

Abstract
Rotavirus nonstructural protein NSP1 can inhibit expression of interferon (IFN) and IFN-stimulated gene products by inducing proteasome-mediated degradation of IFN-regulatory factors (IRFs), including IRF3, IRF5, and IRF7. All IRF proteins share an N-terminal DNA-binding domain (DBD), and IRF3, IRF5, and IRF7 contain a similar C-proximal IRF association domain (IAD) that mediates IRF dimerization. An autoinhibitory domain (ID) at the extreme C terminus interacts with the IAD, burying residues necessary for IRF dimerization. Phosphorylation of serine/threonine residues in the ID induces charge repulsions that unmask the IAD, enabling IRF dimerization and subsequent nuclear translocation. To define the region of IRF proteins targeted for degradation by NSP1, we generated IRF3 and IRF7 truncation mutants and transiently expressed each with simian SA11-4F NSP1. These assays indicated that the IAD represented a necessary and sufficient target for degradation. Because NSP1 did not mediate degradation of truncated forms of the IAD, NSP1 likely requires a structurally intact IAD for recognition and targeting of IRF proteins. IRF9, which contains an IAD-like region that directs interactions with signal inducer and activator of transcription (STAT) proteins, was also targeted for degradation by NSP1, while IRF1, which lacks an IAD, was not. Analysis of mutant forms of IRF3 unable to undergo dimerization or that were constitutively dimeric showed that both were targeted for degradation by NSP1. These results indicate that SA11-4F NSP1 can induce degradation of inactive and activated forms of IAD-containing IRF proteins (IRF3 to IRF9), allowing a multipronged attack on IFN-based pathways that promote antiviral innate and adaptive immune responses.
INTRODUCTION
The interferon (IFN)-regulatory factor (IRF) family of transcription factors consists of nine members (IRF1 to IRF9) that have crucial roles in activating innate and adaptive immune responses to viral infection (1, 2). Several of the IRF proteins, notably IRF3, IRF5, and IRF7, are particularly important for triggering the expression of type I IFN and IFN-stimulated gene (ISG) products (3, 4). Of these three IRF proteins, IRF3 is constitutively synthesized in most cells types (e.g., fibroblasts), where it accumulates in an inactive form within the cytoplasm (1). In contrast, IRF5 and IRF7 are constitutively synthesized in only a limited number of cell types (e.g., plasmacytoid dendritic cells [DCs]); more typically their expression is induced by type I IFN (5). Nonetheless, inactive forms of IRF5 and IRF7, like IRF3, accumulate in the cytoplasm. Interaction of viral RNAs with cytosolic pattern recognition receptors (PRRs) (6, 7), such as retinoic acid-inducible gene I (RIG-I) and melanoma differentiation-associated gene 5 (MDA5), triggers a complex signaling cascade that leads to the phosphorylation and dimerization of IRF proteins (8, 9). The dimers translocate to the nucleus, where they bind to IFN-stimulated response element (ISRE) promoter sequences, stimulating the transcription of genes encoding IFN and ISG products (10, 11).
The IRF proteins all share an N-terminal DNA-binding domain (DBD) with a unique helix-turn-helix signature that includes five tryptophan repeats (Fig. 1A) (12–15). The C-terminal regions of IRF proteins are more diverse and can include elements that have regulatory function (1, 5). The C-terminal regions of IRF3 to IRF9 contain a structurally related IRF association domain (IAD), which mediates homodimer and, in some cases, heterodimer (e.g., IRF3/IRF7) formation (12, 16). Instead of functioning in IRF dimerization, the role of the C-terminal region of IRF9 is to promote interactions with the signal transducer and activator of transcription proteins, STAT1 and STAT2, enabling the formation of the ISGF3 heterotrimeric complex (17, 18). In the nucleus, ISGF3 uses the IRF9 DBD to interact with ISRE promoter sequences.
Fig 1.

Shared domains of representative IRF proteins. (A) All IRF family members contain a well-conserved DNA-binding domain (DBD; green) at the N terminus of the protein. Most IRF proteins contain an IRF association domain (IAD; teal), which facilitates dimerization, and an inhibitory domain (ID; magenta), which is a regulatory element that keeps IRFs in an inactive monomeric state until activated by phosphorylation. Sites of phosphorylation (P) are indicated in yellow. IRF9 contains a STAT binding domain (STAT-BD; white) that is similar to the IAD. (B) Schematic of IRF activation. Ribbon diagram of the C-terminal domain of IRF3 (Protein Data Bank [PBD] code 1QWT) (16) in the inactive monomeric state. Phosphorylation causes the ID to extend away from the IAD, and this extended domain participates in formation of the active dimeric IRF (IRF5 shown; PDB 3DSH) (23). Domains are colored as described for panel A. The illustration was prepared using Chimera (44).
An autoinhibitory domain (ID) at the extreme C terminus of IRF3, IRF5, and IRF7 suppresses the transactivation function of the IAD (19–21). The ID interacts with the IAD to form a condensed hydrophobic core that masks key residues of the IAD required for IRF dimerization (Fig. 1B) (16, 22). Phosphorylation of conserved serine and threonine residues within the ID introduces charge repulsions, causing the ID to adopt an extended conformation. This structural change unmasks the IAD, allowing contacts to be made between IRF monomers, creating a dimer with a functional DBD that, upon translocation to the nucleus, can bind to IFN and ISG enhancer elements (23, 24).
Rotaviruses, members of the family Reoviridae, have genomes consisting of 11 double-stranded RNA segments that are encapsidated within a nonenveloped icosahedral virion (25). The group A rotaviruses (RVAs) are an important cause of gastroenteritis in many animal species, including humans (25, 26), with the villous tips of the small intestine representing the primary site of virus replication. Replication of RVA RNAs is sensed by RIG-I and MDA5, which triggers an IFN-mediated innate immune response that leads to the activation of IRF3 and expression of ISG products (27–29). The RVA nonstructural protein, NSP1, antagonizes the response by interacting with and, in some cases, inducing the degradation of proteins connected to IFN and ISG expression (30–32). Through these mechanisms, NSP1 may function as a virulence determinant, influencing the ability of the virus to replicate and cause disease in its host and, possibly, to spread from the gut to secondary sites of replication (27, 30, 32). Most human RVA NSP1 proteins contain a C-terminal recognition motif (DSGxS) for β-transducing repeat-containing protein (β-TrCP) (33), suggesting that this type of NSP1 antagonizes IFN signaling pathways by interacting with a factor that controls NF-κB activation (34). In contrast, many animal (nonhuman) RVAs encode NSP1 proteins that lack the β-TrCP recognition motif and that induce the degradation of IRF3, IRF5, and IRF7; these include the simian NSP1s of the SA11-4F and RRV strains and the bovine NSP1s of the NCDV and UK strains (35–40).
The variation in NSP1 activities is correlated with the extensive sequence diversity noted for the C-terminal end of the protein (41–43). Indeed, as revealed by yeast two-hybrid assays, the NSP1 C terminus includes the recognition domain for cellular protein targets (35). The importance of the C terminus in target interactions is further supported by studies of animal RVAs that encode C-truncated forms of NSP1; these viruses fail to induce IRF degradation or prevent IFN expression (37, 38). IRF degradation assays performed in vitro with NSP1 proteins bearing C-terminal deletions or domain swaps have further verified the contribution of the C terminus to the interaction of NSP1 and its targets (40). Because all NSP1 proteins share an N-terminal RING domain and treatment of cells with proteasome inhibitors prevents NSP1-mediated degradation of its targets, NSP1 has been proposed to function as an E3 ubiquitin ligase (34, 38).
The capacity of NSP1 proteins of many animal RVs to induce the degradation of IRF3, IRF5, and IRF7 suggests that their NSP1s recognize a common element within these targets (36, 39). To explore this possibility, mutagenesis was used to define the region of IRF3 and IRF7 recognized and targeted for degradation by SA11-4F NSP1. The results indicate that a structurally intact IAD represents the minimal target for NSP1, and that the IAD is subject to degradation irrespective of its conformation (monomeric or dimeric). Given that NSP1 has induced the degradation of all IRF proteins with IAD-like regions tested so far (IRF3, IRF5, IRF7, and IRF9) (38, 39) but the IAD-free IRF1 protein has not, it is possible that NSP1 targets the entire subset of IAD-containing IRF family members (IRF3 to IRF9). Through this mechanism, RVA may inhibit the antiviral activities of multiple antiviral pathways, including those resulting from IFN-stimulated ISG expression, apoptosis induction, or CD4+ and CD8+ lymphocyte differentiation.
MATERIALS AND METHODS
Cells and antibodies.
Human 293T cells were maintained in Dulbecco's modified Eagle's medium (DMEM) (Quality Biological) supplemented with 10% fetal bovine serum (FBS) (Invitrogen) and 1% nonessential amino acids (Lonza). Rabbit polyclonal antiserum to simian SA11-5S NSP1 was used at a dilution of 1:1,000 (39). Rabbit polyclonal antibodies to human IRF1 (1:250; sc-497), IRF3 (1:2,000; sc-9082), and IRF9 (1:250; sc-496), as well as PCNA (1:1,000; sc-7907) and green fluorescent protein (GFP) (1:1,000; sc-8334), were purchased from Santa Cruz Biotechnology.
Expression vectors.
To generate vectors expressing truncated IRF3 proteins, start and stop codons and restriction sites were introduced into a human IRF3 gene sequence at specific sites by PCR amplification using AccuPrime Pfx Supermix (Invitrogen). Reaction mixtures included pEGFP-IRF3 (kindly provided by John Hiscott, McGill University) (37) as the template and primers designed to amplify the fragment of interest while adding terminal EcoRI and MluI restriction sites. PCR products were digested with EcoRI and MluI and ligated into the pAcGFP vector (Clontech) digested with the same enzymes. To generate full-length GFP-IRF3 with S396 and S398 mutated to alanine (pEGFP-IRF3-2A), outward PCR amplification was performed using pEGFP-IRF3 as the template and suitable primers. To generate full-length IRF3 with S396, S398, S402, S405, and T404 each mutated to alanine (pEGFP-IRF3-5A), outward PCR amplification was performed using pEGFP-IRF3-2A as the template and suitable primers. To generate full-length GFP-IRF3 with residues S396, S398, S402, S405, and T404 replaced with aspartic acid, pCMV-IRF3-5D (provided by John Hiscott) was digested with the restriction enzymes XbaI and PvuII. The fragment containing the aspartic acid mutations was ligated into the pEGFP-IRF3 (1-427) vector digested with the same enzymes. To generate full-length GFP-IRF3-5D with R285 changed to glutamic acid or alanine (pEGFP-IRF3-5D R285E or R285A), outward PCR amplification was performed using pEGFP-IRF3-5D as the template and suitable primers. To generate full-length GFP-IRF3-5D with L362 changed to aspartic acid or alanine (pEGFP-IRF3-5D L362D or L362A), outward PCR amplification was performed using pEGFP-IRF3-5D as the template and suitable primers.
Vectors expressing truncated human IRF7 proteins were constructed in a manner similar to that for vectors expressing truncated IRF3 proteins (described above). PCR mixtures included pCMVSport-IRF7H (kindly provided by Paula Pitha-Rowe, Johns Hopkins University) (34) as the template and primers that amplified defined regions of the IRF7 gene sequence and that added terminal HindIII and BamHI restriction sites. PCR products were digested with HindIII and BamHI and ligated into pAcGFP that had been digested with the same enzymes. Sequences of plasmid inserts were verified using an Applied Biosystems 3100 genetic analyzer. Primer sequences used in generating constructs are available upon request.
Expression vectors containing the NSP1 open reading frame (ORF) of RVA strains SA11-4F(pCI-NSP1:4F), SA11-5S(pCI-NSP1ΔC17:5S), and human WI61(pcDNA-NSP1:WI61) have been previously described (39). The WI61 NSP1ΔC13 expression vector was prepared by PCR amplification of pcDNA-NSP1:WI61 using a reverse primer that introduced a stop codon 13 amino acids upstream from the end of the protein. The PCR product was digested with the applicable restriction enzymes and inserted into pcDNA3.1 vector (Invitrogen). The expression vectors for human IRF1 (pCMV6-XL5-IRF1) and human IRF9 (pCMV6-AC-IRF9) were purchased from Origene (Rockville, MD).
Transfections.
Approximately 2.0 × 106 293T cells in 6-well plates were transfected with vector DNA using 8 μl of Lipofectamine 2000 (Invitrogen) per well. To analyze IRF3 proteins containing C-terminal deletions and point mutations, transfection mixtures contained 2 μg of NSP1 expression vector and 0.2 μg of an IRF3 expression vector. To analyze IRF3 proteins containing N-terminal deletions, transfection mixtures contained 2.0 μg of NSP1 expression vector and 2.0 μg of an IRF3 expression vector. To analyze IRF1, IRF7, and IRF9 proteins, transfection mixtures contained 2.0 μg of NSP1 expression vector and 0.5 μg of an IRF1, 0.2 μg of an IRF7, and 0.5 μg of an IRF9 expression vector. At 24 h posttransfection (p.t.), cells were harvested in 200 μl radioimmunoprecipitation assay (RIPA) buffer containing 150 mM NaCl, 150 mM Tris-HCl (pH 8.0), 1.0% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate (SDS), and 1× Complete protease inhibitor cocktail (Roche) and then were assayed for protein content by immunoblot analysis.
Quantitative immunoblot analysis.
Whole-cell lysates were briefly sonicated and then diluted 1:1 in 2× Novex Tris-glycine SDS sample buffer (Invitrogen). Proteins were resolved by electrophoresis on 10% Tris-glycine gels (Invitrogen) and transferred onto nitrocellulose membranes. The membranes were blocked with Odyssey blocking buffer (LI-COR Biosciences) and then incubated with primary antibody in Odyssey blocking buffer containing 0.1% Tween 20. Afterwards, blots were washed with Tris-buffered saline (50 mM Tris-HCl [pH 7.5], 150 mM NaCl) containing 0.1% Tween 20 (TBS-T) and incubated with IRDye 680- or IRDye800-conjugated secondary antibodies in TBS-T and 1% milk. Blots were then washed with TBS-T, and fluorescent bands were imaged and quantified with the Odyssey infrared imaging system. Bands of interest were normalized to PCNA levels to control for loading.
Native PAGE analysis.
At 24 h p.t., 293T cells were washed and resuspended using phosphate-buffered saline (PBS), transferred into a microcentrifuge tube, and pelleted. Cell pellets were lysed on ice in buffer containing 20 mM Tris-HCl (pH 8.0), 137 mM NaCl, 10% glycerol, 1% Nonidet P-40, and 1× Complete protease inhibitor cocktail. Afterwards, cellular debris was pelleted and the supernatant was transferred to a new centrifuge tube. Sodium deoxycholate was added to a 1% final concentration, and 2× Tris-glycine sample buffer (SDS free) was added to a 1× final concentration. No heating step was performed. Proteins were resolved by electrophoresis on Novex 8% Tris-glycine gels in Novex 1× Tris-glycine native running buffer (Invitrogen) and transferred onto nitrocellulose membranes. Monomeric and dimeric forms of EGFP-IRF3 proteins were probed with anti-GFP antibody, and signal was detected using an Odyssey infrared imaging system.
Molecular graphics.
Molecular graphics and analyses were performed with the UCSF Chimera package (version 1.7) (44). Chimera was developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (supported by NIGMS P41-GM103311).
Nucleotide sequence accession numbers.
Sequences of the human IRF1 (NM_002198), IRF3 (NM_001571), IRF5 (NM_032643), IRF7 (AF076494), and IRF9 (NM_002198) genes examined in this study are available in GenBank.
RESULTS
IAD of IRF3 is targeted for degradation by NSP1.
To locate the region of IRF3 necessary for NSP1-mediated degradation, vectors were constructed that expressed human IRF3 proteins with N- or C-terminal truncations of various lengths fused to an N-terminal GFP tag (Fig. 2). The use of the GFP tag allowed quantitation of transiently expressed EGFP-IRF3 proteins by immunoblot analysis and resolution of endogenous forms of IRF3 from transiently expressed forms. Each GFP-IRF3 expression vector was cotransfected into human 293T cells with an expression vector for simian wild-type NSP1 (strain SA11-4F) or an isogenic form of the same protein that contains a C-terminal 17-amino-acid truncation (ΔC17 NSP1 of strain SA11-5S) (37). Previous studies have shown that SA11-4F NSP1 induces the degradation of IRF3, IRF5, and IRF7 but not β-TrCP, and it restricts IFN expression, while SA11-5S ΔC17 NSP1 fails to induce IRF degradation or prevent IFN expression (37, 38). The SA11-5S isolate was recovered from a high-passage-number stock of the SA11-4F virus (45, 46). 293T cells expressing GFP-IRF3 and NSP1 constructs were harvested at 24 h p.t., and their lysates were subjected to SDS-PAGE, followed by immunoblot analysis with anti-GFP antibody. For each construct, protein levels were quantified and calculated as a percentage of GFP-IRF3 coexpressed with SA11-4F NSP1 relative to GFP-IRF3 coexpressed with SA11 ΔC17 NSP1 (which was set to 100%). SDS-PAGE and immunoblot analysis were used to verify the expression of appropriately sized GFP-IRF3 products (Fig. 2) and NSP1 proteins (data not shown) from constructs.
Fig 2.
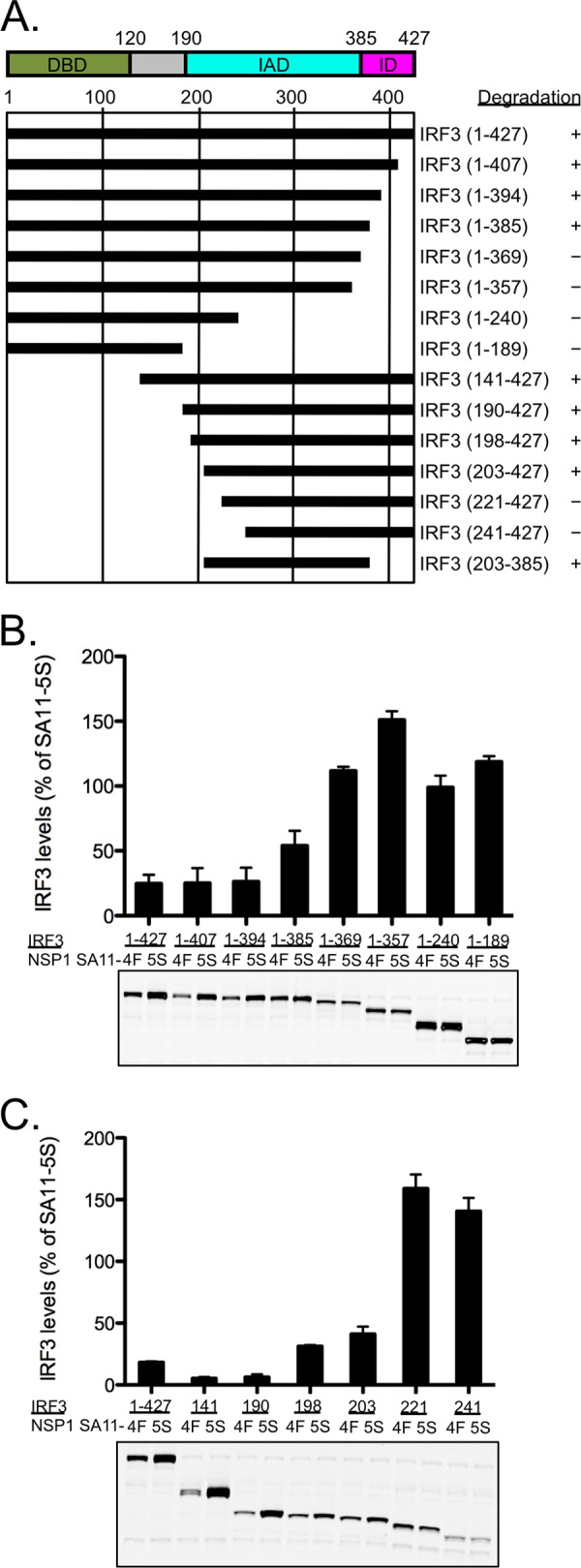
Region of IRF3 necessary for NSP1-induced degradation. (A) Summary of IRF3 deletion constructs. The full-length IRF3 protein is indicated by a horizontal black bar spanning amino acids 1 to 427 (positions are numbered above the bars). IRF3 contains a DNA-binding domain (DBD), an IRF-interactive domain (IAD), and an inhibitory domain (ID), as indicated at the top. IRF3 truncation mutants are also represented by black bars spanning the approximate portion that each represents. The outcome of IRF3 expression in the presence of SA11-4F NSP1 is indicated as degraded (+) or not degraded (−). (B and C) 293T cells were cotransfected with a vector encoding GFP-tagged IRF3 protein with C-terminal (B) or N-terminal (C) truncation and a vector encoding wild-type (SA11-4F; 4F) or defective (SA11-5S; 5S) NSP1. Proteins in cell lysates were resolved by SDS-PAGE and analyzed by immunoblotting for GFP and PCNA. Proteins were quantified with the Odyssey infrared imaging system. Band intensities were normalized to the loading control PCNA and are given as the percentage of protein in cells expressing defective NSP1. Bar graphs represent the mean values from two experiments; ranges are indicated with error bars. Representative immunoblots are shown below each bar graph.
Protein levels of full-length GFP-IRF3 (construct 1-427) coexpressed with SA11-4F NSP1 were reduced by greater than 50% than during coexpression with ΔC17 NSP1, indicating that GFP-tagged IRF3 was degraded in the presence of wild-type NSP1 (Fig. 2B). C-terminal truncations of GFP-IRF3 of up to 42 amino acids (corresponding to IRF3 1-385) had similarly low levels of protein in the presence of SA11-4F NSP1 (Fig. 2B). This suggests that the ID of IRF3 (amino acids 382 to 414), including the sites of phosphorylation, is not required for degradation by wild-type NSP1. When truncations from the C terminus of GFP-IRF3 were greater than 64 amino acids (such as IRF3 1-357), SA11-4F NSP1 no longer reduced the levels of GFP-IRF3 to less than that of ΔC17 NSP1, suggesting the target was no longer degraded. In all experiments, loss of the endogenous form of IRF3 was also detected, indicating that NSP1 actively targeted both the full-length and C-truncated forms of IRF3 for degradation (data not shown). Interestingly, expression of SA11-4F NSP1 caused some nondegraded GFP-IRF3 targets to accumulate to levels above that reached when the targets were expressed with ΔC17 NSP1 (Fig. 2B, 1-357 target). This phenomenon was described before in studies examining the effects of NSP1 on a β-TrCP target (39), and it raises the possibility that full-length NSP1 has an as-yet undefined activity that can upregulate protein expression.
Levels of GFP-IRF3 with N-terminal truncations of up to 202 amino acids (including IRF3 203-427) were reduced by greater than 50% when coexpressed with SA11-4F NSP1 (Fig. 2C). When truncations from the N terminus extended beyond 203 amino acids (such as IRF3 221-427), levels of GFP-IRF3 were approximately equivalent when coexpressed with wild-type or defective NSP1. This suggests that the DBD of IRF3 (amino acids 1 to 150) and the region linking the DBD to the IAD are not required for degradation by NSP1. Thus, by deletion analysis we identified the minimal region of human IRF3 necessary for degradation by NSP1 to be amino acids 203 to 385, corresponding to the IAD, which is the domain responsible for dimerization of IRF proteins.
The IAD of human IRF3 and IRF7 is sufficient for degradation by NSP1.
To determine if the region of IRF3 encompassing the IAD was sufficient for degradation by NSP1, a GFP-tagged IRF3 gene construct was engineered to express IRF3 residues 203 to 385 (Fig. 3A). This was cotransfected with a plasmid expressing the wild-type SA11-4F NSP1 or the defective SA11-5S NSP1 into 293T cells. Cells were harvested at 24 h p.t. and lysates were subjected to SDS-PAGE, followed by quantitative immunoblot analysis with anti-GFP antibodies. The results showed that IRF3 residues 203 to 385 were not sufficient for degradation in the presence of wild-type NSP1 (Fig. 3B). When a GFP-tagged IRF3 expressing residues 190 to 385 was coexpressed with the wild-type NSP1, GFP-IRF3 protein levels were reduced by greater than 50%, suggesting a region of IRF3 including the IAD was sufficient for degradation by wild-type NSP1. The basis for the slight difference in the N-terminal boundary of the IRF3 target defined by the two deletion analyses (Fig. 2, 203-385 and 3, 190-385) is not clear but may result from a partially misfolded determinant in the 203-385 target that is corrected when residues 190 to 203 are present. Alternatively, it is possible that the NSP1-binding site extends slightly upstream and downstream of the 203-385 region of IRF3, and when both are missing, as in the IRF3 203-385 target, NSP1 can no longer efficiently interact with and induce its degradation. In contrast, the presence of extra downstream sequence in the IRF3 203-427 target (Fig. 2B) could enhance NSP1 interaction, thereby promoting target degradation.
Fig 3.
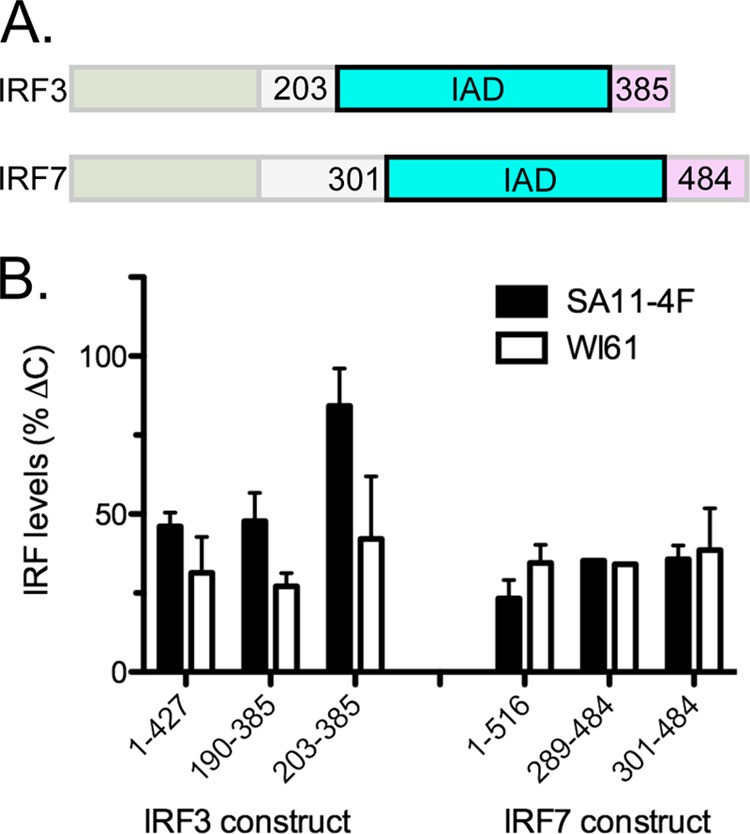
IAD of IRF3 and IRF7 is sufficient for NSP1-induced degradation. (A) Schematic of minimal regions of IRF3 and IRF7 tested. (B) 293T cells were cotransfected with a vector encoding GFP-tagged wild-type IRF3 (construct 1-427) or IRF7 (1-516), IRF3 mutant 190-385 or 203-385, or IRF7 mutant 289-484 or 301-484 and a vector encoding SA11-4F (wild-type), SA11-5S (defective), WI61 (wild-type), or WI61ΔC13 (defective) NSP1. Proteins in cell lysates were resolved by SDS-PAGE and analyzed by immunoblotting for GFP and PCNA. Proteins were quantified with the Odyssey infrared imaging system. Band intensities were normalized to the loading control PCNA and are given as the percentage of the protein in cells expressing defective NSP1. Shown are the mean values from two experiments; ranges are indicated with error bars.
Because the activity of NSP1 can vary between proteins isolated from different RVA strains (34, 39), GFP-IRF3 (190-385) and GFP-IRF3 (203-385) were also coexpressed with human WI61 NSP1, which is known to induce the degradation of IRF3, IRF5, and IRF7 proteins. Protein levels were quantified and calculated as a percentage of GFP-IRF3 coexpressed with the active wild-type WI61 NSP1 relative to that in the presence of a C-terminally truncated, i.e., defective, WI61 NSP1 (WI61 NSP1ΔC13). Protein levels of the GFP-tagged IRF3 construct expressing residues 203 to 385 and 190 to 385 were reduced by greater than 50% in the presence of wild-type WI61 NSP1 (Fig. 3B), indicating that the IAD of IRF3 is the region targeted by NSP1 for degradation.
IRF3 and IRF7 share common biological features, as well as structural homology, and heterodimerize in cells when stimulated by pathogens to drive IFN-β expression (16, 22). To determine whether the region of IRF7 homologous to IRF3 residues 203 to 385 was also degraded in the presence of wild-type NSP1, a human IRF7 gene construct was engineered to express residues 301 to 484 N-terminally tagged with GFP (Fig. 3A). This was cotransfected with a plasmid expressing either wild-type SA11-4F NSP1 or defective SA11-5S NSP1 into 293T cells. The cell lysates were subjected to SDS-PAGE, followed by quantitative immunoblotting. In comparison to cells cotransfected with the defective NSP1, protein levels of IRF7 residues 301 to 484 were reduced by more than 50% when cotransfected with wild-type NSP1 (Fig. 3B). The results showed that IRF7 residues 301 to 484 were sufficient for degradation, indicating that SA11-4F NSP1 targets the homologous IAD of IRF7. Similar results were found with WI61 NSP1 (Fig. 3B). Taken together, these results indicate that the IRF DBD and inhibitory domain are not required, and the IAD is sufficient for NSP1-mediated degradation.
IRF9, but not IRF1, is targeted by NSP1.
All nine IRF proteins contain a DBD, but IRF1 and IRF2 lack the characteristic IAD of IRF3-IRF9 (12). To confirm that the IAD was the region targeted by NSP1, a human IRF1 expression construct was cotransfected with a plasmid expressing either wild-type NSP1 (SA11-4F or WI61) or defective NSP1 (SA11-5S or WI61ΔC13) into 293T cells. The cell lysates were subjected to SDS-PAGE, followed by immunoblotting. IRF1 protein levels were equivalent in cells expressing the wild-type or defective NSP1 protein (Fig. 4A), indicating that, as predicted, IRF1 was not targeted for degradation by NSP1. The C-terminal region of IRF9 is known to bind STAT proteins and form a heterotrimeric complex known as ISGF3. When a human IRF9 expression construct was cotransfected with a plasmid expressing either wild-type NSP1 (SA11-4F or WI61) or defective NSP1 (SA11-5S or WI61ΔC13) into 293T cells, the wild-type NSP1 proteins induced degradation of IRF9 (Fig. 4B). These findings support the conclusion that the IAD is important for NSP1-mediated degradation of IRF proteins.
Fig 4.
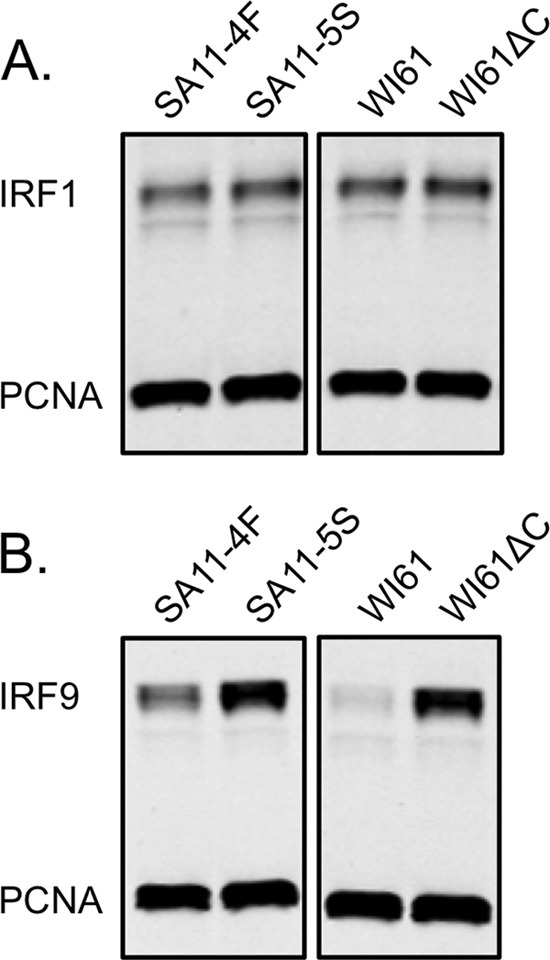
IRF9 is degraded in the presence of NSP1, but IRF1 is not. 293T cells were cotransfected with a vector encoding full-length IRF1 (A) or IRF9 (B) and a vector encoding SA11-4F (wild type), SA11-5S (defective), WI61 (wild-type), or WI61ΔC13 (defective) NSP1. Proteins in cell lysates were resolved by SDS-PAGE and analyzed by immunoblotting for IRF1 or IRF9 and PCNA. Protein levels were visualized and quantified with the Odyssey infrared imaging system. The samples shown in panel A were run on the same gel, as were the samples in panel B.
The monomeric and dimeric forms of IRF3 are both targeted for degradation by NSP1.
In uninfected cells, IRF3 resides in the cytoplasm in an inactive, monomeric state. Upstream stimulation by pathogen-activated signaling pathways activates IRF3 by posttranscriptional phosphorylation and subsequent dimerization. To determine which form of IRF3, monomeric or dimeric, is targeted for degradation by NSP1, we mutated the key sites of phosphorylation (S396, S398, S402, T404, and S405) in the ID (Fig. 5A) (24, 47). Alanine substitution at these sites prevents phosphorylation, resulting in the production of an inactive monomeric form of IRF3 (GFP-IRF3-5A) (11, 24, 47). Alteration at the sites of phosphorylation with aspartic acid, a phosphomimetic, drives the formation of a constitutively activated dimeric form of IRF3 (GFP-IRF3-5D) (11, 22, 24, 47).
Fig 5.
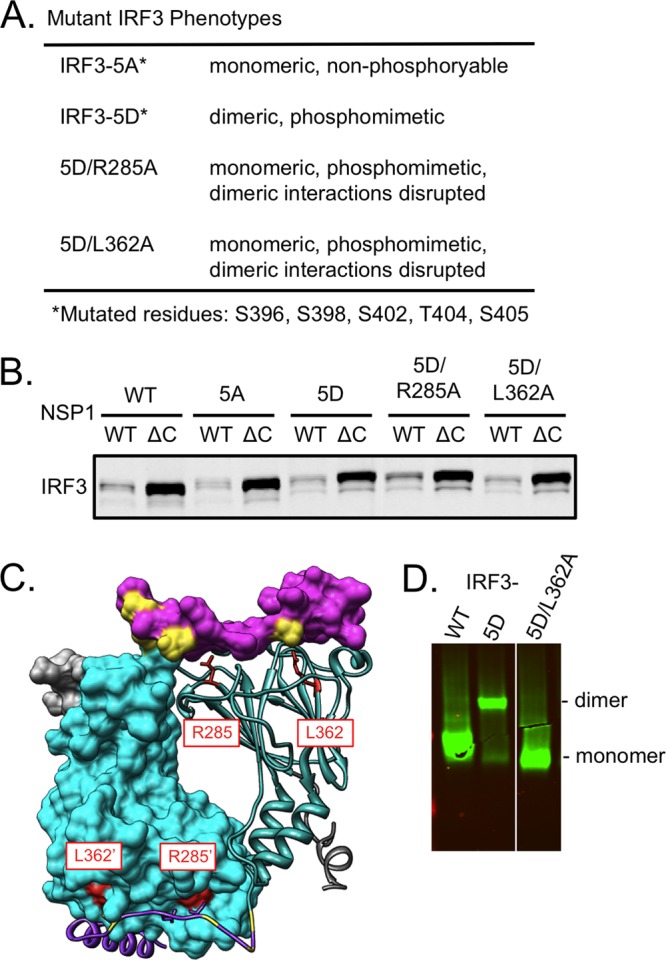
Monomeric and dimeric IRF3 are targets of NSP1-induced degradation. (A) Phenotype of mutant IRF3 proteins and location of critical residues. (B) 293T cells were cotransfected with a vector encoding GFP-tagged IRF3 or mutant IRF3-5A, IRF3-5D, IRF3-5D/R285A, or IRF3-5D/L362A and a vector encoding SA11-4F (wild type; WT) or SA11-5S (defective; ΔC) NSP1. Proteins in cell lysates were resolved by SDS-PAGE and analyzed by immunoblotting for GFP and PCNA. Proteins were quantified with the Odyssey infrared imaging system. (C) Depiction of IRF dimer with one monomer diagrammed as a ribbon and the other as a surface-filled model (PDB code 3DSH). The location of conserved IRF3 R285 and L362 residues are noted in red. (D) 293T cells were transfected with a vector encoding GFP-tagged IRF3, IRF3-5D or IRF3-5D/L362A. Cell lysates were resolved by native PAGE; GFP-IRF3 proteins were detected by immunoblot assay with anti-GFP antibody.
To determine if monomeric IRF3 was targeted for degradation by NSP1, the GFP-IRF3-5A construct was cotransfected with a plasmid expressing either wild-type SA11-4F NSP1 or defective SA11-5S NSP1 into 293T cells. The cell lysates were subjected to SDS-PAGE, followed by quantitative immunoblot analysis. The results showed that GFP-IRF3-5A protein levels were reduced by greater than 50% when coexpressed with wild-type NSP1, indicating that monomeric IRF3 is a degradation target of NSP1 (Fig. 5B). To determine if dimeric IRF3 was similarly targeted by NSP1, the GFP-IRF3-5D construct was likewise cotransfected with plasmids expressing SA11-4F NSP1 or defective SA11-5S NSP1. The results showed that GFP-IRF3-5D protein levels were reduced by greater than 50% when coexpressed with wild-type NSP1, indicating that dimeric IRF3 is also targeted by NSP1 (Fig. 5B).
X-ray crystallography of the IRF5 dimer has defined the extensive intermolecular interface that mediates interactions between monomeric subunits (23). Several key interface residues are conserved among the IRF3 to IRF9 proteins, including the surface-exposed residues IRF3 R285 and L362 (Fig. 5C) (23). Mutation of either residue prevents IRF3 dimerization, even when conserved serine and threonine residues of the ID are replaced with dimer-favoring phosphomimetics (23). To disrupt dimer formation in the context of IRF3 proteins containing phosphomimetics, constructs for GFP-IRF3-5D were generated that contained an R285A or an L362A mutation. As confirmed by native gel electrophoresis (Fig. 5D), the IRF3-5D protein accumulated in transfected cells as a dimer, while IRF3-5D with an L362A mutation accumulated as a monomer. Constructs expressing IRF3-5D/R285A or IRF3-5D/L362A were cotransfected with plasmids expressing either wild-type SA11-4F NSP1 or defective SA11-5S NSP1 into 293T cells. Analysis of the cell lysates showed that wild-type NSP1 reduced levels of both forms of the IRF3-5D by greater than 50% relative to defective SA11-5S NSP1 (Fig. 5B). These results show that NSP1 mediates the degradation of both monomeric and dimeric forms of IRF3.
DISCUSSION
The signaling cascade induced by activated host PRRs culminates in the phosphorylation of IRF proteins, which then form homo- or heterodimeric complexes that translocate to the nucleus to induce IFN and ISG expression (1). RVA NSP1 inhibits the expression of these antiviral products by interacting with and, in some cases, inducing the proteasome-mediated degradation of IRFs and/or β-TrCP (34, 37, 38, 40). Those NSP1 proteins that target β-TrCP are suggested by the presence of a C-terminal β-TrCP recognition motif, and likely phosphodegron, DSGxS (33). The NSP1 proteins of many animal RVAs lack the DSGxS motif and appear to rely on the degradation of IRFs as a principal mechanism for inhibiting IFN and ISG expression (unpublished results). In this study, we have examined the targeting activity of NSP1 proteins (strains SA11-4F and WI61) that are known to induce the degradation of IRFs. The results identified the IAD, or dimerization domain, of IRF3 and IRF7 as the region both necessary and sufficient for these NSP1 proteins to induce degradation. Consistent with these results, NSP1 also mediated the degradation of two other IAD-containing IRF proteins (IRF5 and IRF9) but failed to induce the degradation of IRF1, a transcription factor that lacks an IAD-like dimerization domain. Because the IAD domains of IRF3 to IRF9 are structurally similar (12, 16, 22), it may be predicted that NSP1 targets the entire subset of IAD-containing IRF proteins for degradation, including the untested IRF4, IRF6, and IRF8 proteins. Given the importance of IRF4 and IRF8 to the differentiation of CD4+ DCs and to CD8a+ and plasmacytoid DCs, respectively, the degradation of these targets by NSP1 could significantly delay adaptive immune responses to RVA infection (1, 48).
Interestingly, a recent study by Holloway et al. (49) indicated that RVA interferes with host antiviral responses by preventing the nuclear accumulation of STAT1 and STAT2, despite the fact that activated forms of these factors are present in the cytoplasm. We propose that the failure of activated STAT to translocate from the cytoplasm to the nucleus in RVA-infected cells stems from NSP1-mediated degradation of IRF9. As a result, STAT1 and STAT2 cannot interact with IRF9 to form the heterotrimeric ISGF3 complex, which, when present, translocates to the nucleus and induces ISG transcription. Holloway et al. (49) suggested that NSP1 was not responsible for the failure of STAT to accumulate in the nucleus, but results were not provided for assessing the potential of their form of NSP1 to induce IRF9 degradation.
The formation of the ISGF3 complex via the Jak/STAT pathway is triggered not only by interaction of type I IFNs (IFN-α and IFN-β) with the cellular heterodimeric IFN-αR1/R2 receptor complex but also by interaction of type III IFN (IFN-λ) with the IFN-1R1/interleukin-10R2 receptor complex (50). Because RVA infection stimulates the production of both type I and III IFNs (51 and unpublished results), receptor interactions with either type of IFN could lead to the formation of ISGF3 complexes in RVA-infected cells. However, stemming from the capacity of NSP1 to degrade IRF9 and thereby prevent ISGF3 formation, it is possible that the virus can undermine the capacity of either type of IFN (I or III) to stimulate ISG expression. Moreover, the IFN-λ promoter region, like those of IFN-α and -β, contains binding sites for IRF3 (52). The fact that IRF3 is a target of NSP1 provides the virus with yet another mechanism (IRF3 degradation) for antagonizing the signaling pathways of type I and III IFNs. The essential role for IRF3 in controlling RVA replication was demonstrated in a study showing that viral RNA accumulates to much higher levels in IRF3−/− mouse embryo fibroblasts than in IRF3+/+ fibroblasts (29).
There are many different circulating and laboratory-adapted strains of RVA, and the capacity of NSP1 proteins to induce IRF degradation has been shown to differ depending on the strain (39, 40). In order to determine if more than one NSP1 protein induced degradation of the IAD, we examined the NSP1 activity from two RVA strains, SA11-4F and WI61. Both were capable of inducing degradation of the IAD of IRF3 and IRF7 (Fig. 3), suggesting that the mechanism of recognition and degradation is common among types of NSP1 proteins that target IRFs. We did observe some variation in the size of the fragment of IRF3 recognized by the different NSP1 proteins. NSP1 from the SA11-4F strain was capable of inducing degradation of IRF3 (190-385) but not IRF3 (203-385), suggesting that NSP1 partially interacts with IRF3 in the region spanning amino acids 190 to 203. SA11-4F NSP1 is 10 amino acids longer than WI61 NSP1 (496 versus 486 amino acids, respectively). The additional length of SA11-4F NSP1 is found at the C terminus of the protein, corresponding to the region that is known to interact with IRF3 (35, 37); thus, it is possible that the extra amino acids are important to binding IRF3. The region of IRF proteins sufficient for NSP1-induced degradation was narrowed down to approximately 180 amino acids (Fig. 3), which is probably larger than the actual binding site of NSP1.
The inactive form of IRF proteins is held in a monomeric state by the autoinhibitory domain, which folds back onto the IAD (Fig. 1). Phosphorylation of key serine residues causes the autoinhibitory domain to unfold, exposing key residues that are required for two monomers to interact and form a dimer. Deletion of the IRF3 ID did not alter the capacity of NSP1 to induce degradation (Fig. 2 and 3), suggesting that monomeric (ID present) and possibly dimeric (ID absent) forms of IRF3 both were recognized by NSP1. Alanine replacement of the key phosphorylation sites in the C-terminal autoinhibitory domain of IRF3 (IRF3-5A) prevents dimerization, while aspartic acid replacement of the same amino acids results in a constitutively dimeric form of IRF3 (IRF3-5D) (24). Expression of proteins with such mutations confirmed that NSP1 targeted both monomeric and dimeric forms of IRF3 for degradation (Fig. 5). We also introduced into the constitutively dimeric IRF3-5D a point mutation that disrupted dimer formation and found again that this monomeric form of IRF3 was degraded by NSP1. An earlier study showed that NSP1-mediated degradation of IRF3 was enhanced by poly(I·C) activation, suggesting that the stage of IRF3 signaling interrupted by NSP1 is postphosphorylation (40). In contrast, our experiments did not reveal an obvious preference for NSP1 to induce degradation of monomeric or dimeric IRF proteins. However, we did not perform a quantitative assessment that would have better revealed whether NSP1 preferred one form over another. Given that IRF9 is not known to form a dimer via its IAD-like domain, the ability of NSP1 to induce the IRF9 degradation supports the conclusion that monomeric forms of IRF proteins are targets of NSP1.
We speculate that NSP1 broadly affects IRF protein levels during RVA infection by targeting any IAD-containing IRF that it comes into contact with, regardless of form (monomeric or dimeric). NSP1 has several characteristics that suggest it functions as a viral E3 ubiquitin ligase. Primarily, NSP1 contains a RING domain similar to that of some E3 ligases (41, 53, 54), and the protein induces the proteasomal degradation of target proteins, such as IRF3 and IRF7 (37, 53). Although the features of NSP1 indicate it functions as an E3 ubiquitin ligase, direct evidence in support of this has not yet been obtained. This study supports the hypothesis that NSP1 proteins from different RVA strains share a similar mechanism of activity; however, we are unable to reconcile why NSP1 proteins can show variation in their ability to degrade the various IRF proteins given the similarity (but not identity) of their IAD domains (16, 22).
Identification of the region of IRF3 targeted for degradation by NSP1 lays the groundwork for structural studies of NSP1, possibly in association with IRF3. The structural and electrostatic surface properties of the IAD are similar to those of the MH2 domain of Smad proteins (16, 55). In response to transforming growth factor β (TGF-β) signaling, Smad proteins are activated by phosphorylation and heterotrimerize before translocating to the nucleus, where they regulate transcription of target genes (56–58). One recent report revealed a direct physical and functional interaction between Smad3 and IRF7, suggesting IRF7 could replace one of the Smad3 subunits to form a Smad3/Smad4/IRF7 complex (59). The similarity between the Smad MH2 domain and the IRF IAD suggests that NSP1 can interfere with transcription factors other than IRFs, broadening their effect on host responses.
Indeed, recent reports indicate that NSP1 can interfere with innate immune responses through other activities. Perhaps most notable are results indicating that animal RVA NSP1 proteins can interact with and induce the degradation of p53, a transcription factor that promotes apoptosis in stressed cells (60). Animal NSP1 proteins may also interact with the p85 subunit of the phosphoinositide 3-kinase (P13K), promoting activation of the prosurvival P13K/Akt pathway (61, 62). Moreover, NSP1 appears to have affinity for RIG-I, an interaction that interferes with the ability of RIG-I to stimulate IFN-β expression (63). The fact that NSP1 has RNA-binding activity raises the possibility that the protein also antagonizes interactions between viral RNAs and PRRs or ISG products (64). Based on the discovery that RVA VP3 has a specialized domain that functions to suppress the antiviral oligoadenylate synthetase (OAS)/RNase L pathway, viral proteins in addition to NSP1 may play key roles in suppressing innate immune responses (65).
ACKNOWLEDGMENTS
We are grateful to Kristen Ogden, Shane Trask, and Aitor Navarro for critical review of the manuscript and for helpful discussions.
This work was supported by the Intramural Research Program of the National Institute of Allergy and Infectious Diseases, National Institutes of Health.
Footnotes
Published ahead of print 3 July 2013
REFERENCES
- 1.Savitsky D, Tamura T, Yanai H, Taniguchi T. 2010. Regulation of immunity and oncogenesis by the IRF transcription factor family. Cancer Immunol. Immunother. 59:489–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yanai H, Mizutani T, Inuzuka T, Honda K, Takaoka A, Taniguchi T. 2005. IRF family transcription factors in type I interferon induction. Int. Congr. Ser. 1285:104–113 [Google Scholar]
- 3.Sato M, Suemori H, Hata N, Asagiri M, Ogasawara K, Nakao K, Nakaya T, Katsuki M, Noguchi S, Tanaka N, Taniguchi T. 2000. Distinct and essential roles of transcription factors IRF-3 and IRF-7 in response to viruses for IFN-alpha/beta gene induction. Immunity 13:539–548 [DOI] [PubMed] [Google Scholar]
- 4.Yanai H, Chen HM, Inuzuka T, Kondo S, Mak TW, Takaoka A, Honda K, Taniguchi T. 2007. Role of IFN regulatory factor 5 transcription factor in antiviral immunity and tumor suppression. Proc. Natl. Acad. Sci. U. S. A. 104:3402–3407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Paun A, Pitha PM. 2007. The IRF family, revisited. Biochimie 89:744–753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thompson MR, Kaminski JJ, Kurt-Jones EA, Fitzgerald KA. 2011. Pattern recognition receptors and the innate immune response to viral infection. Viruses 3:920–940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jensen S, Thomsen AR. 2012. Sensing of RNA viruses: a review of innate immune receptors involved in recognizing RNA virus invasion. J. Virol. 86:2900–2910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yoneyama M, Fujita T. 2010. Recognition of viral nucleic acids in innate immunity. Rev. Med. Virol. 20:4–22 [DOI] [PubMed] [Google Scholar]
- 9.Schlee M, Barchet W, Hornung V, Hartmann G. 2007. Beyond double-stranded RNA-type I IFN induction by pppRNA and other viral nucleic acids. Curr. Top. Microbiol. Immunol. 316:207–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Randall RE, Goodbourn S. 2008. Interferons and viruses: an interplay between induction, signalling, antiviral responses and virus countermeasures. J. Gen. Virol. 89:1–47 [DOI] [PubMed] [Google Scholar]
- 11.Meraro D, Hashmueli S, Koren B, Azriel A, Oumard A, Kirchhoff S, Hauser H, Nagulapalli S, Atchison ML, Levi BZ. 1999. Protein-protein and DNA-protein interactions affect the activity of lymphoid-specific IFN regulatory factors. J. Immunol. 163:6468–6478 [PubMed] [Google Scholar]
- 12.Yanai H, Negishi H, Taniguchi T. 2012. The IRF family of transcription factors: inception, impact and implications in oncogenesis. Oncoimmunology 1:1376–1386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fujii Y, Shimizu T, Kusumoto M, Kyogoku Y, Taniguchi T, Hakoshima T. 1999. Crystal structure of an IRF-DNA complex reveals novel DNA recognition and cooperative binding to a tandem repeat of core sequences. EMBO J. 18:5028–5041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Panne D, Maniatis T, Harrison SC. 2007. An atomic model of the interferon-beta enhanceosome. Cell 129:1111–1123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Escalante CR, Yie J, Thanos D, Aggarwal AK. 1998. Structure of IRF-1 with bound DNA reveals determinants of interferon regulation. Nature 391:103–106 [DOI] [PubMed] [Google Scholar]
- 16.Qin BY, Liu C, Lam SS, Srinath H, Delston R, Correia JJ, Derynck R, Lin K. 2003. Crystal structure of IRF-3 reveals mechanism of autoinhibition and virus-induced phosphoactivation. Nat. Struct. Biol. 10:913–921 [DOI] [PubMed] [Google Scholar]
- 17.Brierley MM, Fish EN. 2005. Stats: multifaceted regulators of transcription. J. Interferon Cytokine Res. 25:733–744 [DOI] [PubMed] [Google Scholar]
- 18.Reich NC. 2002. Nuclear/cytoplasmic localization of IRFs in response to viral infection or interferon stimulation. J. Interferon Cytokine Res. 22:103–109 [DOI] [PubMed] [Google Scholar]
- 19.Barnes BJ, Kellum MJ, Field AE, Pitha PM. 2002. Multiple regulatory domains of IRF-5 control activation, cellular localization, and induction of chemokines that mediate recruitment of T lymphocytes. Mol. Cell. Biol. 22:5721–5740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lin R, Mamane Y, Hiscott J. 1999. Structural and functional analysis of interferon regulatory factor 3: localization of the transactivation and autoinhibitory domains. Mol. Cell. Biol. 19:2465–2474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lin R, Genin P, Mamane Y, Hiscott J. 2000. Selective DNA binding and association with the CREB binding protein coactivator contribute to differential activation of alpha/beta interferon genes by interferon regulatory factors 3 and 7. Mol. Cell. Biol. 20:6342–6353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Takahasi K, Suzuki NN, Horiuchi M, Mori M, Suhara W, Okabe Y, Fukuhara Y, Terasawa H, Akira S, Fujita T, Inagaki F. 2003. X-ray crystal structure of IRF-3 and its functional implications. Nat. Struct. Biol. 10:922–927 [DOI] [PubMed] [Google Scholar]
- 23.Chen W, Lam SS, Srinath H, Jiang Z, Correia JJ, Schiffer CA, Fitzgerald KA, Lin K, Royer WE., Jr 2008. Insights into interferon regulatory factor activation from the crystal structure of dimeric IRF5. Nat. Struct. Mol. Biol. 15:1213–1220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lin R, Heylbroeck C, Pitha PM, Hiscott J. 1998. Virus-dependent phosphorylation of the IRF-3 transcription factor regulates nuclear translocation, transactivation potential, and proteasome-mediated degradation. Mol. Cell. Biol. 18:2986–2996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Greenberg HB, Estes MK. 2009. Rotaviruses: from pathogenesis to vaccination. Gastroenterology 136:1939–1951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tate JE, Burton AH, Boschi-Pinto C, Steele AD, Duque J, Parashar UD, WHO-Coordinated Global Rotavirus Surveillance Network 2012. 2008 Estimate of worldwide rotavirus-associated mortality in children younger than 5 years before the introduction of universal rotavirus vaccination programmes: a systematic review and meta-analysis. Lancet Infect. Dis. 12:136–141 [DOI] [PubMed] [Google Scholar]
- 27.Angel J, Franco MA, Greenberg HB. 2012. Rotavirus immune responses and correlates of protection. Curr. Opin. Virol. 2:419–425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Broquet AH, Hirata Y, McAllister CS, Kagnoff MF. 2011. RIG-I/MDA5/MAVS are required to signal a protective IFN response in rotavirus-infected intestinal epithelium. J. Immunol. 186:1618–1626 [DOI] [PubMed] [Google Scholar]
- 29.Sen A, Pruijssers AJ, Dermody TS, García-Sastre A, Greenberg HB. 2011. The early interferon response to rotavirus is regulated by PKR and depends on MAVS/IPS-1, RIG-I, MDA-5, and IRF3. J. Virol. 85:3717–3732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Arnold MM, Sen A, Greenberg HB, Patton JT. 2013. The battle between rotavirus and its host for control of the interferon signaling pathway. PLoS Pathog. 9:e1003064. 10.1371/journal.ppat.1003064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sherry B. 2009. Rotavirus and reovirus modulation of the interferon response. J. Interferon Cytokine Res. 29:559–567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hu L, Crawford SE, Hyser JM, Estes MK, Prasad BV. 2012. Rotavirus non-structural proteins: structure and function. Curr. Opin. Virol. 2:380–388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mansur DS, Maluquer de Motes C, Unterholzner L, Sumner RP, Ferguson BJ, Ren H, Strnadova P, Bowie AG, Smith GL. 2013. Poxvirus targeting of E3 ligase β-TrCP by molecular mimicry: a mechanism to inhibit NF-κB activation and promote immune evasion and virulence. PLoS Pathog. 9:e1003183. 10.1371/journal.ppat.1003183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Graff JW, Ettayebi K, Hardy ME. 2009. Rotavirus NSP1 inhibits NF-kB activation by inducing proteasome-dependent degradation of beta-TrCP: a novel mechanism of IFN antagonism. PLoS Pathog. 5:e1000280. 10.1371/journal.ppat.1000280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Graff JW, Mitzel DN, Weisend CM, Flenniken ML, Hardy ME. 2002. Interferon regulatory factor 3 is a cellular partner of rotavirus NSP1. J. Virol. 76:9545–9550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Feng N, Sen A, Nguyen H, Vo P, Hoshino Y, Deal EM, Greenberg HB. 2009. Variation in antagonism of the interferon response to rotavirus NSP1 results in differential infectivity in mouse embryonic fibroblasts. J. Virol. 83:6987–6994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Barro M, Patton JT. 2005. Rotavirus nonstructural protein 1 subverts innate immune response by inducing degradation of IFN regulatory factor 3. Proc. Natl. Acad. Sci. U. S. A. 102:4114–4119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Barro M, Patton JT. 2007. Rotavirus NSP1 inhibits expression of type I interferon by antagonizing the function of interferon regulatory factors IRF3, IRF5, and IRF7. J. Virol. 81:4473–4481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Arnold MM, Patton JT. 2011. Diversity of interferon antagonist activities mediated by NSP1 proteins of different rotavirus strains. J. Virol. 85:1970–1979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sen A, Feng N, Ettayebi K, Hardy ME, Greenberg HB. 2009. IRF3 inhibition by rotavirus NSP1 is host cell and virus strain dependent but independent of NSP1 proteasomal degradation. J. Virol. 83:10322–10335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hua J, Mansell EA, Patton JT. 1993. Comparative analysis of the rotavirus NS53 gene: conservation of basic and cysteine-rich regions in the protein and possible stem-loop structures in the RNA. Virology 196:372–378 [DOI] [PubMed] [Google Scholar]
- 42.Xu L, Tian Y, Tarlow O, Harbour D, McCrae MA. 1994. Molecular biology of rotaviruses. IX. Conservation and divergence in genome segment 5. J. Gen. Virol. 75:3413–3421 [DOI] [PubMed] [Google Scholar]
- 43.Dunn SJ, Cross TL, Greenberg HB. 1994. Comparison of the rotavirus nonstructural protein NSP1 (NS53) from different species by sequence analysis and northern blot hybridization. Virology 203:178–183 [DOI] [PubMed] [Google Scholar]
- 44.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. 2004. UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 25:1605–1612 [DOI] [PubMed] [Google Scholar]
- 45.Patton JT, Taraporewala Z, Chen D, Chizhikov V, Jones M, Elhelu A, Collins M, Kearney K, Wagner M, Hoshino Y, Gouvea V. 2001. Effect of intragenic rearrangement and changes in the 3′ consensus sequence on NSP1 expression and rotavirus replication. J. Virol. 75:2076–2086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Small C, Barro M, Brown TL, Patton JT. 2007. Genome heterogeneity of SA11 rotavirus due to reassortment with “O” agent. Virology 359:415–424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mori M, Yoneyama M, Ito T, Takahashi K, Inagaki F, Fujita T. 2004. Identification of Ser-386 of interferon regulatory factor 3 as critical target for inducible phosphorylation that determines activation. J. Biol. Chem. 279:9698–9702 [DOI] [PubMed] [Google Scholar]
- 48.Lu R. 2008. Interferon regulatory factor 4 and 8 in B-cell development. Trends Immunol. 29:487–492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Holloway G, Truong TT, Coulson BS. 2009. Rotavirus antagonizes cellular antiviral responses by inhibiting the nuclear accumulation of STAT1, STAT2, and NF-kB, J. Virol. 83:4942–4951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Donnelly RP, Kotenko SV. 2010. Interferon-lambda: a new addition to an old family. J. Interferon Cytokine Res. 30:555–564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pott J, Mahlakõiv T, Mordstein M, Duerr CU, Michiels T, Stockinger S, Staeheli P, Hornef MW. 2011. IFN-lambda determines the intestinal epithelial antiviral host defense. Proc. Natl. Acad. Sci. U. S. A. 108:7944–7949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Iversen MB, Paludan SR. 2010. Mechanisms of type III interferon expression. J. Interferon Cytokine Res. 30:573–578 [DOI] [PubMed] [Google Scholar]
- 53.Aravind L, Iyer LM, Koonin EV. 2003. Scores of RINGS but no PHDs in ubiquitin signaling. Cell Cycle 2:123–126 [DOI] [PubMed] [Google Scholar]
- 54.Pina-Vazquez C, De Nova-Ocampo M, Guzman-Leon S, Padilla-Noriega L. 2007. Post-translational regulation of rotavirus protein NSP1 expression in mammalian cells. Arch. Virol. 152:345–368 [DOI] [PubMed] [Google Scholar]
- 55.Eroshkin A, Mushegian A. 1999. Conserved transactivation domain shared by interferon regulatory factors and Smad morphogens. J. Mol. Med. (Berlin) 77:403–405 [DOI] [PubMed] [Google Scholar]
- 56.Kawabata M, Inoue H, Hanyu A, Imamura T, Miyazono K. 1998. Smad proteins exist as monomers in vivo and undergo homo- and hetero-oligomerization upon activation by serine/threonine kinase receptors. EMBO J. 17:4056–4065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chacko BM, Qin B, Correia JJ, Lam SS, de Caestecker MP, Lin K. 2001. The L3 loop and C-terminal phosphorylation jointly define Smad protein trimerization. Nat. Struct. Biol. 8:248–253 [DOI] [PubMed] [Google Scholar]
- 58.Qin BY, Chacko BM, Lam SS, de Caestecker MP, Correia JJ, Lin K. 2001. Structural basis of Smad1 activation by receptor kinase phosphorylation. Mol. Cell 8:1303–1312 [DOI] [PubMed] [Google Scholar]
- 59.Qing J, Liu C, Choy L, Wu RY, Pagano JS, Derynck R. 2004. Transforming growth factor beta/Smad3 signaling regulates IRF-7 function and transcriptional activation of the beta interferon promoter. Mol. Cell. Biol. 24:1411–1425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bhowmick R, Halder UC, Chattopadhyay S, Nayak MK, Chawla-Sarkar M. 2013. Rotavirus-encoded nonstructural protein 1 modulates cellular apoptotic machinery by targeting tumor suppressor protein p53. J. Virol. 87:6840–6850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bagchi P, Dutta D, Chattopadhyay S, Mukherjee A, Halder UC, Sarkar S, Kobayashi N, Komoto S, Taniguchi K, Chawla-Sarkar M. 2010. Rotavirus nonstructural protein 1 suppresses virus-induced cellular apoptosis to facilitate viral growth by activating the cell survival pathways during early stages of infection. J. Virol. 84:6834–6845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bagchi P, Nandi S, Nayak MK, Chawla-Sarkar M. 2013. Molecular mechanism behind rotavirus NSP1-mediated PI3 kinase activation: interaction between NSP1 and the p85 subunit of PI3 kinase. J. Virol. 87:2358–2362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Qin L, Ren L, Zhou Z, Lei X, Chen L, Xue Q, Liu X, Wang J, Hung T. 2011. Rotavirus nonstructural protein 1 antagonizes innate immune response by interacting with retinoic acid inducible gene I. Virol. J. 8:526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hua J, Chen X, Patton JT. 1994. Deletion mapping of the rotavirus metalloprotein NS53 (NSP1): the conserved cysteine-rich region is essential for virus-specific RNA binding. J. Virol. 68:3990–4000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhang R, Jha BK, Ogden KM, Dong B, Zhao L, Elliott R, Patton JT, Silverman RH, Weiss SR. Homologous 2′, 5′-phosphodiesterases from disparate RNA viruses antagonize antiviral innate immunity. Proc. Natl. Acad. Sci. U. S. A., in press [DOI] [PMC free article] [PubMed] [Google Scholar]