

Abstract
Exchange of the native Corynebacterium glutamicum promoter of the aceE gene, encoding the E1p subunit of the pyruvate dehydrogenase complex (PDHC), with mutated dapA promoter variants led to a series of C. glutamicum strains with gradually reduced growth rates and PDHC activities. Upon overexpression of the l-valine biosynthetic genes ilvBNCE, all strains produced l-valine. Among these strains, C. glutamicum aceE A16 (pJC4 ilvBNCE) showed the highest biomass and product yields, and thus it was further improved by additional deletion of the pqo and ppc genes, encoding pyruvate:quinone oxidoreductase and phosphoenolpyruvate carboxylase, respectively. In fed-batch fermentations at high cell densities, C. glutamicum aceE A16 Δpqo Δppc (pJC4 ilvBNCE) produced up to 738 mM (i.e., 86.5 g/liter) l-valine with an overall yield (YP/S) of 0.36 mol per mol of glucose and a volumetric productivity (QP) of 13.6 mM per h [1.6 g/(liter × h)]. Additional inactivation of the transaminase B gene (ilvE) and overexpression of ilvBNCD instead of ilvBNCE transformed the l-valine-producing strain into a 2-ketoisovalerate producer, excreting up to 303 mM (35 g/liter) 2-ketoisovalerate with a YP/S of 0.24 mol per mol of glucose and a QP of 6.9 mM per h [0.8 g/(liter × h)]. The replacement of the aceE promoter by the dapA-A16 promoter in the two C. glutamicum l-lysine producers DM1800 and DM1933 improved the production by 100% and 44%, respectively. These results demonstrate that C. glutamicum strains with reduced PDHC activity are an excellent platform for the production of pyruvate-derived products.
INTRODUCTION
Corynebacterium glutamicum is a Gram-positive, facultative anaerobic organism that grows on a variety of sugars and organic acids and is the workhorse for the production of a number of amino acids, e.g., l-glutamate, l-lysine, and also l-valine (1–4). Recent studies also showed the successful employment of C. glutamicum for the production of the diamines putrescine and cadaverine (5–10), the organic acids d-lactate, succinate, 2-ketoisovalerate, and pyruvate (11–15), the biofuels ethanol and isobutanol (16–18), xylitol (19), and heterologous proteins (20, 21).
Since the common precursor of the products mentioned above is pyruvate (with the exception of xylitol and proteins), the optimization of its availability has a high potential to improve microbial production processes. Radmacher et al. (22) showed that inactivation of d-pantothenate biosynthesis by deleting the panBC genes in combination with plasmid-bound overexpression of the genes encoding acetohydroxyacid synthase (AHAS) (ilvBN gene product), acetohydroxyacid isomeroreductase (AHAIR) (ilvC gene product), dihydroxyacid dehydratase (DHAD) (ilvD gene product), and/or transaminase B (TA) (ilvE gene product) (Fig. 1) led to increased l-valine production of C. glutamicum when cultivated under d-pantothenate-limiting conditions. Later on, Bartek et al. (23) showed that this limitation results in a drastically increased cytoplasmic pyruvate pool, due to reduced coenzyme A (CoA) availability for the reaction of the pyruvate dehydrogenase complex (PDHC). Schreiner et al. (24) identified and functionally characterized the E1p subunit of the PDHC in C. glutamicum and showed that the activity of this multienzyme complex is essential for growth of this organism on glucose, pyruvate, or l-lactate. Deletion of the aceE gene, encoding the E1p subunit, caused PDHC deficiency, and the resulting strain C. glutamicum ΔaceE strain required either acetate or ethanol as additional carbon source for growth (24, 25). Further characterization of the PDHC-deficient strain C. glutamicum ΔaceE showed that the mutant formed significant amounts of l-valine, l-alanine, and pyruvate from glucose when acetate was exhausted from the medium and growth stopped (26). Plasmid-bound overexpression of the l-valine biosynthesis genes ilvBNCE shifted the product spectrum toward l-valine (26), and inactivation of the pyruvate:quinone oxidoreductase (PQO) (pqo gene product; Fig. 1) and phosphoglucose isomerase (pgi gene product) in C. glutamicum ΔaceE (pJC4 ilvBNCE) resulted in even more efficient l-valine production (up to 410 mM, with a maximum yield of 0.86 mol per mol of glucose in the production phase [27]). Based on these results, we engineered the wild type (WT) of C. glutamicum for the aerobic, growth-decoupled production of 2-ketoisovalerate (KIV) from glucose by deletion of the aceE, pqo, and ilvE genes and additional overexpression of the ilvBNCD genes (11). In fed-batch fermentations at high cell densities, C. glutamicum ΔaceE Δpqo ΔilvE (pJC4 ilvBNCD) produced up to 188 mM KIV and showed a volumetric productivity of about 4.6 mM KIV per h in the overall production phase (11). In further approaches, we used the PDHC-deficient C. glutamicum strain as a platform for the efficient production of about 500 mM pyruvate (i.e., 45 g/liter) or 175 mM isobutanol (i.e., 13 g/liter) and also to improve l-lysine production with C. glutamicum (15, 16, 28).
Fig 1.

Enzymes of the central metabolism with the biosynthetic pathway of 2-ketoisovalerate/l-valine and l-lysine in C. glutamicum. Abbreviations: AHAIR, acetohydroxyacid isomeroreductase; AHAS, acetohydroxyacid synthase; AK, acetate kinase; DHAD, dihydroxyacid dehydratase; PCx, pyruvate carboxylase; PDHC, pyruvate dehydrogenase complex; PEP, phosphoenolpyruvate; PEPCk, PEP carboxykinase; PEPCx, PEP carboxylase; PK, pyruvate kinase; PTA, phosphotransacetylase; PQO, pyruvate:quinone oxidoreductase; TA, transaminase B; TCA, tricarboxylic acid.
As stated above, a common feature of all PDHC-deficient C. glutamicum strains is the start of the production phase only after complete consumption of the acetate required for growth. Although the addition of maltose instead of glucose, the use of ethanol instead of acetate, or the inactivation of the transcriptional regulator SugR led to growth-coupled production of l-valine, all strains still require the use of acetate (or ethanol) as an essential carbon source (25, 29), resulting in a production process more laborious than approaches with glucose as a single carbon source. This holds true especially in large-scale production processes, for which rather simple approaches are always preferred.
Promoter engineering is a suitable metabolic engineering strategy to carefully modulate transcription and to alter the resulting enzyme activity avoiding deletion of a corresponding gene and its unwanted effects such as auxotrophies. Holátko et al. (30) reduced activity of the ilvA (encoding threonine deaminase) and leuA (encoding isopropylmalate synthase) promoters and increased the activities of the ilvD and ilvE promoters by site-directed mutagenesis of the respective (extended) −10 regions. These modifications in combination with the deletion of panB (see above) and expression of ilvBN alleles encoding a feedback-resistant variant of the AHAS resulted in an l-isoleucine bradytrophy and improved production strain, which is, however, still auxotrophic for d-pantothenate (30). To improve l-lysine production with C. glutamicum, several studies employed the strong promoters of the superoxide dismutase gene or of the elongation factor TU gene to replace the native chromosomal promoters of target genes (31–33).
By site-directed mutagenesis of the (extended) −10 region of the dihydrodipicolinate synthase gene (dapA) promoter, Vasicová et al. (34) engineered a library with gradually differing promoter activity. Here we made use of this dapA promoter library and show that the replacement of the native promoter of the aceE gene by mutated dapA promoters leads to a series of C. glutamicum strains with gradually reduced PDHC activity and growth on glucose, without a requirement for additional carbon sources. These strains were investigated for their ability to produce l-valine, and the most promising strain was then further engineered for high-titer l-valine and 2-ketoisovalerate production. Finally, the novel C. glutamicum platform with reduced PDHC activity was employed for improving l-lysine production.
MATERIALS AND METHODS
Bacterial strains and plasmids.
All bacterial strains, plasmids, and oligonucleotides used and their relevant characteristics, sequences and sources or purposes are listed in Table 1.
Table 1.
Strains, plasmids, and oligonucleotides used in this study
| Strain, plasmid, or oligonucleotide | Relevant characteristic(s) or sequence | Source, reference, or purpose |
|---|---|---|
| Strains | ||
| E. coli DH5α | F− ϕ80lacZΔM15 Δ(lacZYA-argF)U169 endA1 recA1 hsdR17 (rK− mK+) supE44 thi-1 gyrA96 relA1 phoA | 62 |
| C. glutamicum WT | Wild-type strain ATCC 13032, biotin auxotrophic | American Type Culture Collection |
| C. glutamicum ΔaceE | C. glutamicum WT with deletion of the E1p gene (aceE) of the pyruvate dehydrogenase complex | 24 |
| C. glutamicum DM1800 | l-Lysine producer; pycP485S, lysCT311I, derived from C. glutamicum WT | 63 |
| C. glutamicum DM1800 aceE A16 | C. glutamicum DM1800 in which the native aceE promoter was replaced by the dapA-A16 promoter | This work |
| C. glutamicum DM1933 | l-Lysine producer; Δpck, pycP458S; homV59A; 2× lysCT311I, 2× asd, 2× dapA, 2× dapB, 2× ddh, 2× lysA, 2× lysE, derived from C. glutamicum WT | 64 |
| C. glutamicum DM1933 aceE A16 | C. glutamicum DM1933 in which the native aceE promoter was replaced by the dapA-A16 promoter | This work |
| C. glutamicum aceE L1, A23, A25, A16 | C. glutamicum WT in which the native aceE promoter was replaced by the dapA-L1, -A23, -A25, or -A16 promoter, respectively | This work |
| C. glutamicum aceE A16 Δpqo | C. glutamicum aceE A16 with deleted pqo gene, encoding pyruvate:quinone oxidoreductase | This work |
| C. glutamicum aceE A16 Δpqo Δppc | C. glutamicum aceE A16 Δpqo with deleted ppc gene, encoding phosphoenolpyruvate carboxylase | This work |
| C. glutamicum aceE A16 Δpqo Δppc ΔilvE | C. glutamicum aceE A16 Δpqo Δppc with deleted ilvE gene, encoding transaminase B | This work |
| Plasmids | ||
| pK18/19mobsacB | Kmr, mobilizable (oriT), oriV | 40 |
| pK18mobsacBaceE-rAc | pK18mobsacB carrying a truncated promoter region of the aceE gene, encoding the E1 subunit of the PDHC | This work |
| pK18mobsacB PaceE dapA-L1, -A23, -A25, -A16 | pK19mobsacB PaceE carrying the dapA-L1, -A23, -A25, or -A16 promoter, respectively | This work |
| pK19mobsacB Δppc | pK19mobsacB carrying a truncated ppc gene, encoding phosphoenolpyruvate carboxylase | This work |
| pK19mobsacB Δpqo | pK19mobsacB carrying a truncated pqo gene, encoding pyruvate:quinone oxidoreductase | 41 |
| pK19mobsacB ΔilvE | pK19mobsacB carrying a truncated ilvE gene, encoding transaminase B | 61 |
| pK19mobsacB dPgltA540-PdapA-L1, -A23, -A25, -A16 | pK19mobsacB carrying a truncated promoter region of the gltA gene and the dapA-L1, -A23, -A25, or -A16 promoter, respectively | 60 |
| pJC4 ilvBNCD | Kanr; plasmid carrying the ilvBNCD genes encoding the l-valine biosynthetic enzymes acetohydroxyacid synthase, isomeroreductase, and dihydroxyacid dehydratase | 65 |
| pJC4 ilvBNCE | Kanr; plasmid carrying the ilvBNCE genes encoding the l-valine biosynthetic enzymes acetohydroxyacid synthase, isomeroreductase, and transaminase B | 22 |
| Oligonucleotides | ||
| dapAfow | 5′-AACTGCAGAACCAATGCATTGGTTCTGCAGTTATCACA CCC-3′ | Amplification of dapA promoters, NsiI site underlined |
| dapArev2 | 5′-GGGAATTCCATATGAGGCTCCTTTTAAATCGAGCGGCT CCGGTCTTAGCTGTTAAACC-3′ | Amplification of dapA promoters/verification of promoter exchange, NdeI site underlined |
| ace1 | 5′-CCCAAGCTTGCACATTACCGTCCAACC-3′ | Sequencing of dapA promoters/verification of promoter exchange |
| ace2 | 5′-CGCGGATCCCGACGGTAACGCTTCTCC-3′ | Sequencing of dapA promoters |
| ilvE1 | 5′-GCGTTGACTGATTCTTGGTC-3′ | Primer to verify deletion of ilvE (11) |
| ilvE2 | 5′-CGAGTTCGATGGAATCTTCG-3′ | Primer to verify deletion of ilvE (11) |
| pqodel1 | 5′-AAGGAATTCGTTTTCGAGGCGACCAGACAG-3′ | Primer to verify deletion of pqo (41) |
| pqodel4 | 5′-TGGCACAAGCTTGTTAAGCGCTCGCGGTCAATG-3′ | Primer to verify deletion of pqo (41) |
| ppc1 | 5′-CCCAAGCTTGAGTTGCGCAGCGCAGTG-3′ | Primer for deletion of ppc, HindIII site underlined |
| ppc2 | 5′-GTGCTGCGCAATGCTGAGGGCATTAGAGCAGTGGATT GG-3′ | Primer for deletion of ppc, crossover overlap underlined |
| ppc3 | 5′-CCTCAGCATTGCGCAGCACATCGGCCACAGCTTCTGC-3′ | Primer for deletion of ppc, crossover overlap underlined |
| ppc4 | 5′-CGCGGATCCCGATGACATCAGGTTCCTC-3′ | Primer for deletion of ppc, BamHI site underlined |
| ppcdel1 | 5′-GGAATAGACTCGCTCGGC-3′ | Primer to verify deletion of ppc |
| ppcdel2 | 5′-GTGAACAGGCTCTCGATGC-3′ | Primer to verify deletion of ppc |
| aceEup-fw | 5′-CGGGATCCCGACCCAATGCGTACCGATGTG-3′ | Primer for deletion of aceE promoter region, BamHI site underlined |
| aceE-intrev | 5′-GCGCTAGCGCCACCATCGGAGGTGTTGTTC-3′ | Primer for deletion of aceE promoter region, NheI site underlined |
| aceE-rAC-soeleft | 5′-TTGATCGGCCATATGTATTATGCATCTCTCACGTTTGACG CGAATCG-3′ | Primer for deletion of aceE promoter region, crossover overlap underlined, NsiI and NdeI in italic |
| aceE-rAC-soeright | 5′-CAAACGTGAGAGATGCATAATACATATGGCCGATCAAGC AAAACTTGG-3′ | Primer for deletion of aceE promoter region, crossover overlap underlined, NsiI and NdeI in italic |
DNA preparation and transformation.
Isolation of plasmids from Escherichia coli was performed as described previously (35). Plasmid DNA transfer into C. glutamicum was carried out by electroporation, and recombinant strains were selected on Luria-Bertani brain heart infusion (LB-BHI) agar plates containing 0.5 M sorbitol, 85 mM potassium acetate, and appropriate concentrations of kanamycin (50 μg/ml) (36). Isolation of chromosomal DNA from C. glutamicum was performed as described previously (36). Electroporation of E. coli was carried out with competent cells according to the method of Dower et al. (37).
Culture conditions.
E. coli was grown aerobically in 2× TY complex medium (38) at 37°C as 50-ml cultures in 500-ml baffled Erlenmeyer flasks on a rotary shaker at 120 rpm. Precultures of the different C. glutamicum strains were grown in 2× TY medium containing 0.5% (wt/vol) potassium acetate. The l-lysine producer strains C. glutamicum DM1800 and DM1933 and their derivatives were grown in BHI medium (Becton Dickinson) (37 g/liter) containing 1% (wt/vol) glucose. For growth and amino acid fermentations in shake flasks, cells of an overnight preculture were washed with 0.9% (wt/vol) NaCl and inoculated into CGXII minimal medium (pH 7.4) (39) with 222 mM glucose to give an optical density at 600 nm (OD600) of about 1. The plasmid-carrying strains were grown in the presence of kanamycin (50 μg/ml). C. glutamicum was grown aerobically at 30°C as 50-ml cultures in 500-ml baffled Erlenmeyer flasks on a rotary shaker at 120 rpm.
Fed-batch fermentations for l-valine production were performed at 30°C in 300-ml cultures in a Fedbatch Pro fermentation system from DASGIP (Jülich, Germany), and those for 2-ketoisovalerate production were performed in 1,500-ml cultures in a Bioengineering (Wald, Switzerland) stirred tank reactor with a head pressure of 1.5 × 105 Pa. Batch fermentations for l-lysine production were performed in glass reactors as 600-ml cultures. The pH was maintained at 7.3 by online measurement using a standard pH probe (Mettler Toledo, Giessen, Germany) and addition of 10% NH3 and 5 M H2SO4 (for l-valine and l-lysine) or 5 M KOH and 5 M H2SO4 (for 2-ketoisovalerate). Foam development was prevented by manual injection of about 20 μl of Struktol 674 antifoam (Schill und Seilacher, Hamburg, Germany). Dissolved oxygen was measured online using a polarometric oxygen electrode (Mettler Toledo, Giessen, Germany) and adjusted to ≥20% of saturation in a cascade by stirring at 300 to 1,500 rpm and aeration up to 1 volume per volume per minute. The fermentations were carried out in CGXII minimal medium (pH 7.4) (39) initially containing 222 mM or 333 mM glucose. For 2-ketoisovalerate fermentations, 1% (wt/vol) yeast extract and 10 mM l-valine, l-isoleucine, and l-leucine were additionally added, and for l-lysine fermentations, 0.5% (wt/vol) BHI powder was additionally added. Antibiotics were added as appropriate (kanamycin, 50 μg/ml). During the fed-batch processes, adequate amounts of 50% (wt/vol) glucose were injected.
Construction of C. glutamicum deletion and promoter exchange mutants.
Chromosomal inactivation of the phosphoenolpyruvate carboxylase gene ppc in C. glutamicum aceE A16 Δpqo was performed using crossover PCR and the suicide vector pK19mobsacB. DNA fragments were generated using the primer pairs ppc1/ppc2 and ppc3/ppc4. The two fragments were purified, mixed in equal amounts, and subjected to crossover PCR using primers ppc1 and ppc4. The resulting fusion product (containing the ppc gene with an internal deletion of 1,922 bp) was ligated into BamHI/HindIII-restricted plasmid pK19mobsacB and transformed into E. coli. After isolation and sequencing (MWG Biotech), the recombinant plasmid was electroporated into C. glutamicum aceE A16 Δpqo. Using the method described by Schäfer et al. (40), the intact chromosomal ppc gene was replaced by the truncated ppc gene via homologous recombination (double crossover). The screening of the ppc mutants was done on 2× TY agar plates containing 10% (wt/vol) sucrose and 0.5% (wt/vol) potassium acetate. The replacement at the chromosomal locus was verified by PCR using primers ppcdel1/ppcdel2.
Chromosomal inactivation of the pyruvate:quinone oxidoreductase gene pqo in C. glutamicum aceE A16 and of the transaminase B gene ilvE in C. glutamicum aceE A16 Δpqo Δppc was performed as described before (11, 41). The replacement at the chromosomal locus was verified by PCR using primers pqodel1/pqodel4 or ilvE1/ilvE2, respectively.
Chromosomal replacement of the native aceE promoter by mutated dapA promoters in C. glutamicum WT was performed using the suicide vector pK18mobsacB. DNA fragments were generated using primer pairs aceEup-fw/aceE-rAC-soeleft and aceE-intrev/aceE-rAC-soeright. The two fragments were purified, mixed in equal amounts, and subjected to crossover PCR using primers aceEup-fw and aceE-intrev. The resulting fusion product (containing the aceE promoter region shortened by 338 bp) was ligated into BamHI/XbaI-restricted plasmid pK18mobsacB and transformed into E. coli. After isolation, the nucleotide sequence of the insert region in the recombinant plasmid pK18mobsacBaceE-rAC was verified (MWG Biotec), and then the mutated dapA promoters (A16, A23, A25, and L1) were amplified via PCR with the primers dapAfow and dapArev2 from plasmids pK19mobsacB dPgltA540-PdapA-L1, -A23, -A25, and -A16, respectively, and ligated into NsiI/NdeI-restricted pK18mobsacBaceE-rAC. All nucleotide sequences of the inserts in the newly constructed plasmids were verified by sequencing (MWG Biotech). Double crossover and screening for the correct mutants were performed as described above. The replacement of the native aceE promoter by the dapA-L1, -A16, -A23, and -A25 promoters, respectively, at the chromosomal locus was verified by PCR using primers ace1/dapArev2.
Determination of PDHC activities.
For determination of PDHC activities, the relevant strains were cultivated aerobically in shake flasks to an OD600 of about 5. The cells were harvested by centrifugation for 10 min at 4,500 × g and 4°C, washed once with 25 ml 0.2 M Tris-HCl (pH 7.4), centrifuged again, and resuspended in 0.5 ml 0.2 M Tris-HCl (pH 7.4), 10 mM MgCl2, 3 mM l-cysteine, and 10% (vol/vol) glycerol. The cell suspension was transferred into 2-ml screw-cap vials together with 250 mg of glass beads (0.1-mm diameter; Roth) and subjected to mechanical disruption three times for 30 s at 6,500 rpm with a Precellys 24 instrument (Peqlab) at room temperature (RT) with intermittent cooling on ice for 5 min. Intact cells and cell debris were removed by centrifugation for 15 min at 12,100 × g and 4°C. The resulting cell extract was then subjected to ultracentrifugation for 45 min at 45,000 × g and 4°C, and the supernatant was directly used to determine the PDHC activity as described by Guest and Creaghan (42).
For all tested strains, three biological and two technical replicates were performed. The protein concentration was quantified with the Pierce bicinchoninic acid (BCA) protein assay (Thermo Scientific) with bovine serum albumin as a standard. Assay results were linear over time and proportional to the protein concentration. One unit of activity is defined as 1 μmol NADH formed per min at 30°C.
Analytics.
Biomass formation was followed by determining either the OD600 or the cell dry weight (CDW) (in g/liter) at a given time point. Both techniques were correlated for several independent fermentations, resulting in CDW = OD600 × 0.3.
For determination of glucose and organic and amino acid concentrations in the culture fluid, 1 ml of the culture was harvested by centrifugation (12,100 × g, 10 min, RT) and the supernatant was analyzed. Glucose concentrations were determined by enzymatic tests (Roche Diagnostics). The phosphate concentration was analyzed with the phosphate kit LCK 348 (Hach Lange). The pyruvate concentrations were determined enzymatically as described by Lamprecht and Heinz (43) or by high-pressure liquid chromatography (HPLC) (see below).
The amino acid concentration was determined by reversed-phase HPLC (on an HP 1100 instrument; Hewlett-Packard) with fluorimetric detection (excitation at 230 nm and emission at 450 nm) after automatic precolumn derivatization with ortho-phthaldialdehyde (44). Separation was carried out at 40°C on a Multohyp octyldecylsilane column (particle size, 5 μm; 125 by 4 mm; CS-Chromatographie). The elution buffer consisted of a polar phase (0.1 M sodium acetate, pH 7.2) and methanol as a nonpolar phase. Quantification was done by calculation of the concentration using an internal standard (l-ornithine at 100 μM) and with a 10-point calibration curve for each amino acid. Amino acid concentrations were also determined using an Agilent 1200 series apparatus (Agilent Technologies) equipped with an Agilent Zorbax Eclipse Plus C18 column (250 by 4.6 mm, 5 μm) protected by an Agilent Zorbax Eclipse Plus C18 guard column (12.5 by 4.6 mm, 5 μm). Fluorometric detection (excitation at 230 nm and emission at 450 nm) was carried out after automatic precolumn derivatization with ortho-phthaldialdehyde. The elution buffer consisted of a polar phase (10 mM Na2HPO4, 10 mM Na2B4O7, 0.5 mM NaN3, pH 8.2) and a nonpolar phase (45% [vol/vol] acetonitrile, 45% [vol/vol] methanol). Protocol details were as given by Henderson and Brooks (45). Quantification of the analytes was conducted by using l-norvaline as an internal standard to correct for analyte variability and with an 8-point calibration curve for each component as an external reference standard.
Organic acid concentrations were measured via HPLC using an Agilent 1200 series apparatus equipped with a Rezex ROA organic acid H+ (8%) column (300 by 7.8 mm, 8 μm; Phenomenex) protected by a Rezex ROA organic acid H+ (8%) guard column (50 by 7.8 mm). A protocol for phosphate precipitation was applied to each sample and standard prior to measurement. Thus, 45 μl 4 M NH3 and 100 μl 1.2 M MgSO4 were added to 1,000 μl sample. After 5 min of incubation, the sample was centrifuged for 5 min at 18,000 × g and RT, and 500 μl supernatant was then transferred to 500 μl 0.1 M H2SO4. After thorough mixing and 15 min of incubation at RT, samples were finally centrifuged for 15 min at 18,000 × g at RT. Subsequently, the supernatant was provided for HPLC injection (10-μl injection volume). Separation was performed under isocratic conditions at 50°C (column temperature) for 45 min with 5 mM H2SO4 as the mobile phase at a constant flow rate of 0.4 ml/min. Detection of glucose and organic acids was achieved via an Agilent 1200 series refractive index detector at 32°C. Quantification of the analytes was conducted by using l-rhamnose as an internal standard to correct for analyte variability and with an 8-point calibration curve for each component as an external reference standard.
RESULTS
Replacement of the aceE promoter by mutated dapA promoters results in reduced growth and PDHC activity.
Recently, we identified and functionally characterized the E1p subunit of the PDHC in C. glutamicum, located the promoter region, and identified the transcriptional start site of the aceE gene at 121 nucleotides upstream of the translational start (24). To replace the native aceE promoter, we first cloned the flanking promoter regions into the suicide vector pK18mobsacB, deleting 338 nucleotides upstream of the translational start site. We then cloned the four dapA promoter variants A16, A23, A25, and L1 (34) between the flanking regions. Applying homologous recombination, we were able to replace the native aceE promoter in C. glutamicum WT by the mutated dapA promoters (see Materials and Methods). Subsequently, the growth of the resulting strains C. glutamicum aceE A16, A23, A25, and L1 was compared with that of C. glutamicum WT and the PDHC-deficient strain C. glutamicum ΔaceE in minimal medium with 4% (wt/vol) glucose (Fig. 2A). As already shown by Schreiner et al. (24), C. glutamicum ΔaceE showed almost no growth (OD600 of 2.8 after 24 h), whereas C. glutamicum WT reached an OD600 of about 61 after 24 h, with a growth rate of 0.32 ± 0.01 h−1. C. glutamicum strains with the exchanged dapA promoters A16, A25, A23, and L1 reached significantly lower final OD600s of about 14.5, 12.3, 7.4, and 7.0, respectively. Compared to that of the parental WT strain, the growth rates of the mutant strains were gradually decreased to μ = 0.22, 0.20, 0.15, and 0.14 h−1 for strains A16, A25, A23, and L1, respectively.
Fig 2.
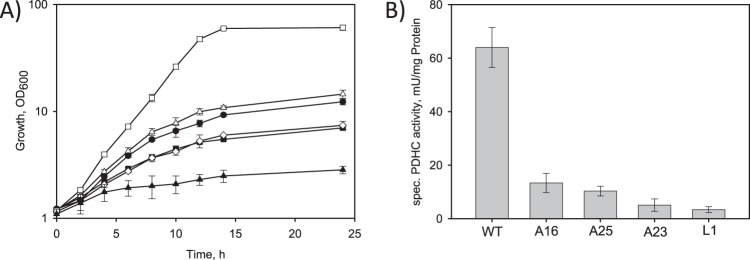
(A) Growth of C. glutamicum WT and its derivatives with reduced (A16, A23, A25, and L1) or abolished (C. glutamicum ΔaceE) PDHC activity in shake flasks containing CGXII medium with 222 mM glucose. □, C. glutamicum WT; ▲, C. glutamicum ΔaceE; △, C. glutamicum aceE A16; ●, C. glutamicum aceE A25; ♢, C. glutamicum aceE A23; ■, C. glutamicum aceE L1. (B) Specific PDHC activities of C. glutamicum WT and its derivatives with reduced PDHC activity grown in shake flasks containing CGXII medium with 222 mM glucose. Three independent fermentations were performed. Error bars show standard deviations.
To further analyze the replacement of the native aceE promoter by mutated dapA promoters, we determined the overall PDHC activities in the respective strains grown in minimal medium with 4% (wt/vol) glucose. C. glutamicum WT showed in the exponential growth phase a PDHC activity of 64 ± 7 mU per mg protein. In agreement with the decreased growth rates, C. glutamicum aceE A16, A25, A23, and L1 showed significantly lower PDHC activities of 13, 10, 5, and 3 mU per mg protein, respectively (Fig. 2B).
Overexpression of the ilvBNCE genes in strains with reduced PDHC activity results in improved l-valine overproduction.
To analyze the suitability of the engineered C. glutamicum strains with reduced PDHC activity for l-valine production, we transformed C. glutamicum aceE A16, A25, A23, and L1 and the WT strain with plasmid pJC4 ilvBNCE, carried out shake flask cultivations in minimal medium with 222 mM glucose, and investigated growth, substrate consumption, and amino acid formation (Table 2). Overexpression of the l-valine biosynthetic pathway genes resulted in l-alanine and l-valine overproduction in all strains. C. glutamicum WT (pJC4 ilvBNCE) showed a substrate-specific biomass yield (YX/S) of about 0.066 g CDW per mmol of glucose, a biomass-specific glucose consumption rate (qS) of about 0.80 g glucose per g CDW and h, a substrate-specific l-alanine yield of about 0.02 mol l-alanine per mol of glucose, and a substrate-specific l-valine yield (YP/S) of about 0.11 mol l-valine per mol of glucose consumed. Interestingly, among the modified C. glutamicum strains, the one with lowest YX/S but with the uppermost qS did not exhibit the highest YP/S. With stepwise increasing YX/S (L1, A23, A25, and A16), the YP/S increased from about 0.21 (L1) to 0.37 (A16) mol l-valine per mol of glucose, and the substrate-specific l-alanine yield dropped from about 0.06 (L1) to 0.02 (A16) mol l-alanine per mol of glucose (Table 2).
Table 2.
Substrate-specific biomass yield (YX/S), biomass-specific glucose consumption rate (qS), and substrate-specific l-alanine and l-valine yields (YP/S) of C. glutamicum l-valine producer strains cultivated in CGXII medium with 4% (wt/vol) glucose in shake flasksa
| Strain | YX/S(g CDW/mmol glucose) | qS [g glucose/(g CDW × h)] |
YP/S (mol/mol glucose) |
|
|---|---|---|---|---|
| l-Alanine | l-Valine | |||
| C. glutamicum WT (pJC4 ilvBNCE) | 0.066 ± 0.012 | 0.80 ± 0.15 | 0.02 ± 0.01 | 0.11 ± 0.04 |
| C. glutamicum aceE L1 (pJC4 ilvBNCE) | 0.024 ± 0.004 | 1.00 ± 0.17 | 0.06 ± 0.01 | 0.21 ± 0.01 |
| C. glutamicum aceE A23 (pJC4 ilvBNCE) | 0.027 ± 0.003 | 0.91 ± 0.10 | 0.07 ± 0.01 | 0.25 ± 0.01 |
| C. glutamicum aceE A25 (pJC4 ilvBNCE) | 0.040 ± 0.003 | 0.80 ± 0.06 | 0.04 ± 0.01 | 0.35 ± 0.02 |
| C. glutamicum aceE A16 (pJC4 ilvBNCE) | 0.042 ± 0.002 | 0.77 ± 0.04 | 0.02 ± 0.01 | 0.37 ± 0.01 |
| C. glutamicum aceE A16 Δpqo (pJC4 ilvBNCE) | 0.042 ± 0.003 | 0.72 ± 0.05 | 0.02 ± 0.01 | 0.40 ± 0.03 |
| C. glutamicum aceE A16 Δpqo Δppc (pJC4 ilvBNCE) | 0.039 ± 0.005 | 0.80 ± 0.11 | 0.02 ± 0.01 | 0.42 ± 0.02 |
Values represent the arithmetic mean ± standard deviation from at least three independent experiments.
Inactivation of the pyruvate:quinone oxidoreductase and phosphoenolpyruvate carboxylase further improves l-valine production.
Among the engineered C. glutamicum l-valine producers, C. glutamicum aceE A16 (pJC4 ilvBNCE) showed the best performance, i.e., the highest YX/S and YP/S combined with the lowest substrate-specific l-alanine yield (Table 2). In shake flask experiments, this strain consumed 245 mM glucose within 30 h and produced about 90 mM l-valine after 48 h (Fig. 3). Thus, C. glutamicum aceE A16 (pJC4 ilvBNCE) formed the optimal basis for further improvement, and therefore we stepwise deleted the pqo and ppc genes to further increase pyruvate availability. Both modifications did not significantly alter the YX/S, qS, or substrate-specific l-alanine yield, but they cumulatively led to about a 14% increased YP/S, from 0.37 to 0.42 mol l-valine per mol of glucose (Table 2).
Fig 3.
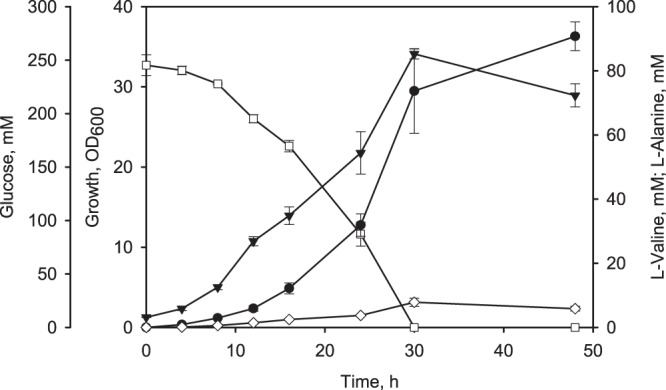
Growth, glucose consumption, and l-alanine and l-valine formation by C. glutamicum aceE A16 (pJC4 ilvBNCE) cultivated in shake flasks with CGXII medium containing 222 mM glucose. ▼, OD600; □, glucose; ♢, l-alanine; ●, l-valine. Three independent fermentations were performed. Error bars show standard deviations.
Fed-batch fermentations with C. glutamicum aceE A16 Δpqo Δppc (pJC4 ilvBNCE).
In order to test the suitability of C. glutamicum aceE A16 Δpqo Δppc (pJC4 ilvBNCE) for an improved l-valine production process, we carried out fed-batch fermentations in minimal medium initially containing about 333 mM glucose (Fig. 4). Under these conditions, C. glutamicum aceE A16 Δpqo Δppc (pJC4 ilvBNCE) grew exponentially with a growth rate of 0.15 h−1 to an OD600 of about 158 (i.e., 47.4 g CDW/liter) and produced about 378 mM l-valine (i.e., 44.3 g/liter) within 33.5 h. Phosphate in the medium then became limiting; however, despite the transition to the stationary phase, the cells continued to metabolize glucose and to produce l-valine (Fig. 4). Until the end of the fermentation after 55 h, C. glutamicum aceE A16 Δpqo Δppc (pJC4 ilvBNCE) produced about 41 mM l-alanine and up to 738 mM l-valine (86.5 g/liter), with a YP/S of 0.36 mol l-valine per mol of glucose and a volumetric productivity of about 13.6 mM per h [1.6 g/(l × h)]. Taken together, the results show that C. glutamicum aceE A16 Δpqo Δppc (pJC4 ilvBNCE) with reduced PDHC activity represents an excellent production platform. In contrast to the previously developed producer strains, C. glutamicum aceE A16 Δpqo Δppc (pJC4 ilvBNCE) did not require any additional carbon source (such as acetate or ethanol) for growth (see the introduction).
Fig 4.
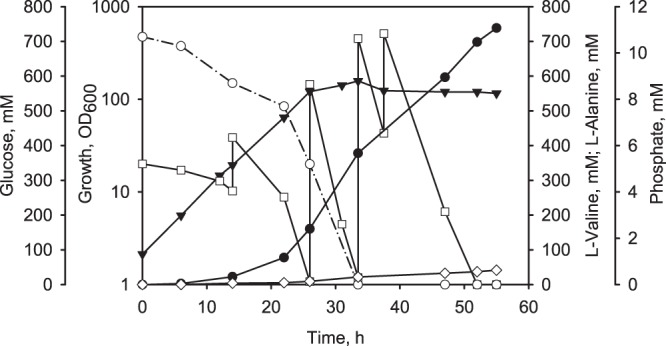
Representative fed-batch fermentation of C. glutamicum aceE A16 Δpqo Δppc (pJC4 ilvBNCE) in CGXII medium initially containing about 333 mM glucose. ▼, OD600; □, glucose; ♢, l-alanine; ●, l-valine; ○, phosphate.
Inactivation of transaminase B and overexpression of ilvBNCD in C. glutamicum aceE A16 Δpqo Δppc results in efficient 2-ketoisovalerate production.
Recently, we demonstrated the ability of PDHC-deficient C. glutamicum strains to produce 2-ketoisovalerate from glucose when acetate was exhausted from the culture broth (11). To investigate the applicability of the novel production platform for 2-ketoisovalerate production, we inactivated the transaminase B by deletion of the ilvE gene in C. glutamicum aceE A16 Δpqo Δppc and transformed the resulting strain with plasmid pJC4 ilvBNCD. With the final strain, C. glutamicum aceE A16 Δpqo Δppc ΔilvE (pJC4 ilvBNCD), we carried out fed-batch fermentations in minimal medium initially containing 333 mM glucose, 1% (wt/vol) yeast extract, and 10 mM l-valine, l-isoleucine, and l-leucine. In contrast to the corresponding (transaminase B-positive) l-valine producer, C. glutamicum aceE A16 Δpqo Δppc ΔilvE (pJC4 ilvBNCD) showed no exponential growth but showed a steadily decreasing growth rate. After 29 h and at an OD600 of about 84 (25.2 g CDW/liter) (Fig. 5) growth stopped, although neither phosphate nor l-valine, l-isoleucine, or l-leucine became limiting in the culture broth (not shown). In the growth phase, C. glutamicum aceE A16 Δpqo Δppc ΔilvE (pJC4 ilvBNCD) produced about 228 mM (26 g/liter) 2-ketoisovalerate. After the growth arrest, the strain continued to consume glucose and further excreted 2-ketoisovalerate into the medium. At the end of the fermentation after 44 h, the strain reached an OD600 of about 88 (26.4 g CDW/liter) and produced about 30 mM pyruvate, 33 mM l-valine, and up to 303 mM (35 g/liter) 2-ketoisovalerate (Fig. 5), with a YK/S of 0.24 mol per mol of glucose and a volumetric productivity of about 6.9 mM per h [0.8 g (l × h)].
Fig 5.
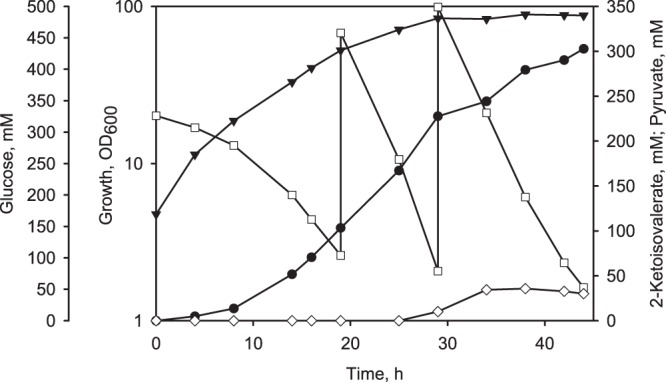
Representative fed-batch fermentation of C. glutamicum aceE A16 Δpqo Δppc ΔilvE (pJC4 ilvBNCD) in CGXII medium initially containing about 333 mM glucose, 1% (wt/vol) yeast extract, and 10 mM l-valine, l-isoleucine, and l-leucine. ▼, OD600; □, glucose; ♢, pyruvate; ●, 2-ketoisovalerate.
Reducing PDHC activity improves l-lysine production.
Since pyruvate is also the precursor for l-lysine synthesis (see Fig. 1), we analyzed the effect of reduced PDHC activity on l-lysine production with C. glutamicum. For this purpose, we replaced the native aceE promoter in the defined l-lysine producer C. glutamicum DM1800 by the dapA-A16 promoter. With the resulting strain, C. glutamicum DM1800 aceE A16, we carried out shake flask cultivations in minimal medium with 222 mM glucose and 0.5% (wt/vol) BHI and analyzed growth and l-lysine production. In comparison to the parental strain C. glutamicum DM1800, the newly constructed C. glutamicum DM1800 aceE A16 showed a 34% reduced YX/S of about 0.048 g CDW per mmol glucose; however, it produced two times more l-lysine and showed a 34% increased biomass-specific production rate (qP) (Table 3). We also introduced the dapA-A16 promoter in C. glutamicum DM1933, which produces about 60% more l-lysine than strain DM1800, and carried out batch fermentations in aerated and stirred bioreactors. Under these conditions, C. glutamicum DM1933 aceE A16 reached a YX/S of about 0.043 g CDW per mmol glucose, secreted the by-products l-valine (5 mM) and l-alanine (3 mM), and showed a YP/S of 0.23 mol l-lysine per mol of glucose and a qP of 0.138 g l-lysine per g CDW and h (Table 3), which are about 44% and 77% higher than the values for the parental C. glutamicum DM1933.
Table 3.
Substrate-specific biomass yield (YX/S), final titer, substrate-specific l-lysine yield (YP/S), and biomass-specific production rate (qP) of C. glutamicum l-lysine producers cultivated in CGXII medium with 4% (wt/vol) glucose and 0.5% (wt/vol) BHI in shake flasks (DM1800) or in bioreactors (DM1933)a
| Strain | YX/S (g CDW/mmol glucose) | Titer (mM l-lysine) | YP/S (mol l-lysine/mol glucose) | qP [g l-lysine/(g CDW × h)] |
|---|---|---|---|---|
| C. glutamicum DM1800 | 0.072 ± 0.001 | 22 ± 2 | 0.10 ± 0.01 | 0.068 ± 0.007 |
| C. glutamicum DM1800 aceE A16 | 0.048 ± 0.001 | 45 ± 2 | 0.20 ± 0.02 | 0.091 ± 0.009 |
| C. glutamicum DM1933 | 0.069 ± 0.004 | 35 ± 3 | 0.16 ± 0.03 | 0.078 ± 0.016 |
| C. glutamicum DM1933 aceE A16 | 0.043 ± 0.006 | 52 ± 6 | 0.23 ± 0.04 | 0.138 ± 0.028 |
Values represent the arithmetic mean ± standard deviation from three independent experiments.
Platform comparison: PDHC deficiency versus reduction of PDHC activity.
Table 4 gives an overview of the production characteristics of l-valine and 2-ketoisovalerate producers based on PDHC deficiency or on PDHC with reduced activity. The genetic backgrounds of the respective strains are not completely identical; however, as shown above, the additional deletion of the ppc gene improved the YP/S of l-valine production with C. glutamicum aceE A16 Δpqo Δppc (pJC4 ilvBNCE) by only 5% compared to that with the parental strain with an active phosphoenolpyruvate carboxylase. With the given improvements taken into account, the contribution of the ppc deletion is low, and therefore, a comparison of the respective producers is acceptable.
Table 4.
Final product concentrations (cP), substrate-specific product yields (YP/S), and volumetric productivities (QP) of fed-batch fermentations of C. glutamicum l-valine and 2-ketoisovalerate producers with either inactivated PDHC or PDHC with reduced activitya
| Condition and strain | cP (g/liter) | YP/S (mol C/mol C) | QP [g/(liter × h)] |
|---|---|---|---|
| l-Valine overproduction | |||
| C. glutamicum ΔaceE Δpqo (pJC4 ilvBNCE)b | 24.6 ± 2.6 | 0.23 ± 0.02 | 0.5 ± 0.1 |
| C. glutamicum aceE A16 Δpqo Δppc (pJC4 ilvBNCE) | 83.6 ± 2.6 | 0.33 ± 0.01 | 1.5 ± 0.1 |
| 2-Ketoisovalerate overproduction | |||
| C. glutamicum ΔaceE Δpqo ΔilvE (pJC4 ilvBNCD)c | 21.8 ± 3.2 | 0.26 ± 0.02 | 0.5 ± 0.1 |
| C. glutamicum aceE A16 Δpqo Δppc ΔilvE (pJC4 ilvBNCD) | 33.7 ± 1.0 | 0.21 ± 0.04 | 0.7 ± 0.1 |
For l-valine production, the reduction of the PDHC activity in C. glutamicum in combination with the deletion of the pqo and ppc genes and overexpression of the ilvBNCE genes led to growth-coupled l-valine production of 83.6 g/liter in fed-batch fermentations. Compared to the value for C. glutamicum ΔaceE Δpqo (pJC4 ilvBNCE) (27), this titer is 3.4 times higher. Furthermore, C. glutamicum aceE A16 Δpqo Δppc (pJC4 ilvBNCE) showed a 43% higher overall YP/S and a 3-times-higher volumetric productivity (QP) (Table 4).
For 2-ketoisovalerate, the effect is not as drastic as for l-valine production. Compared to the YP/S of the PDHC-deficient producer strain, that of C. glutamicum aceE A16 Δpqo Δppc ΔilvE (pJC4 ilvBNCD) was reduced by about 19%; however, the final titer and the QP were improved by 55% and 40%, respectively (Table 4).
Taken together, our results demonstrate that the novel engineered C. glutamicum platform with reduced PDHC activity has characteristics which are superior compared to its PDHC-deficient counterpart.
DISCUSSION
Several recent studies with C. glutamicum aimed to improve the pyruvate availability for l-valine, l-lysine, l-alanine, 2-ketoisovalerate, or isobutanol production or to engineer C. glutamicum for the production of pyruvate itself. These approaches included (i) inactivating enzymes of d-pantothenate synthesis to limit CoA availability for the PDHC reaction (22, 30, 46), (ii) applying anaerobic conditions to abolish/reduce oxidative tricarboxylic acid (TCA) flux (47–49), (iii) using an ATPase-defective mutant leading to increased pyruvate availability (50, 51), and (iv) deleting the aceE gene, encoding the E1p subunit of the PDHC (11, 15, 16, 25–28). However, all these approaches either resulted in a requirement for d-pantothenate, ethanol, or acetate, did not allow an adequate adjustment of the PDHC activity and the carbon flux into the TCA cycle, and/or required a cleverly devised redox state of the cell (under anaerobic conditions). In this study, we applied promoter engineering to develop C. glutamicum strains with gradually decreased PDHC activity by exchange of the native aceE promoter by a series of mutated dapA promoters, allowing the screening of the most promising variant for further optimization by metabolic engineering. Since the PDHC is a multienzyme complex consisting of three subunits (AceE, AceF, and Lpd), which are arranged in a tightly defined stoichiometric composition (52, 53), it is difficult to predict the effects of tuning the promoter activity of one of the respective genes (aceE, aceF, or lpd) on the overall PDHC activity. Therefore, we randomly choose four dapA promoter variants (A16, A23, A25, and L1) (34) to replace the native aceE promoter and analyzed growth and overall PDHC activity. Shake flask cultivations of the different engineered strains (C. glutamicum aceE A16, A23, A25, and L1) revealed that with decreasing PDHC activity the growth rate coherently decreased but the final biomass concentration also did so (Fig. 2A and B), although glucose was still present in the medium (data not shown). The reduced biomass formation is surprising, but it might be attributed to metabolic inhibition since all strains excreted significant amounts of l-valine, l-alanine, and pyruvate (data not shown). However, overexpression of the ilvBNCE genes in these strains, leading to a drain of pyruvate toward l-valine, did not improve the final biomass concentration (data not shown). Moreover, compared to the wild type, C. glutamicum aceE A23 and L1 (pJC4 ilvBNCE) showed 14% and 25% increased qS (Table 2), indicating energy limitation, which might have led to the observed growth.
For the promoter variants dapA-A16, -A23, and -A25, we found a linear correlation of the overall PDHC and the respective promoter activity (data not shown). However, the dapA-A16 promoter showed the highest activity (34), but interestingly, this promoter was unable to completely compensate for the activity of the native aceE promoter, since C. glutamicum aceE A16 showed 79% lower PDHC activity and a 31% decreased growth rate compared to those of C. glutamicum WT (Fig. 2). It is noteworthy that the −10 region (TATCCT) of the native aceE promoter corresponds to that of the dapA-A14 promoter, which shows lower activity than the dapA-A16 promoter (34). As a consequence, other enhancing elements such as the extended −10 region or the action of transcriptional regulators such as RamB (54) give the aceE promoter its strength. Since C. glutamicum aceE A16 showed the highest YP/S and YX/S, it might be anticipated that other promoters, stronger than the dapA-A16 promoter but weaker than the native aceE promoter, might be even more suitable for production purposes. Alternatively to or in combination with our approach, modulation of translation initiation by altering the sequence length between the ribosome binding site (RBS) and the translational start codon (TSC), using different TSCs, or deleting the RBS as recently done to improve putrescine production with C. glutamicum (9) as well as self-cloning (55) or deletion of repressor/activator binding sites (for, e.g., RamB) might be promising strategies to adjust expression of the aceE gene more exactly. However, taking into account the numbers for current aerobic production processes for l-valine with E. coli (61 g/liter) (56) or C. glutamicum (48 g/liter) (27) and for 2-ketoisovalerate with C. glutamicum (22 g/liter) (11), the novel engineered C. glutamicum platform with reduced PDHC activity (84 g/liter l-valine and 34 g/liter 2-ketoisovalerate) (Table 4) is highly competitive or even superior.
In contrast to the l-valine producer (Fig. 4), the 2-ketoisovalerate producer C. glutamicum aceE A16 Δpqo Δppc ΔilvE (pJC4 ilvBNCD) showed no constant growth but a steadily decreasing growth rate (Fig. 5). This phenomenon might be attributed to an inhibiting effect of the product, since neither phosphate nor one of the supplemented amino acids (l-valine, l-isoleucine, or l-leucine) became limiting (data not shown). However, the decreasing growth rate of the 2-ketoisovalerate producer shows the general advantage of the PDHC-deficient platform (production does not start before acetate is depleted and growth stops) for the production of cytotoxic products. In any case, despite the continuously decreasing growth rate, C. glutamicum aceE A16 Δpqo Δppc ΔilvE (pJC4 ilvBNCD) reached a significantly higher 2-ketoisovalerate titer than the PDHC-deficient counterpart (Table 4). For the production of more toxic/growth-inhibiting products, the situation might be different, and a growth-decoupled production process will then be advantageous.
C. glutamicum is the workhorse for industrial l-lysine production, and several improvements have been made by metabolic engineering (reviewed in references 57, 58, and 59). Recently, we inactivated the PDHC in the defined l-lysine producer C. glutamicum DM1729, which led to an auxotrophy for acetate; however, it also led to 44% improved l-lysine production compared to that of the parental strain (28). Here we found that introduction of the dapA-A16 promoter in C. glutamicum DM1800, which shows an YP/S identical to that of DM1729, led to prototrophic growth and increased the YP/S by 100%, indicating that reduction of PDHC activity might be more useful to optimize l-lysine production with C. glutamicum than a complete shutdown of the PDHC. The reason for reduced improvement of the PDHC-deficient counterpart might be reduced expression of relevant genes under nongrowing conditions, limiting efficient l-lysine production.
Van Ooyen et al. (60) also made use of the dapA promoter library from Vasicová et al. (34) to adjust citrate synthase (encoded by gltA) flux for optimized l-lysine production with C. glutamicum. The most promising variant, C. glutamicum DM1800 ΔprpC1 ΔprpC2 with gltA under the control of the C7-dapA promoter, improved l-lysine production by 82% compared to that in the parental strain (60), which is in the same range as the improvement by reduction of PDHC activity (see above). In any case, both examples demonstrate that reducing the TCA flux by optimizing pyruvate and oxaloacetate supply is highly beneficial for efficient l-lysine production with C. glutamicum. C. glutamicum DM1933 aceE A16 excreted l-alanine and l-valine in significant amounts in batch fermentations. l-Alanine formation is catalyzed mainly by the aminotransferase AlaT from pyruvate (61). This reaction is in equilibrium, and therefore formation of l-alanine and also l-valine is a good indicator for an increased intracellular pyruvate concentration, suggesting that l-lysine production in this strain is limited by the reactions from pyruvate to l-lysine and not by pyruvate availability. The PDHC-deficient l-lysine producer DM1729 ΔaceE also excreted l-alanine, l-valine, and pyruvate into the medium, and the additional overexpression of the ddh gene (encoding diaminopimelate dehydrogenase) reduced the formation of these by-products and increased the YP/S by 60% (28). Here, we found that introducing a second copy of the ddh gene and additionally of all l-lysine biosynthetic genes (lysCT311I, asd, dapA, dapB, ddh, lysA, and lysE) in C. glutamicum DM1800 aceE A16 resulted in only about a 15% improved YP/S (DM1800 aceE A16 versus DM1933 aceE A16), indicating that either ddh expression is still too low or other obstacles such as a sufficient NADPH availability have to be overcome in C. glutamicum DM1933 aceE A16. However, the surplus of the precursor pyruvate in C. glutamicum DM1933 aceE A16 opens the possibility for further optimization of l-lysine production by metabolic engineering.
ACKNOWLEDGMENTS
Plasmids pJC4 ilvBNCD and pJC4 ilvBNCE were kindly provided by Lothar Eggeling (Research Center Jülich). We thank Andreas Freund, Salaheddine Laghrami, Mira-Lenfers Lücker, Ulrike Hillemann, and Yasmin Kalmbach for technical assistance.
The support of the Fachagentur Nachwachsende Rohstoffe (FNR) of the BMELV (FNR grant 220-095-08A; BioProChemBB project in the frame of the ERA-IB program) is gratefully acknowledged.
Footnotes
Published ahead of print 8 July 2013
REFERENCES
- 1.Leuchtenberger W, Huthmacher K, Drauz K. 2005. Biotechnological production of amino acids and derivates: current status and prospects. Appl. Microbiol. Biotechnol. 69:1–8 [DOI] [PubMed] [Google Scholar]
- 2.Liebl W. 1991. The genus Corynebacterium—nonmedical, p 1157–1171 In Balows A, Trüper HG, Dworkin M, Harder W, Schleifer KH. (ed), The prokaryotes, vol 2 Springer, New York, NY [Google Scholar]
- 3.Nishimura T, Vertès AA, Shinoda Y, Inui M, Yukawa H. 2007. Anaerobic growth of Corynebacterium glutamicum using nitrate as a terminal electron acceptor. Appl. Microbiol. Biotechnol. 75:889–897 [DOI] [PubMed] [Google Scholar]
- 4.Takors R, Bathe B, Rieping M, Hans S, Kelle R, Huthmacher K. 2007. Systems biology for industrial strains and fermentation processes—example: amino acids. J. Biotechnol. 129:181–190 [DOI] [PubMed] [Google Scholar]
- 5.Kind S, Jeong WK, Schröder H, Wittmann C. 2010. Sytems-wide metabolic pathway engineering in Corynebacterium glutamicum for bio-based production of diaminopentane. Metab. Eng. 12:341–351 [DOI] [PubMed] [Google Scholar]
- 6.Kind S, Jeong WK, Schröder H, Zelder O, Wittmann C. 2010. Identification and elimination of the competing N-acetyldiaminopentane pathway for improved production of diaminopentane by Corynebacterium glutamicum. Appl. Environ. Microbiol. 76:5175–5180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mimitsuka T, Sawai H, Hatsu M, Yamada K. 2007. Metabolic engineering of Corynebacterium glutamicum for cadaverine fermentation. Biosci. Biotechnol. Biochem. 71:2130–2135 [DOI] [PubMed] [Google Scholar]
- 8.Schneider J, Wendisch VF. 2010. Putrescine production by engineered Corynebacterium glutamicum. Appl. Microbiol. Biotechnol. 88:859–868 [DOI] [PubMed] [Google Scholar]
- 9.Schneider J, Eberhardt D, Wendisch VF. 2012. Improving putrescine production by Corynebacterium glutamicum by fine-tuning ornithine transcarbamoylase activity using a plasmid addiction system. Appl. Microbiol. Biotechnol. 95:169–178 [DOI] [PubMed] [Google Scholar]
- 10.Tateno T, Okada Y, Tsuchidate T, Tanaka T, Fukuda H, Kondo A. 2009. Direct production of cadaverine from soluble starch using Corynebacterium glutamicum coexpressing alpha-amylase and lysine decarboxylase. Appl. Microbiol. Biotechnol. 82:115–121 [DOI] [PubMed] [Google Scholar]
- 11.Krause FS, Blombach B, Eikmanns BJ. 2010. Metabolic engineering of Corynebacterium glutamicum for 2-ketoisovalerate production. Appl. Environ. Microbiol. 76:8053–8061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Okino S, Inui M, Yukawa H. 2005. Production of organic acids by Corynebacterium glutamicum under oxygen deprivation. Appl. Microbiol. Biotechnol. 68:475–480 [DOI] [PubMed] [Google Scholar]
- 13.Okino S, Noburyu R, Suda M, Jojima T, Inui M, Yukawa H. 2008. An efficient succinic acid production process in a metabolically engineered Corynebacterium glutamicum strain. Appl. Microbiol. Biotechnol. 81:459–464 [DOI] [PubMed] [Google Scholar]
- 14.Okino S, Suda M, Fujikura K, Inui M, Yukawa H. 2008. Production of d-lactic acid by Corynebacterium glutamicum under oxygen deprivation. Appl. Microbiol. Biotechnol. 78:449–454 [DOI] [PubMed] [Google Scholar]
- 15.Wieschalka S, Blombach B, Eikmanns BJ. 2012. Engineering Corynebacterium glutamicum for the production of pyruvate. Appl. Microbiol. Biotechnol. 94:449–459 [DOI] [PubMed] [Google Scholar]
- 16.Blombach B, Riester T, Wieschalka S, Ziert C, Youn J-W, Wendisch VF, Eikmanns BJ. 2011. Corynebacterium glutamicum tailored for efficient isobutanol production Appl. Environ. Microbiol. 77:3300–3310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Inui M, Kawaguchi H, Murakami S, Vertès AA, Yukawa H. 2004. Metabolic engineering of Corynebacterium glutamicum for fuel ethanol production under oxygen-deprivation conditions. J. Mol. Microbiol. Biotechnol. 8:243–254 [DOI] [PubMed] [Google Scholar]
- 18.Smith K, Cho K, Liao JC. 2010. Engineering Corynebacterium glutamicum for isobutanol production. Appl. Microbiol. Biotechnol. 87:1045–1055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sasaki M, Jojima T, Inui M, Yukawa H. 2010. Xylitol production by recombinant Corynebacterium glutamicum under oxygen deprivation. Appl. Microbiol. Biotechnol. 86:1057–1066 [DOI] [PubMed] [Google Scholar]
- 20.An SJ, Yim SS, Jeong KJ. 2013. Development of a secretion system for the production of heterologous proteins in Corynebacterium glutamicum using the porin B signal peptide. Protein Expr. Purif. 89:251–257 [DOI] [PubMed] [Google Scholar]
- 21.Scheele S, Oertel D, Bongaerts J, Evers S, Hellmuth H, Maurer KH, Bott M, Freudl R. 2013. Secretory production of an FAD cofactor-containing cytosolic enzyme (sorbitol-xylitol oxidase from Streptomyces coelicolor) using the twin-arginine translocation (Tat) pathway of Corynebacterium glutamicum. Microb. Biotechnol. 6:202–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Radmacher E, Vaitsiková A, Burger U, Krumbach K, Sahm H, Eggeling L. 2002. Linking central metabolism with increased pathway flux: l-valine accumulation by Corynebacterium glutamicum. Appl. Environ. Microbiol. 68:2246–2250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bartek T, Makus P, Klein B, Lang S, Oldiges M. 2008. Influence of l-isoleucine and pantothenate auxotrophy for l-valine formation in Corynebacterium glutamicum revisited by metabolome analyses. Bioprocess Biosyst. Eng. 31:217–225 [DOI] [PubMed] [Google Scholar]
- 24.Schreiner ME, Fiur D, Holátko J, Pátek M, Eikmanns BJ. 2005. E1 enzyme of the pyruvate dehydrogenase complex in Corynebacterium glutamicum: molecular analysis of the gene and phylogenetic spects. J. Bacteriol. 187:6005–6018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Blombach B, Arndt A, Auchter M, Eikmanns BJ. 2009. l-Valine production during growth of pyruvate dehydrogenase complex-deficient Corynebacterium glutamicum in the presence of ethanol or by inactivation of the transcriptional regulator SugR. Appl. Environ. Microbiol. 75:1197–1200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Blombach B, Schreiner ME, Holatko J, Bartek T, Oldiges M, Eikmanns BJ. 2007. l-Valine production with pyruvate dehydrogenase complex-deficient Corynebacterium glutamicum. Appl. Environm. Microbiol. 73:2079–2084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Blombach B, Schreiner ME, Bartek T, Oldiges M, Eikmanns BJ. 2008. Corynebacterium glutamicum tailored for high-yield l-valine production. Appl. Microbiol. Biotechnol. 79:471–479 [DOI] [PubMed] [Google Scholar]
- 28.Blombach B, Schreiner ME, Moch M, Oldiges M, Eikmanns BJ. 2007. Effect of pyruvate dehydrogenase complex deficiency on l-lysine production with Corynebacterium glutamicum. Appl. Microbiol. Biotechnol. 76:615–623 [DOI] [PubMed] [Google Scholar]
- 29.Krause F, Henrich A, Blombach B, Krämer R, Eikmanns BJ, Seibold GM. 2010. Increased glucose utilization in Corynebacterium glutamicum by use of maltose, and its application for the improvement of l-valine productivity. Appl. Environ. Microbiol. 76:370–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Holátko J, Elišáková V, Prouza M, Sobotka M, Nešvera J, Pátek M. 2009. Metabolic engineering of the l-valine biosynthesis pathway in Corynebacterium glutamicum using promoter activity modulation. J. Biotechnol. 139:203–210 [DOI] [PubMed] [Google Scholar]
- 31.Becker J, Klopprogge C, Zelder O, Heinzle E, Wittmann C. 2005. Amplified expression of fructose 1,6-bisphosphatase in Corynebacterium glutamicum increases in vivo flux through the pentose phosphate pathway and lysine production on different carbon sources. Appl. Environ. Microbiol. 71:8587–8596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Becker J, Klopprogge C, Herold A, Zelder O, Bolten CJ, Wittmann C. 2007. Metabolic flux engineering of l-lysine production in Corynebacterium glutamicum—over expression and modification of G6P dehydrogenase. J. Biotechnol. 132:99–109 [DOI] [PubMed] [Google Scholar]
- 33.Becker J, Zelder O, Häfner S, Schröder H, Wittmann C. 2011. From zero to hero—design-based systems metabolic engineering of Corynebacterium glutamicum for l-lysine production. Metab. Eng. 13:159–168 [DOI] [PubMed] [Google Scholar]
- 34.Vasicová P, Pátek M, Sahm H, Eikmanns BJ. 1999. Analysis of the Corynebacterium glutamicum dapA promoter. J. Bacteriol. 181:6188–6191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Eikmanns BJ, Thum-Schmitz N, Eggeling L, Ludtke KU, Sahm H. 1994. Nucleotide sequence, expression and transcriptional analysis of the Corynebacterium glutamicum gltA gene encoding citrate synthase. Microbiology 140:1817–1828 [DOI] [PubMed] [Google Scholar]
- 36.van der Rest ME, Lange C, Molenaar D. 1999. A heat shock following electroporation induces highly efficient transformation of Corynebacterium glutamicum with xenogenic plasmid DNA. Appl. Microbiol. Biotechnol. 52:541–545 [DOI] [PubMed] [Google Scholar]
- 37.Dower WJ, Miller JF, Ragsdale CW. 1988. High efficiency transformation of E. coli by high voltage electroporation. Nucleic Acids Res. 16:6127–6145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sambrook J, Russel DW, Irwin N, Janssen UA. 2001. Molecular cloning: a laboratory manual, 3rd ed. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY [Google Scholar]
- 39.Eikmanns BJ, Metzger M, Reinscheid D, Kircher M, Sahm H. 1991. Amplification of three threonine biosynthesis genes in Corynebacterium glutamicum and its influence on carbon flux in different strains. Appl. Microbiol. Biotechnol. 34:617–622 [DOI] [PubMed] [Google Scholar]
- 40.Schäfer A, Tauch A, WJäger Kalinowski J, Thierbach G, Pühler A. 1994. Small mobilizable multi-purpose cloning vectors derived from the E. coli plasmids pK18 and pK19: selection of defined deletions in the chromosome of Corynebacterium glutamicum. Gene 145:69–73 [DOI] [PubMed] [Google Scholar]
- 41.Schreiner ME, Riedel C, Holatko J, Patek M, Eikmanns BJ. 2006. Pyruvate:quinone oxidoreductase in Corynebacterium glutamicum: molecular analysis of the pqo gene, significance of the enzyme, and phylogenetic aspects. J. Bacteriol. 188:1341–1350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Guest JR, Creaghan IT. 1974. Further studies with lipoamide dehydrogenase mutants of Escherichia coli K12. J. Gen. Microbiol. 81:237–245 [DOI] [PubMed] [Google Scholar]
- 43.Lamprecht W, Heinz F. 1983. Pyruvate, p 570–577 In Bergmeyer HU. (ed), Methods of enzymatic analysis, 3rd ed. Verlag Chemie, Weinheim, Germany [Google Scholar]
- 44.Lindroth P, Mopper K. 1979. High performance liquid chromatographic determination of subpicomole amounts of amino acids by precolumn fluorescence derivatization with o-phthalaldehyde. Anal. Chem. 51:1667–1674 [Google Scholar]
- 45.Henderson JW, Brooks A. 2010. Improved amino acid methods using Agilent ZORBAX Eclipse Plus C18 columns for a variety of Agilent LC instrumentation and separation goals. Agilent Technologies, Wilmington, DE: http://www.chem.agilent.com/Library/applications/5990-4547EN.pdf [Google Scholar]
- 46.Elišáková V, Pátek M, Holátko J, Nešvera J, Leyval D, Goergen JL, Delaunay S. 2005. Feedback-resistant acetohydroxy acid synthase increases valine production in Corynebacterium glutamicum. Appl. Environ. Microbiol. 71:207–213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hasegawa S, Uematsu K, Natsuma Y, Suda M, Hiraga K, Jojima T, Inui M, Yukawa H. 2012. Improvement of the redox balance increases l-valine production by Corynebacterium glutamicum under oxygen deprivation conditions. Appl. Environ. Microbiol. 78:865–875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hasegawa S, Suda M, Uematsu K, Natsuma Y, Hiraga K, Jojima T, Inui M, Yukawa H. 2013. Engineering Corynebacterium glutamicum for high-yield l-valine production under oxygen deprivation conditions. Appl. Environ. Microbiol. 79:1250–1257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yamamoto S, Gunji W, Suzuki H, Toda H, Suda M, Jojima T, Inui M, Yukawa H. 2012. Overexpression of genes encoding glycolytic enzymes in Corynebacterium glutamicum enhances glucose metabolism and alanine production under oxygen deprivation conditions. Appl. Environ. Microbiol. 78:4447–4457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Aoki R, Wada M, Takesue N, Tanaka K, Yokota A. 2005. Enhanced glutamic acid production by a H+-ATPase-defective mutant of Corynebacterium glutamicum. Biosci. Biotechnol. Biochem. 69:1466–1472 [DOI] [PubMed] [Google Scholar]
- 51.Wada M, Hijikata N, Aoki R, Takesue N, Yokota A. 2008. Enhanced valine production in Corynebacterium glutamicum with defective H+-ATPase and C-terminal truncated acetohydroxyacid synthase. Biosci. Biotechnol. Biochem. 72:2959–2965 [DOI] [PubMed] [Google Scholar]
- 52.de Kok A, Gengeveld AF, Martin A, Westphal AH. 1998. The pyruvate dehydrogenase multi-enzyme complex from Gram-negative bacteria. Biochim. Biophys. Acta 1385:353–366 [DOI] [PubMed] [Google Scholar]
- 53.Neveling U, Bringer-Meyer S, Sahm H. 1998. Gene and subunit organization of bacterial pyruvate-dehydrogenase complexes. Biochim. Biophys. Acta 1385:367–372 [DOI] [PubMed] [Google Scholar]
- 54.Blombach B, Cramer A, Eikmanns BJ, Schreiner M. 2009. RamB is an activator of the pyruvate dehydrogenase complex subunit E1p gene in Corynebacterium glutamicum. J. Mol. Microbiol. Biotechnol. 16:236–239 [DOI] [PubMed] [Google Scholar]
- 55.Kirchner O, Tauch A. 2003. Tools for genetic engineering in the amino acid-producing bacterium Corynebacterium glutamicum. J. Biotechnol. 104:287–299 [DOI] [PubMed] [Google Scholar]
- 56.Park JH, Jang J-S, Lee JW, Lee SY. 2011. Escherichia coli W as a new platform strain for the enhanced production of l-valine by systems metabolic engineering. Biotechnol. Bioeng. 108:934–946 [DOI] [PubMed] [Google Scholar]
- 57.Blombach B, Seibold GM. 2010. Carbohydrate metabolism in Corynebacterium glutamicum and applications for the metabolic engineering of l-lysine production strains. Appl. Microbiol. Biotechnol. 86:1313–1322 [DOI] [PubMed] [Google Scholar]
- 58.Kelle R, Hermann T, Bathe B. 2005. l-Lysine production, p 465–488 In Eggeling L, Bott M. (ed), Handbook of Corynebacterium glutamicum. CRC Press, Boca Raton, FL [Google Scholar]
- 59.Park JH, Lee SY. 2010Metabolic pathways and fermentative production of l-aspartate family amino acids. Biotechnol. J. 5:560–577 [DOI] [PubMed] [Google Scholar]
- 60.van Ooyen J, Noack S, Bott M, Reth A, Eggeling L. 2012. Improved l-lysine production with Corynebacterium glutamicum and systemic insight into citrate synthase flux and activity. Biotechnol. Bioeng. 109:2070–2081 [DOI] [PubMed] [Google Scholar]
- 61.Marienhagen J, Kennerknecht N, Sahm H, Eggeling L. 2005. Functional analysis of all aminotransferase proteins inferred from the genome sequence of Corynebacterium glutamicum. J. Bacteriol. 187:7639–7646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hanahan D. 1983. Studies on transformation of Escherichia coli with plasmids. J. Mol. Biol. 166:557–580 [DOI] [PubMed] [Google Scholar]
- 63.Georgi T, Rittmann D, Wendisch VF. 2005. Lysine and glutamate production by Corynebacterium glutamicum on glucose, fructose and sucrose: roles of malic enzyme and fructose-1,6-bisphosphatase. Metab. Eng. 7:291–301 [DOI] [PubMed] [Google Scholar]
- 64.Blombach B, Hans S, Bathe B, Eikmanns BJ. 2009. Acetohydroxyacid synthase, a novel target for improvement of l-lysine production by Corynebacterium glutamicum. Appl. Environ. Microbiol. 75:411–427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sahm H, Eggeling L. 1999. d-Pantothenate synthsis in Corynebacterium glutamicum and use of panBC and genes encoding l-valine synthsis for d-pantothenate overproduction. Appl. Environ. Microbiol. 65:1973–1979 [DOI] [PMC free article] [PubMed] [Google Scholar]