that contained the T7 RNA polymerase promoter and a sequence corresponding to the beginning of ORF2 and antisense primers that contained an engineered in-frame terminator codon and sequence complementary to the various regions of the 3′ end of ORF2.
The amplified DNA fragments were agarose gel purified and used as templates in a coupled transcription-and-translation reaction (TNT T7 Quick for PCR DNA; Promega, Inc., Madison, WI). For radiolabeling of synthesized proteins, [35S]methionine (>1,000 Ci/mmol) from PerkinElmer (Waltham, MA) was used at a concentration of 0.4 mCi/ml.
Immunoprecipitation of the radiolabeled NV VP1 protein and its truncated versions from in vitro translation mixtures was performed according to protocols described previously (41), with minor modifications. Briefly, after normalization for protein expression levels, 5- to 15-μl aliquots of the TNT T7 Quick translation reaction mixtures were diluted with 50 μl of radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris-HCl [pH 7.5], 0.15 M NaCl, 1% Triton X-100, 0.6% sodium deoxycholate) without SDS. The proteins were incubated for 1 h at RT with either recombinant Fab or MAb (IgG) at a concentration of 10 μg/ml, and immune complexes were precipitated with protein G beads (Amersham Biosciences, Piscataway, NJ). Immunoprecipitation of the Fab complexes was enhanced by an additional 1-h incubation with anti-human Fab IgG at 4 μg/ml (Jackson ImmunoResearch). Binding, washing, and elution steps were carried out as described previously (41). The eluted proteins were resolved by SDS-PAGE in a 4-to-20% gradient Tris-glycine gel (Invitrogen), and the bands corresponding to the radiolabeled immunoprecipitated proteins were detected by autoradiography after exposure of the dried gel to X-ray film (Eastman Kodak Co., Rochester, NY).
Yeast surface display of VP1.
The gene encoding full-length NV VP1 (530 amino acid residues) was amplified by PCR with the addition of an NheI site at the 5′ end and a SalI site at the 3′ end. After digestion with NheI and SalI, the gene was cloned into yeast expression vector pCTCON2 so that the N terminus of VP1 was fused with a Saccharomyces cerevisiae protein, Aga2p, through a short flexible linker and the C terminus was tagged with c-myc. The construct was confirmed by sequencing and was designated NVVP1-pCTCON2.
Yeast transformation, culture, and expression were carried out as described previously (42). In brief, yeast EBY100 cells were transformed with NVVP1-pCTCON2 and selected on SDCAA agar (42) plates. Protein expression was induced by culturing yeast in SGCAA medium (42) at 22°C for 48 h with shaking. Cells were harvested, washed, and incubated with anti-c-myc antibody followed by Alexa Fluor 488 goat anti-chicken IgG to monitor VP1 expression or incubated with mouse anti-NV MAb NV4 and chimpanzee anti-NV Fabs B7, D9, E4, and G4, respectively, followed by anti-mouse IgG DyLight 649 or anti-human Fab DyLight 649 to determine antibody binding. The labeled cells were analyzed on a BD FACS Canto II flow cytometer.
Site-directed mutagenesis of the VP1 gene of NV.
To introduce 4 mutations found within or adjacent to the VP1 P2 subdomain of the GI.1 SW-2007 strain (GenBank accession number FJ384783) into the corresponding VP1 sequence of NV (43), six primer pairs were designed to create single G365N, V370I, I376V, and Y410F mutations as well as double (G365N/V370I, V370I/I376V, and G365N/I376V) mutations. In addition, a construct was generated with a triple (G365N/V370I/I376V) mutation. Site-directed mutagenesis of pCI-Norwalk (VP1) was performed by using the QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA) and complementary forward and reverse primers that carried the nucleotide mutations (44). The restriction enzyme DpnI (10 U/μl) was used to digest the parental DNA. Each of the mutated products was transformed into Epicurian Coli XL1-Blue supercompetent cells (Stratagene). Transformed cells were grown overnight on LB plates with carbenicillin (50 μg/ml), and individual colonies were used for plasmid amplification. The resulting plasmids were subjected to sequence analysis to verify the entire VP1 coding region and to confirm the presence of the introduced mutations.
Immunofluorescence microscopy.
Vero cells were plated into 96-well plates at 80,000 cells/well and infected with modified vaccinia T7 virus (1 PFU/cell) for 1 h. After infection, cells were transfected with 400 ng/well of each DNA construct with Lipofectamine 2000 (Invitrogen) according to the manufacturer's recommendations. Cells were incubated for 24 h and fixed with cold methanol for 20 min. Each Fab was used at a concentration of 5 μg/ml. Goat anti-human IgG F(ab′)2 conjugated with DyLight 549 (Jackson ImmunoResearch) was used for detection. Expression of VP1 was confirmed with a murine norovirus-specific cross-reactive MAb (1:200 dilution) and goat anti-mouse IgG(H+L) conjugated with Alexa Fluor 594 (Molecular Probes-Invitrogen, Carlsbad, CA). Cells transfected with a vector expressing green fluorescent protein (GFP) served as the negative control for specific Fab binding.
Protein modeling.
The solved structure of VP1 of NV (Protein Data Bank [PDB] accession number 1IHM) was used to identify the residues involved in binding with G4 Fab and visualized by using MacPyMol (DeLano Scientific LLC).
HBGA binding-blocking assay.
The ability of antibodies to block the binding of NV rVLPs to synthetic HBGA H type 1 (H1) trisaccharide was determined as described previously (32). Briefly, IgGs were 2-fold serially diluted and incubated for 2 h with 1.5 μg/ml NV rVLP, and the IgG-NV rVLP mixture was then transferred into NeutriAvidin-coated plates (Pierce, Rockford, IL) containing biotinylated H1 carbohydrate (Glycotech, Gaithersburg, MD) and incubated for 1 h. The binding of captured NV rVLP was determined by incubation with guinea pig anti-NV hyperimmune serum (1:2,000 dilution), followed by incubation with a peroxidase-conjugated goat anti-guinea pig serum (1:2,000 dilution; KPL). The reaction was visualized with a peroxidase substrate, 2,2′-azinobis(3-ethylbenzthiazolinesulfonic acid) (ABTS) (KPL), and read at 405 nm with a Dynex Technologies Revelation 4.25 plate reader (Dynatech). All incubations were performed at room temperature. The 50% blocking antibody titer (BT50) was defined as the reciprocal of the lowest antibody dilution tested that blocked at least 50% of binding compared to that in the absence of antibody pretreatment (control binding).
HAI assay.
Inhibition of the hemagglutination activity of NV rVLPs by antibodies was determined as described previously (45, 46). Briefly, serial dilutions of the MAbs were incubated with Norwalk rVLP (80 ng of VLPs or 16 hemagglutinin [HA] units) in V-bottomed 96-well plates and incubated for 1 h at RT. Fifty microliters of 0.75% type O human erythrocytes was added to the plate and incubated for 2 h at 4°C. The hemagglutination inhibition (HAI) titer was defined as the reciprocal of the highest dilution of antibody that completely inhibited hemagglutination by the viral antigen (Ag).
Antibody-mediated NV neutralization in a chimpanzee model.
Due to limited availability, one chimpanzee was studied in a preliminary experiment. Anti-NV MAb D8 (0.5 mg) at 1 mg/ml in 1× PBS was incubated with 10 CID50 of NV (8fIIa) for 1 h at room temperature, followed by incubation overnight at 4°C in a total volume of 1.5 ml. The following day, a chimpanzee was inoculated intravenously with the antibody-virus mixture. Stool samples from the chimpanzee were collected daily, from 1 day preinoculation to 4 weeks postinoculation. The titer of the NV genome was quantified by norovirus-specific quantitative real-time PCR (qRT-PCR) (32). The chimpanzee was observed for the appearance of clinical signs of gastroenteritis, including vomiting, diarrhea, and malaise, and was monitored weekly for elevation of levels of hepatic enzymes. Whole-blood samples were collected preinoculation and every other week until the termination of the study (4 weeks after inoculation). Anti-NV MAb B7 was tested similarly, except that only 1.0 ml of the inoculum was injected because of an allergic reaction in the chimpanzee. Thus, the chimpanzee actually received 0.35 mg of B7 mixed with 7 CID50 of NV.
RESULTS
Isolation and characterization of NV-specific Fabs.
A phage Fab display library constructed using bone marrow of chimpanzees immunized with NV was panned against NV rVLPs for three cycles. The phage-Fab clones specific to NV were identified by phage ELISA. Sequence analysis of the variable domains of the heavy (VH) and light (VL) chains showed that four unique clones specific to NV, B7, D8, E5, and G4 were recovered. By immortalization of memory B cells, an additional clone, F11, was recovered. These positive clones were subsequently converted to produce soluble Fabs. Each soluble Fab was confirmed for its specific binding to NV, as they bound only to NV rVLPs and not to BSA, phosphorylase b, thyroglobulin, or lysozyme. The ELISA binding profile of a representative Fab is shown in Fig. 1A.
Fig 1.
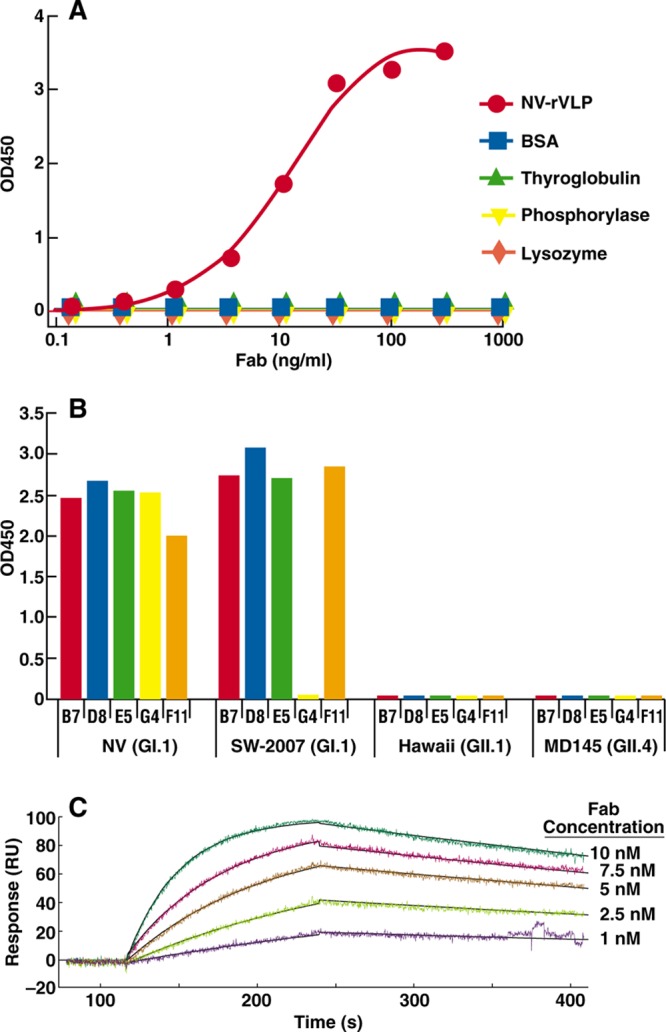
Binding specificities of anti-NV Fabs. (A) Binding in response to different concentrations of Fab D8 (a representative clone) with NV rVLPs or the unrelated proteins bovine serum albumin (BSA), thyroglobulin, lysozyme, and phosphorylase b, as measured by ELISA with antigens directly attached to the solid phase. (B) The binding profiles of Fabs B7, D8, E5, G4, and F11 at a concentration of 100 ng/ml on rVLPs from NV (GI.1), SW-2007 (GI.1), Hawaii (GII.1), and MD-145 (GII.4) that were attached to the solid phase at 1.5 μg/ml were measured by ELISA. OD, optical density. (C) Representative sensorgram of Fab B7 analyzed by SPR, showing raw data and curves fitted by using a 1:1 model.
To determine the binding spectrum of the anti-NV Fabs, we performed binding assays using an ELISA with rVLPs derived from representative human NoVs: NV (GI.1), NV-2007 (GI.1), Hawaii (GII.1), and MD-145 (GII.4). As shown in Fig. 1B, four of the five Fabs were reactive with both prototype NV (circulating in 1968) and NV-2007 (circulating in 2007) (43), whereas Fab G4 was reactive only with NV. In a separate experiment, B7 was shown to also react with GI.6 rVLPs (data not shown).The five Fabs were not reactive with rVLPs from any of the GII viruses tested. The absence of cross-reactivity with strains of GII was consistent with the serotypic differences between NV and Hawaii virus (HV) established in cross-challenge studies (13).
The interaction between Fab and immobilized rVLP was measured by SPR and recorded within a sensorgram; a representative tracing for Fab B7 is shown in Fig. 1C. The kinetic data were evaluated via fitting of the sensorgram data with BIAevaluation software using a 1:1 binding model. Chi-square values of 0.95, 2.94, 1.13, and 27.8 were estimated for Fabs D8, B7, G4, E5, and F11, respectively, indicating relatively good fitting. The kinetic constants, including the equilibrium dissociation constant (Kd), association rate constant (kon), and dissociation rate constant (koff), were determined for each Fab and are summarized in Table 1. High affinities, with a Kd range of 0.8 to 1.9 nM, were observed for all anti-NV clones. In general, anti-NV Fabs had relatively high on-rates and medium off-rates.
Table 1.
Affinities of anti-Norwalk virus Fabs determined by SPRa
| Fab | koff (1/s) | kon (M−1s−1) | Kd (M) |
|---|---|---|---|
| D8 | 1.56 × 10−3 | 2.02 × 106 | 7.72 × 10−10 |
| B7 | 1.53 × 10−3 | 1.89 × 106 | 8.09 × 10−10 |
| G4 | 1.42 × 10−3 | 1.46 × 106 | 9.74 × 10−10 |
| E5 | 4.18 × 10−3 | 2.23 × 106 | 1.89 × 10−9 |
| F11 | 6.55 × 10−4 | 4.42 × 105 | 1.48 × 10−9 |
Kinetics of binding of antibody Fab fragments to Norwalk virus-like particles were assessed by SPR using a Biacore 1000 instrument.
Analysis of the H and L chain repertoire within our panel of Abs showed that a single IgHV3 family with a biased usage of the JH4 gene segment was present in the heavy chains and that a single IGKV1 family was present in the light chains (Table 2). This marked restriction in V gene usage suggests that significant structural constraints may be operating in the selection of antibodies to NV. A similar restriction in gene usage was reported for Abs against other antigens, including rotavirus antigen (47), hepatitis C virus (HCV) antigen (48), rhesus D antigen (49), and Haemophilus influenzae capsule polysaccharide (50). Sequences in the variable regions of both heavy and light chains were mutated in frame without stop codons, as expected from affinity-matured plasma cells. All sequences had unique VDJ joint regions (Fig. 2), with mutation frequencies of 5 to 13% for heavy chains and 1 to 6% for light chains (Table 2). This range of mutation rates is consistent with an antigen-driven immune response (51) and may account for the high affinities of these Fabs (Table 1). Although mutations were detected in the framework regions, most differences were within the complementarity-determining regions (CDRs) (Fig. 2), consistent with other reported Ab-Ag interactions (52).
Table 2.
Assignment of genes coding for NV-neutralizing MAbs to their closest human germ line counterparts, based on nucleotide sequence homologya
| Fab | Germ line gene in: |
||||||
|---|---|---|---|---|---|---|---|
| Heavy chain |
Light chain |
||||||
| V gene | Identity (%)b | J gene | D gene | V gene | Identity (%) | J gene | |
| B7 | IGHV3-7 | 92 | IGHJ4 | IGHD6-25 | IGKV1-39 | 94 | IGKJ5 |
| D8 | IGHV3-74 | 90 | IGHJ4 | IGHD5-12 | IGKV1-27 | 95 | IGKJ3 |
| E5 | IGHV3-23 | 89 | IGHJ4 | IGHD5-12 | IGKV1-27 | 99 | IGKJ4 |
| G4 | IGHV3-23 | 87 | IGHJ4 | IGHD1-26 | IGKV1-5 | 94 | IGKJ4 |
| F11 | IGHV3-66 | 95 | IGHJ6 | IGHD6-13 | IGKV1-37 | 95 | IGKJ2 |
The closest human V gene germ lines were identified by a search of the IMGT database (http://www.imgt.org/).
The VH and VL genes before CDR3 were used to calculate the percent nucleotide identity. Mutations in the first 20 bp were excluded, since these mutations could be introduced by PCR primers.
Fig 2.

Deduced amino acid sequences of variable domains of heavy (A) and light (B) chains of chimpanzee anti-NV Fabs. Complementarity-determining regions (CDR1 to CDR3), shown in boxes, and framework regions (FWR1 to FWR4) are assigned according to the nomenclature of Wu and Kabat (68). Relative to the B7 sequence, substitutions are expressed as single letters denoting amino acids, and identical residues are indicated by asterisks. Dashes denote the absence of corresponding residues relative to the longest sequence.
Anti-NV Fabs recognize conformational epitopes on NV VP1.
We first determined that none of the anti-NV Fabs was reactive with denatured NV rVLP protein in Western blots, indicating that the Fabs likely recognize conformational epitopes present in hetero-oligomeric forms of the capsid protein. To map the conformational epitopes, a series of constructs that expressed full-length or truncated forms of NV VP1 was generated to localize the binding sites of the antibodies by a radioimmunoprecipitation assay (RIPA). Since Fab F11 was generated months later by immortalizing memory B cells, only Fabs B7, D8, E5, and G4 were included in the assay. All the tested Fabs displayed the same binding pattern shown in Fig. 3A. Deletion of the N-terminal region of VP1 through most of the S domain did not affect Fab binding. However, deletion into the P domain from either the N or C terminus resulted in complete abolishment of antibody binding. This result suggests that anti-NV Fabs bind to conformational epitopes that require presentation in the context of virtually the entire P domain of VP1. The specificity of Fabs B7, D8, E5, and G4 for the NV P domain was confirmed with chimeric VLPs containing P and S domain swaps from GI or GII VP1 proteins (34) (data not shown).
Fig 3.

Epitope mapping. (A) Summary of epitope mapping of anti-NV Fabs B7, D8, E5, and G4 by radioimmunoprecipitation assay. 35S-labeled VP1 polypeptides, prepared in vitro, were incubated with anti-NV Fabs. The immune complexes were captured by protein G-coupled agarose beads and were separated by SDS-PAGE. The immunoprecipitated VP1 polypeptides were detected by exposing the dried gel to an X-ray film (not shown). Numbers denote the starting and ending amino acids. The polypeptides that reacted with Fab and, hence, were detected on an X-ray film were considered to be positive (+). (B) Yeast cells expressing the c-myc-tagged monomeric VP1 fusion protein were analyzed by FACS analysis for VP1 expression and binding of VP1 by mouse anti-NV MAb NV4 and by chimpanzee anti-NV Fabs B7, D8, E5, and G4. Percentages indicate the numbers of cells gated by positive VP1 expression that were bound by antibody.
To address whether the conformational epitopes are present in the monomeric form of VP1, we applied a strategy using yeast surface display. In principle, yeast-displayed VP1 is expressed in a monomeric form, since the VP1 is expressed fused with the yeast Aga2 protein and displayed on the yeast cell surface. The expression of VP1 and the binding of each MAb were monitored by flow cytometry for the detection of Alexa Fluor 488, which is directed to the c-myc tag at the C terminus of VP1 (display fluorescence), and DyLight 649, which is directed to the MAb/Fabs (Ab binding fluorescence). Display fluorescence was detectable in cells transformed with the VP1 construct, indicating positive expression of VP1 on the yeast surface. Anti-NV Fabs B7, D8, E5, and G4 along with a control MAb, mouse anti-NV MAb NV4, were tested for binding. MAb NV4 recognizes a conformational epitope within the NV P domain (G. I. Parra, unpublished data). As shown in Fig. 3B, 36% of c-myc-positive cells, when incubated with mouse MAb NV4, were positive for Ab binding fluorescence, indicating that this antibody can recognize an epitope present in the monomeric form. However, c-myc-positive cells treated with any one of the four chimpanzee anti-NV Fabs were negative for Ab binding fluorescence, suggesting that they recognize conformational epitopes present in hetero-oligomeric forms, such as dimers. The negative results with the Fabs were confirmed by testing the full-length IgG forms of MAbs D8 and B7 (data not shown).
To assess the spatial relationship of antigenic sites in the P domain, the five anti-NV Fabs were examined in a competitive SPR assay. The assay is based on the assumption that the full occupancy of a specific epitope in the P domain by a given Fab would compete with a second Fab that recognizes the homologous epitope. Four distinguishable epitopes were identified based on the competition patterns (Table 3). Among them, Fabs B7, G4, and F11 each recognized a distinct epitope because their binding did not inhibit the binding of any other Fabs. In contrast, D8 and E5 shared an epitope that was distinct from other epitopes.
Table 3.
Epitope groups recognized by anti-Norwalk virus Fabs revealed by competitive SPR
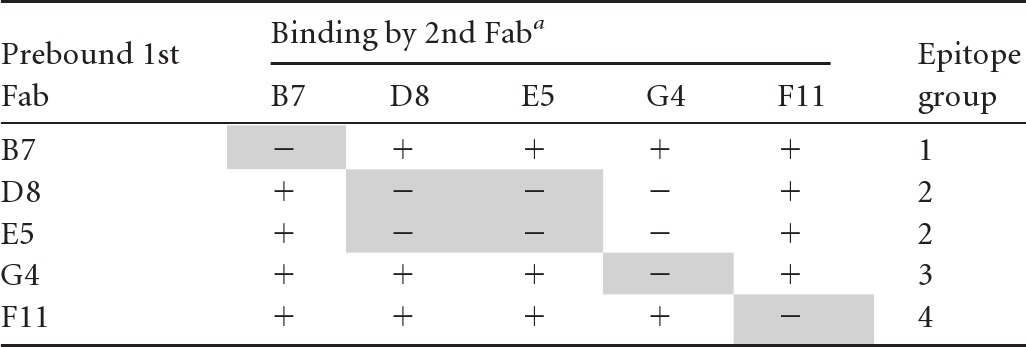
Shown are data for binding of the second Fab at a saturating concentration to Norwalk virus-like particles that was prebound with the first Fab at a saturating concentration. −, <10% increase in signal; +, >10% increase in signal. Epitope groups are identified by shading.
Fab G4 recognizes a specific site on VP1 involving amino acid residue G365.
Amino acid sequence comparison of the VP1 protein of Norwalk virus and that of SW-2007 virus revealed that there are 8 amino acid substitutions with no additional insertions or deletions. Among them, three changes (G365N, V370I, and I376V) were found in the VP1 P2 subdomain, and one (Y410F) was found in the VP1 P1 subdomain next to P2 (Fig. 4A). Since P2 constitutes the most exposed surface of the viral particle and is involved in interactions with antibodies and HBGA oligosaccharides, the impact of each of these four changes on G4 binding was tested. Cells transfected with an expression vector carrying VP1 of NV with single, double, or triple mutations were incubated with Fabs B7, D8, E5, and G4 and examined by an immunofluorescence assay. The data showed that G365 in P2 is a critical residue for G4 binding since the G365N mutation, whether presented as a single mutation or combined with other mutations, abolished G4 binding (Fig. 4B and data not shown). Mutation of the other three residues did not affect G4 binding. The G365N mutant was still reactive with the other three Fabs (Fig. 4B). Examination of the X-ray structure of NV VP1 revealed that G365 is located near, but not in, the HBGA binding site (Fig. 4C).
Fig 4.

Fine epitope mapping of Norwalk virus Fab G4. (A) Comparison of VP1 sequences of GI.1 Norwalk and SW-2007 virus strains. A schematic diagram representing VP1 protein is shown at the top. The locations of 530 amino acids are labeled, and S, P1, and P2 subdomains are shown in tan, light blue, and blue, respectively. The 8 amino acid differences between the Norwalk virus and SW-2007 strains, resulting in different reactivities to G4, are shown at the bottom. The boundaries between the S domain and the P1 and P2 subdomains are shown as green dotted lines. The 4 amino acid changes under study are underlined. (B) Immunofluorescence staining results from Vero cells transfected with different DNA constructs. Mutations of VP1 of Norwalk virus were introduced by using specific primers and a QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA) according to the manufacturer's recommendations. (C) X-ray structure of the VP1 dimer of NV (PDB accession number 1IHM). The different amino acids between Norwalk and SW-2007 viruses are shown in red. The localization of the key mutation G365N in the Norwalk virus VP1 P2 subdomain structure is indicated by arrows. The shell domain is shown in tan, and the P1 and P2 subdomains are shown in light blue and blue, respectively. Amino acids responsible for the HBGA binding sites are shown in light pink.
Anti-NV MAbs block HBGA binding and inhibit hemagglutination by NV.
Anti-NV IgGs were tested with available functional assays for norovirus antibodies. First, the IgGs were tested in an HBGA carbohydrate binding-blocking assay, which has been proposed as a surrogate neutralization assay (17). As shown in Fig. 5, all the MAbs blocked the binding of NV rVLPs to H1 HBGA carbohydrate in a dose-dependent manner. The estimated concentrations yielding 50% blocking were 2.5 nM for D8, 7.3 nM for B7, 4.7 nM for E5, 2.8 nM for G4, and 10.7 nM for F11. It was recently reported that the hemagglutination inhibition (HAI) activity of antibody correlates with protection from gastroenteritis in persons infected with NV (45). Therefore, we tested the HAI ability of the MAbs. We found that all five MAbs can efficiently inhibit viral hemagglutination activity with an antibody concentration of approximately 8 nM (data not shown).
Fig 5.

Detection of HBGA binding-blocking activities of anti-NV IgGs. Decreasing concentrations of monoclonal antibodies B7, D8, E5, G4, and F11 (IgG) were tested for their ability to block the interaction between recombinant NV rVLPs and H1 carbohydrates.
Anti-NV MAbs prevent NV infection of chimpanzees.
The cumulative data obtained from experimental inoculation of chimpanzees with the same NV challenge pool permitted us to calculate the CID50, which had not been calculated previously (32). We determined that the pool contained NV at a concentration of 102 CID50 per ml. We then tested whether HBGA blocking activity correlated with in vivo neutralization in chimpanzees (32). Due to the limited availability of chimpanzees for the experiment, MAbs D8 and B7 were chosen for testing in the animal model, given their different binding properties. An antibody-negative chimpanzee was administered an infectious NV inoculum (7 to 10 CID50), a known dose for 100% infection, that was preincubated with either MAb D8 or B7, and the chimpanzee was monitored for NV infection. The results showed that the chimpanzee did not shed virus in either case, consistent with neutralization of NV by the D8 and B7 antibodies.
DISCUSSION
Noroviruses are important agents of human gastroenteritis, and infections are associated with a significant disease burden, yet there are no disease interventions available. Despite substantial advances in the development of NoV vaccines based on VLPs (23), there are several remaining challenges to address in NoV vaccine development, including the duration of immunity, rapidly evolving new variant strains, and an incomplete understanding of the immune correlates of protection. Limited cross-protection between NoV genotypes within the same genogroup (GII.4 variant) and a lack of cross-protection between genogroups are observed (13).
As an alternative or complement to vaccines, passive immunization with anti-NoV MAbs could provide immediate protection against NoV infection in high-risk populations. For this purpose, we have successfully isolated five NV-specific antibodies from immunized chimpanzees and have demonstrated that these antibodies were able to block the binding of NV to HBGA carbohydrates, a known binding ligand for NV in the intestinal mucosa. We completed one study of antibody efficacy in a chimpanzee with two MAbs that recognize different epitopes prior to the suspension of such animal studies in U.S. research (53), and this investigation provided promising preliminary evidence that the antibody may exert a protective effect. Such antibodies may be useful in emergency prophylaxis and treatment of NoV infection, but additional efficacy data are needed. Because chimpanzees were the only permissive animal model for the evaluation of vaccines and antiviral compounds for GI NV (32), studies in adult volunteers may be required for the continued evaluation of these and other candidate therapeutic antibodies.
Here we describe the isolation of chimeric chimpanzee-human anti-NV neutralizing antibodies by combining hyperimmunization of chimpanzees with phage Fab display library technology, a method previously used for isolation of humanized MAbs against a variety of viral and bacterial pathogens (36, 37, 54–56). In addition, from PBMCs, we have isolated the F11 antibody against NV and several antibodies against NoV MD145 (GII.4) (data not shown) using B-cell immortalization with EBV (39). F11 had affinity and surrogate in vitro neutralizing activity (HBGA binding blocking and HAI) comparable to those of antibodies isolated from bone marrow B cells by phage display. Therefore, with this new method, it is possible to use PBMCs from any primed or infected chimpanzee or human for making therapeutic antibodies. Indeed, this method was recently used for isolation of human antibodies against GII.4 norovirus (57).
Pathogenic RNA viruses such as human immunodeficiency virus (HIV), hepatitis C virus (HCV), and influenza virus are markedly diverse (58–61). In recent years, several promising broadly neutralizing MAbs against HIV, HCV, and influenza viruses have been reported (62–66). These MAbs may prove useful for immunoprophylaxis, and knowledge of their binding sites can inform universal vaccine design. Analysis of the binding spectrum of antibodies isolated from chimpanzees immunized with NV in this study showed that four of the five Fabs were specific for GI.1 viruses and that the remaining one was exquisitely specific for NV, with no cross-reaction with NoVs of other genotypes. This finding is consistent with previous observations (13, 32) and may partly explain why there is little evidence of heterotypic immunity for NoVs. It will be important to expand our panel to include broadly reactive antibodies that recognize the diverse range of norovirus genogroups and genotypes. This may be achieved by additional panning against multiple NoV rVLPs.
We have shown that four anti-NV MAbs recognized conformational epitopes located within the P domain of the capsid protein. The entire P domain in an oligomeric form was absolutely required to maintain the correct conformation for antibody binding. Four distinguishable epitopes were identified through competitive SPR. Among them, the G4 epitope was mapped to a specific site involving the G365 residue in the P2 subdomain. Our data suggested that the loss of binding of MAb G4 to the G365N VP1 mutant was not due to a severe overall conformational change in the oligomeric form of the capsid protein, because the other antibodies recognized their cognate conformational epitopes on the mutagenized protein. Further study will be needed to elucidate the precise role of amino acid residue 365 in the formation of the G4 epitope. Our efforts to perform precise epitope mapping for the other epitopes with standard approaches (28), such as screening synthetic overlapping peptides spanning VP1 or phage display peptide libraries of NV VP1, were not successful, likely due to their conformational nature (data not shown). Instead, three-dimensional structural approaches such as X-ray crystallography of antigen-antibody complexes (52) and imaging of antigen-antibody complexes by cryo-electron microscopy (67) may be required.
We have established the NV infectivity titer for chimpanzees of the 8FIIa stool filtrate as 102 CID50 per ml. This pool represents the first identified human gastroenteritis virus and was used previously for infecting volunteers in 1974 and chimpanzees in 1978 and 2011 (13, 31, 32). For the 2011 study, a titer of 10 CID per 1 ml of the 2% stool filtrate was arbitrarily set, and most inocula consisted of a 10% dilution of this filtrate (listed as 1 CID). In reality, 1 CID was 10 CID50 and equivalent to 4 × 107 genome equivalents. Thus, the reported results (32) are more robust than previously reported. As noted by Reed and Muench in their original 1937 paper (35), “This [50%] endpoint is less affected by small chance variations than is any other; the worst in this respect being the 100% point so frequently used.” Chimpanzee studies were discontinued before these NV-specific antibodies could be tested for pre- and postexposure protection in vivo, and further testing will be needed in animal models (or human volunteers) and cell culture systems as they become available. When such systems are available, it may be possible to explore the use of an antibody cocktail that targets several different epitopes.
Noroviruses are a diverse group of viruses (29), and there is increasing recognition of their role in serious diarrheal illness (1, 7). Our proof-of-concept approach for the development of functional antibodies directed against NV should be applicable to other NoVs, including the predominant GII.4 viruses (1). Neutralizing MAbs against NoVs could prove useful for emergency prophylaxis to protect individuals in the proximity of a developing NoV outbreak or when encountering an increased risk of exposure. An effective treatment to alleviate chronic norovirus gastroenteritis in debilitated or immunocompromised individuals would be a welcome breakthrough in the management of these patients.
ACKNOWLEDGMENT
This research was supported by the Intramural Research Program of the NIAID, NIH.
Footnotes
Published ahead of print 19 June 2013
REFERENCES
- 1.Glass RI, Parashar UD, Estes MK. 2009. Norovirus gastroenteritis. N. Engl. J. Med. 361:1776–1785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Centers for Disease Control and Prevention 2007. Norovirus activity—United States, 2006-2007. MMWR Morb. Mortal. Wkly. Rep. 56:842–846 [PubMed] [Google Scholar]
- 3.Bryce J, Boschi-Pinto C, Shibuya K, Black RE. 2005. WHO estimates of the causes of death in children. Lancet 365:1147–1152 [DOI] [PubMed] [Google Scholar]
- 4.Green KY, Lew JF, Jiang X, Kapikian AZ, Estes MK. 1993. Comparison of the reactivities of baculovirus-expressed recombinant Norwalk virus capsid antigen with those of the native Norwalk virus antigen in serologic assays and some epidemiologic observations. J. Clin. Microbiol. 31:2185–2191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sakai Y, Nakata S, Honma S, Tatsumi M, Numata-Kinoshita K, Chiba S. 2001. Clinical severity of Norwalk virus and Sapporo virus gastroenteritis in children in Hokkaido, Japan. Pediatr. Infect. Dis. J. 20:849–853 [DOI] [PubMed] [Google Scholar]
- 6.Zintz C, Bok K, Parada E, Barnes-Eley M, Berke T, Staat MA, Azimi P, Jiang X, Matson DO. 2005. Prevalence and genetic characterization of caliciviruses among children hospitalized for acute gastroenteritis in the United States. Infect. Genet. Evol. 5:281–290 [DOI] [PubMed] [Google Scholar]
- 7.Patel MM, Widdowson MA, Glass RI, Akazawa K, Vinje J, Parashar UD. 2008. Systematic literature review of role of noroviruses in sporadic gastroenteritis. Emerg. Infect. Dis. 14:1224–1231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Atmar RL, Opekun AR, Gilger MA, Estes MK, Crawford SE, Neill FH, Graham DY. 2008. Norwalk virus shedding after experimental human infection. Emerg. Infect. Dis. 14:1553–1557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dolin R, Blacklow NR, DuPont H, Buscho RF, Wyatt RG, Kasel JA, Hornick R, Chanock RM. 1972. Biological properties of Norwalk agent of acute infectious nonbacterial gastroenteritis. Proc. Soc. Exp. Biol. Med. 140:578–583 [DOI] [PubMed] [Google Scholar]
- 10.Dolin R, Blacklow NR, DuPont H, Formal S, Buscho RF, Kasel JA, Chames RP, Hornick R, Chanock RM. 1971. Transmission of acute infectious nonbacterial gastroenteritis to volunteers by oral administration of stool filtrates. J. Infect. Dis. 123:307–312 [DOI] [PubMed] [Google Scholar]
- 11.Graham DY, Jiang X, Tanaka T, Opekun AR, Madore HP, Estes MK. 1994. Norwalk virus infection of volunteers: new insights based on improved assays. J. Infect. Dis. 170:34–43 [DOI] [PubMed] [Google Scholar]
- 12.Parrino TA, Schreiber DS, Trier JS, Kapikian AZ, Blacklow NR. 1977. Clinical immunity in acute gastroenteritis caused by Norwalk agent. N. Engl. J. Med. 297:86–89 [DOI] [PubMed] [Google Scholar]
- 13.Wyatt RG, Dolin R, Blacklow NR, DuPont HL, Buscho RF, Thornhill TS, Kapikian AZ, Chanock RM. 1974. Comparison of three agents of acute infectious nonbacterial gastroenteritis by cross-challenge in volunteers. J. Infect. Dis. 129:709–714 [DOI] [PubMed] [Google Scholar]
- 14.Jiang X, Wang M, Graham DY, Estes MK. 1992. Expression, self-assembly, and antigenicity of the Norwalk virus capsid protein. J. Virol. 66:6527–6532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Prasad BV, Hardy ME, Dokland T, Bella J, Rossmann MG, Estes MK. 1999. X-ray crystallographic structure of the Norwalk virus capsid. Science 286:287–290 [DOI] [PubMed] [Google Scholar]
- 16.Prasad BV, Rothnagel R, Jiang X, Estes MK. 1994. Three-dimensional structure of baculovirus-expressed Norwalk virus capsids. J. Virol. 68:5117–5125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Harrington PR, Lindesmith L, Yount B, Moe CL, Baric RS. 2002. Binding of Norwalk virus-like particles to ABH histo-blood group antigens is blocked by antisera from infected human volunteers or experimentally vaccinated mice. J. Virol. 76:12335–12343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hutson AM, Atmar RL, Graham DY, Estes MK. 2002. Norwalk virus infection and disease is associated with ABO histo-blood group type. J. Infect. Dis. 185:1335–1337 [DOI] [PubMed] [Google Scholar]
- 19.Lindesmith L, Moe C, Marionneau S, Ruvoen N, Jiang X, Lindblad L, Stewart P, LePendu J, Baric R. 2003. Human susceptibility and resistance to Norwalk virus infection. Nat. Med. 9:548–553 [DOI] [PubMed] [Google Scholar]
- 20.Marionneau S, Ruvoen N, Le Moullac-Vaidye B, Clement M, Cailleau-Thomas A, Ruiz-Palacois G, Huang P, Jiang X, Le Pendu J. 2002. Norwalk virus binds to histo-blood group antigens present on gastroduodenal epithelial cells of secretor individuals. Gastroenterology 122:1967–1977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Atmar RL, Bernstein DI, Harro CD, Al-Ibrahim MS, Chen WH, Ferreira J, Estes MK, Graham DY, Opekun AR, Richardson C, Mendelman PM. 2011. Norovirus vaccine against experimental human Norwalk virus illness. N. Engl. J. Med. 365:2178–2187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Feng X, Jiang X. 2007. Library screen for inhibitors targeting norovirus binding to histo-blood group antigen receptors. Antimicrob. Agents Chemother. 51:324–331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Herbst-Kralovetz M, Mason HS, Chen Q. 2010. Norwalk virus-like particles as vaccines. Expert Rev. Vaccines 9:299–307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tacket CO, Sztein MB, Losonsky GA, Wasserman SS, Estes MK. 2003. Humoral, mucosal, and cellular immune responses to oral Norwalk virus-like particles in volunteers. Clin. Immunol. 108:241–247 [DOI] [PubMed] [Google Scholar]
- 25.Cao S, Lou Z, Tan M, Chen Y, Liu Y, Zhang Z, Zhang XC, Jiang X, Li X, Rao Z. 2007. Structural basis for the recognition of blood group trisaccharides by norovirus. J. Virol. 81:5949–5957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen R, Neill JD, Estes MK, Prasad BV. 2006. X-ray structure of a native calicivirus: structural insights into antigenic diversity and host specificity. Proc. Natl. Acad. Sci. U. S. A. 103:8048–8053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Katpally U, Wobus CE, Dryden K, Virgin HW, IV, Smith TJ. 2008. Structure of antibody-neutralized murine norovirus and unexpected differences from viruslike particles. J. Virol. 82:2079–2088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lochridge VP, Jutila KL, Graff JW, Hardy ME. 2005. Epitopes in the P2 domain of norovirus VP1 recognized by monoclonal antibodies that block cell interactions. J. Gen. Virol. 86:2799–2806 [DOI] [PubMed] [Google Scholar]
- 29.Zheng DP, Ando T, Fankhauser RL, Beard RS, Glass RI, Monroe SS. 2006. Norovirus classification and proposed strain nomenclature. Virology 346:312–323 [DOI] [PubMed] [Google Scholar]
- 30.Kapikian AZ. 2000. The discovery of the 27-nm Norwalk virus: an historic perspective. J. Infect. Dis. 181(Suppl 2):S295–S302. 10.1086/315584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wyatt RG, Greenberg HB, Dalgard DW, Allen WP, Sly DL, Thornhill TS, Chanock RM, Kapikian AZ. 1978. Experimental infection of chimpanzees with the Norwalk agent of epidemic viral gastroenteritis. J. Med. Virol. 2:89–96 [DOI] [PubMed] [Google Scholar]
- 32.Bok K, Parra GI, Mitra T, Abente E, Shaver CK, Boon D, Engle R, Yu C, Kapikian AZ, Sosnovtsev SV, Purcell RH, Green KY. 2011. Chimpanzees as an animal model for human norovirus infection and vaccine development. Proc. Natl. Acad. Sci. U. S. A. 108:325–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ehrlich PH, Moustafa ZA, Harfeldt KE, Isaacson C, Ostberg L. 1990. Potential of primate monoclonal antibodies to substitute for human antibodies: nucleotide sequence of chimpanzee Fab fragments. Hum. Antibodies Hybridomas 1:23–26 [PubMed] [Google Scholar]
- 34.Ogata N, Ostberg L, Ehrlich PH, Wong DC, Miller RH, Purcell RH. 1993. Markedly prolonged incubation period of hepatitis B in a chimpanzee passively immunized with a human monoclonal antibody to the a determinant of hepatitis B surface antigen. Proc. Natl. Acad. Sci. U. S. A. 90:3014–3018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Reed LJ, Muench H. 1938. A simple method of estimating fifty per cent endpoints. Am. J. Hyg. 27:493–497 [Google Scholar]
- 36.Chen Z, Earl P, Americo J, Damon I, Smith SK, Yu F, Sebrell A, Emerson S, Cohen G, Eisenberg RJ, Gorshkova I, Schuck P, Satterfield W, Moss B, Purcell R. 2007. Characterization of chimpanzee/human monoclonal antibodies to vaccinia virus A33 glycoprotein and its variola virus homolog in vitro and in a vaccinia virus mouse protection model. J. Virol. 81:8989–8995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen Z, Earl P, Americo J, Damon I, Smith SK, Zhou YH, Yu F, Sebrell A, Emerson S, Cohen G, Eisenberg RJ, Svitel J, Schuck P, Satterfield W, Moss B, Purcell R. 2006. Chimpanzee/human mAbs to vaccinia virus B5 protein neutralize vaccinia and smallpox viruses and protect mice against vaccinia virus. Proc. Natl. Acad. Sci. U. S. A. 103:1882–1887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.National Research Council 1996. Guide for the care and use of laboratory animals. National Academies Press, Washington, DC [Google Scholar]
- 39.Traggiai E, Becker S, Subbarao K, Kolesnikova L, Uematsu Y, Gismondo MR, Murphy BR, Rappuoli R, Lanzavecchia A. 2004. An efficient method to make human monoclonal antibodies from memory B cells: potent neutralization of SARS coronavirus. Nat. Med. 10:871–875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fernandez-Vega V, Sosnovtsev SV, Belliot G, King AD, Mitra T, Gorbalenya A, Green KY. 2004. Norwalk virus N-terminal nonstructural protein is associated with disassembly of the Golgi complex in transfected cells. J. Virol. 78:4827–4837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sosnovtsev SV, Sosnovtseva SA, Green KY. 1998. Cleavage of the feline calicivirus capsid precursor is mediated by a virus-encoded proteinase. J. Virol. 72:3051–3059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Makiya M, Dolan M, Agulto L, Purcell R, Chen Z. 2012. Structural basis of anthrax edema factor neutralization by a neutralizing antibody. Biochem. Biophys. Res. Commun. 417:324–329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nenonen NP, Hannoun C, Olsson MB, Bergstrom T. 2009. Molecular analysis of an oyster-related norovirus outbreak. J. Clin. Virol. 45:105–108 [DOI] [PubMed] [Google Scholar]
- 44.Parra GI, Abente EJ, Sandoval-Jaime C, Sosnovtsev SV, Bok K, Green KY. 2012. Multiple antigenic sites are involved in blocking the interaction of GII.4 norovirus capsid with ABH histo-blood group antigens. J. Virol. 86:7414–7426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Czako R, Atmar RL, Opekun AR, Gilger MA, Graham DY, Estes MK. 2012. Serum hemagglutination inhibition activity correlates with protection from gastroenteritis in persons infected with Norwalk virus. Clin. Vaccine Immunol. 19:284–287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hutson AM, Atmar RL, Marcus DM, Estes MK. 2003. Norwalk virus-like particle hemagglutination by binding to h histo-blood group antigens. J. Virol. 77:405–415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tian C, Luskin GK, Dischert KM, Higginbotham JN, Shepherd BE, Crowe JE., Jr 2008. Immunodominance of the VH1-46 antibody gene segment in the primary repertoire of human rotavirus-specific B cells is reduced in the memory compartment through somatic mutation of nondominant clones. J. Immunol. 180:3279–3288 [DOI] [PubMed] [Google Scholar]
- 48.Carbonari M, Caprini E, Tedesco T, Mazzetta F, Tocco V, Casato M, Russo G, Fiorilli M. 2005. Hepatitis C virus drives the unconstrained monoclonal expansion of VH1-69-expressing memory B cells in type II cryoglobulinemia: a model of infection-driven lymphomagenesis. J. Immunol. 174:6532–6539 [DOI] [PubMed] [Google Scholar]
- 49.Andersen PS, Haahr-Hansen M, Coljee VW, Hinnerfeldt FR, Varming K, Bregenholt S, Haurum JS. 2007. Extensive restrictions in the VH sequence usage of the human antibody response against the rhesus D antigen. Mol. Immunol. 44:412–422 [DOI] [PubMed] [Google Scholar]
- 50.Pinchuk GV, Nottenburg C, Milner EC. 1995. Predominant V-region gene configurations in the human antibody response to Haemophilus influenzae capsule polysaccharide. Scand. J. Immunol. 41:324–330 [DOI] [PubMed] [Google Scholar]
- 51.Tomlinson IM, Walter G, Jones PT, Dear PH, Sonnhammer EL, Winter G. 1996. The imprint of somatic hypermutation on the repertoire of human germline V genes. J. Mol. Biol. 256:813–817 [DOI] [PubMed] [Google Scholar]
- 52.Amit AG, Mariuzza RA, Phillips SE, Poljak RJ. 1986. Three-dimensional structure of an antigen-antibody complex at 2.8 A resolution. Science 233:747–753 [DOI] [PubMed] [Google Scholar]
- 53.Altevogt BM, Pankevich DE, Shelton-Davenport MK, Kahn JP. (ed). 2011. Chimpanzees in biomedical and behavioral research: assessing the necessity, 2012/04/20 ed. National Academies Press, Washington, DC: [PubMed] [Google Scholar]
- 54.Chen Z, Chumakov K, Dragunsky E, Kouiavskaia D, Makiya M, Neverov A, Rezapkin G, Sebrell A, Purcell R. 2011. Chimpanzee-human monoclonal antibodies for treatment of chronic poliovirus excretors and emergency postexposure prophylaxis. J. Virol. 85:4354–4362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chen Z, Moayeri M, Crown D, Emerson S, Gorshkova I, Schuck P, Leppla SH, Purcell RH. 2009. Novel chimpanzee/human monoclonal antibodies that neutralize anthrax lethal factor, and evidence for possible synergy with anti-protective antigen antibody. Infect. Immun. 77:3902–3908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chen Z, Moayeri M, Zhou YH, Leppla S, Emerson S, Sebrell A, Yu F, Svitel J, Schuck P, St Claire M, Purcell R. 2006. Efficient neutralization of anthrax toxin by chimpanzee monoclonal antibodies against protective antigen. J. Infect. Dis. 193:625–633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lindesmith LC, Beltramello M, Donaldson EF, Corti D, Swanstrom J, Debbink K, Lanzavecchia A, Baric RS. 2012. Immunogenetic mechanisms driving norovirus GII.4 antigenic variation. PLoS Pathog. 8:e1002705. 10.1371/journal.ppat.1002705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nobusawa E, Aoyama T, Kato H, Suzuki Y, Tateno Y, Nakajima K. 1991. Comparison of complete amino acid sequences and receptor-binding properties among 13 serotypes of hemagglutinins of influenza A viruses. Virology 182:475–485 [DOI] [PubMed] [Google Scholar]
- 59.Robertson DL, Anderson JP, Bradac JA, Carr JK, Foley B, Funkhouser RK, Gao F, Hahn BH, Kalish ML, Kuiken C, Learn GH, Leitner T, McCutchan F, Osmanov S, Peeters M, Pieniazek D, Salminen M, Sharp PM, Wolinsky S, Korber B. 2000. HIV-1 nomenclature proposal. Science 288:55–56 [DOI] [PubMed] [Google Scholar]
- 60.Simmonds P, Bukh J, Combet C, Deleage G, Enomoto N, Feinstone S, Halfon P, Inchauspe G, Kuiken C, Maertens G, Mizokami M, Murphy DG, Okamoto H, Pawlotsky JM, Penin F, Sablon E, Shin IT, Stuyver LJ, Thiel HJ, Viazov S, Weiner AJ, Widell A. 2005. Consensus proposals for a unified system of nomenclature of hepatitis C virus genotypes. Hepatology 42:962–973 [DOI] [PubMed] [Google Scholar]
- 61.Webster RG, Bean WJ, Gorman OT, Chambers TM, Kawaoka Y. 1992. Evolution and ecology of influenza A viruses. Microbiol. Rev. 56:152–179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Corti D, Voss J, Gamblin SJ, Codoni G, Macagno A, Jarrossay D, Vachieri SG, Pinna D, Minola A, Vanzetta F, Silacci C, Fernandez-Rodriguez BM, Agatic G, Bianchi S, Giacchetto-Sasselli I, Calder L, Sallusto F, Collins P, Haire LF, Temperton N, Langedijk JP, Skehel JJ, Lanzavecchia A. 2011. A neutralizing antibody selected from plasma cells that binds to group 1 and group 2 influenza A hemagglutinins. Science 333:850–856 [DOI] [PubMed] [Google Scholar]
- 63.Johansson DX, Voisset C, Tarr AW, Aung M, Ball JK, Dubuisson J, Persson MA. 2007. Human combinatorial libraries yield rare antibodies that broadly neutralize hepatitis C virus. Proc. Natl. Acad. Sci. U. S. A. 104:16269–16274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Law M, Maruyama T, Lewis J, Giang E, Tarr AW, Stamataki Z, Gastaminza P, Chisari FV, Jones IM, Fox RI, Ball JK, McKeating JA, Kneteman NM, Burton DR. 2008. Broadly neutralizing antibodies protect against hepatitis C virus quasispecies challenge. Nat. Med. 14:25–27 [DOI] [PubMed] [Google Scholar]
- 65.Whittle JR, Zhang R, Khurana S, King LR, Manischewitz J, Golding H, Dormitzer PR, Haynes BF, Walter EB, Moody MA, Kepler TB, Liao HX, Harrison SC. 2011. Broadly neutralizing human antibody that recognizes the receptor-binding pocket of influenza virus hemagglutinin. Proc. Natl. Acad. Sci. U. S. A. 108:14216–14221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wu X, Yang ZY, Li Y, Hogerkorp CM, Schief WR, Seaman MS, Zhou T, Schmidt SD, Wu L, Xu L, Longo NS, McKee K, O'Dell S, Louder MK, Wycuff DL, Feng Y, Nason M, Doria-Rose N, Connors M, Kwong PD, Roederer M, Wyatt RT, Nabel GJ, Mascola JR. 2010. Rational design of envelope identifies broadly neutralizing human monoclonal antibodies to HIV-1. Science 329:856–861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wu W, Chen Z, Cheng N, Watts NR, Stahl SJ, Farci P, Purcell RH, Wingfield PT, Steven AC. 2013. Specificity of an anti-capsid antibody associated with hepatitis B virus-related acute liver failure. J. Struct. Biol. 181:53–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wu TT, Kabat EA. 1970. An analysis of the sequences of the variable regions of Bence Jones proteins and myeloma light chains and their implications for antibody complementarity. J. Exp. Med. 132:211–250 [DOI] [PMC free article] [PubMed] [Google Scholar]