

Abstract
Our understanding of the sources of Mycobacterium avium infection is partially based on genotypic matching of pathogen isolates from cases and environmental sources. These approaches assume that genotypic identity is rare in isolates from unlinked cases or sources. To test this assumption, a high-resolution PCR-based genotyping approach, large-sequence polymorphism (LSP)-mycobacterial interspersed repetitive unit–variable-number tandem repeat (MIRU-VNTR), was selected and used to analyze clinical and environmental isolates of M. avium from geographically diverse sources. Among 127 clinical isolates from seven locations in North America, South America, and Europe, 42 genotypes were observed. Among 12 of these genotypes, matches were seen in isolates from apparently unlinked patients in two or more geographic locations. Six of the 12 were also observed in environmental isolates. A subset of these isolates was further analyzed by alternative strain genotyping methods, pulsed-field gel electrophoresis and MIRU-VNTR, which confirmed the existence of geographically dispersed strain genotypes. These results suggest that caution should be exercised in interpreting high-resolution genotypic matches as evidence for an acquisition event.
INTRODUCTION
Members of the Mycobacterium avium complex (MAC) live in natural soils and water sources, commercial soils and treated water, and other human-made niches (1, 2). At least some strains cause diseases in susceptible humans, including lymphadenitis in children, disseminated disease in severely immunocompromised individuals, and lung disease associated with several clinical profiles (3). MAC comprises two major species, M. avium and M. intracellulare, as well as minor species and members that are not classified into species (4–8). M. avium is further divided into subspecies, M. avium subsp. avium, M. avium subsp. silvaticum, M. avium subsp. paratuberculosis, and M. avium subsp. hominissuis (9). Even within a single group, genomes can vary markedly among strains (10).
High-resolution genotypic fingerprints are taken to be uniquely associated with specific strains of the MAC. Several methods have been used to generate high-resolution genetic fingerprints of the MAC. These include pulsed-field gel electrophoresis (PFGE) (11), restriction-fragment-length polymorphism (RFLP) analysis using insertion sequences (12, 13), repetitive-sequence-based PCR (rep-PCR) analysis (14, 15), randomly amplified polymorphic DNA (RAPD) analysis (6), mycobacterial interspersed repetitive unit–variable-number tandem repeat (MIRU-VNTR) analysis (16), amplified fragment length polymorphism (ALFP) (17), (CCG)4-based PCR analysis (18), and multispacer sequence typing (MST) (19). These methods vary in the quantity and purity of DNA required for analysis and their portability (the ease with which results can be compared across analyses and exchanged among laboratories). The hypothetical gold standard for validating genotypic fingerprinting methods is complete genome sequencing. In practice, however, methods are compared on the basis of their discriminatory index (DI), a quantity that reflects a method's probability of placing any two isolates into separate genotypic groups (20). Some validation is provided by the fact that multiple isolates from the same patient, either from separate anatomic sites or taken over time, often have the same fingerprint (11, 12, 17, 21, 22). When matching or highly similar fingerprints are found between patients (17, 21–23), in the same environmental source over time (22, 24), or between a patient and his environment (22, 23, 25–28), this is often interpreted as support for a link of some sort. These kinds of matches are interpreted to indicate a shared source of acquisition, continuous colonization, or probable source of acquisition, respectively.
Despite the utility of this approach (29), it is noteworthy that several studies using high-resolution genotyping methods have found matches or clusters between cases with no apparent shared sources (12, 15, 21, 30). Previous work in our laboratory used large-sequence polymorphism (LSP) analysis (also known as deligotyping) and rep-PCR to show that the M. avium genome sequence strain 104 was found in clinical isolate collections from southern California and northwestern Washington (15). An earlier study used rep-PCR and IS1245 RFLP to identify a group of clinical isolates from these same two locations that shared another genotype (14). More recently, an MIRU-VNTR analysis of 47 human isolates of M. avium found high phylogenetic proximity between isolates collected over the course of 2 decades from a clinical site in Italy (31). These observations suggest that some M. avium genotypes are stable or have wide geographic distributions.
In order to more rigorously assess the wide geographical distribution of some MAC strains, this study used high-throughput genotyping methods to characterize (179) geographically diverse clinical and environmental isolates of M. avium. Many widely used M. avium genotyping methods, including IS1245 RFLP and PFGE, have limited portability and require large quantities of DNA. Thus, we chose to evaluate PCR-based typing methods with greater portability and smaller DNA requirements, i.e., 3′ hsp65 sequencing, rep-PCR, MIRU-VNTR, and deligotyping, to find one with sufficient reproducibility and discriminatory power for use in the larger analysis. Two methods were then applied to larger sets of isolates for our geographic analysis. Subsets were further analyzed by PFGE and MIRU-VNTR.
MATERIALS AND METHODS
M. avium isolates.
Genotypic analyses were carried out using subsets of a large M. avium isolate and DNA collection assembled at Seattle Biomedical Research Institute (Seattle BioMed) and currently housed at the University of Washington (UW). Permanent cultures or genomic DNA of archived M. avium isolates were received from multiple collaborators. The isolates and DNA samples used in this study were all previously described (14, 15, 22–26, 28, 32–39) and are listed in Tables S1 to S3 in the supplemental material. Most isolates were identified to the species level. In total, 127 clinical M. avium isolates (each from a separate individual case) and 52 environmental M. avium isolates were examined.
Bacterial culture and preparation of genomic DNA.
For some isolates, genomic DNA was received from collaborators. Isolates for which permanent cultures were archived at Seattle BioMed were grown on Middlebrook 7H10 containing 10% oleic acid-albumin-dextrose-catalase (OADC; Fisher Scientific International, Hampton, NJ) at 37°C, and then DNA was extracted by one of three methods: by boiling, using the ArchivePure DNA cell/tissue kit (5 Prime, Gaithersburg, MD), or using the UltraClean microbial DNA isolation kit (MoBio Laboratories, Carlsbad, CA). The ArchivePure protocol was modified for mycobacteria as shown in File S4 in the supplemental material. The MoBio kit was employed using the recommendations for mycobacterial species from Bacterial Barcodes, and for some isolates the MoBio method was modified by adding a freeze-thaw step after suspending cells in bead solution but before vortexing, or as described previously (40). Extracted DNA ranged in concentration from 10 to 250 ng/μl and was not adjusted in the following analyses unless otherwise noted.
PCR and sequencing of hsp65.
A 1,059-bp portion of the variable 3′ end of the hsp65 gene was PCR amplified and sequenced. Amplification primers were MAChsp65F_574 (5′-CGGTTCGACAAGGGTTACAT-3′) and MAChsp65R (5′-ACTGACTCAGAAGTCCATG-3′), as reported in Turenne et al. (41). PCR mixtures consisted of 5 μg of genomic DNA, 1× PCR buffer with MgCl2, 0.2 mM deoxynucleoside triphosphates (dNTPs), 1 U of EconoTaq, (Lucigen, Middleton, WI), 1 M betaine, (Sigma, St. Louis, MO), 5 μM forward and reverse primer, and type 1 purified water (Barnstead Nanopure) to adjust the final volume to 25 μl. Cycling conditions were 95°C for 10 min, followed by 35 cycles of 95°C for 1 min, 55°C for 1 min, and 72°C for 90 s, and then a final 10-min extension at 72°C. Products were visualized by electrophoresis, purified with a QIAquick spin PCR purification kit (Qiagen, Valencia, CA), and submitted for sequencing using an Applied Biosystems 3730XL genetic analyzer. PCR primers and an additional primer (hsp65-1047R, 5′-GTAGTCGGAGTCGGTGTTCT-3′) were used for sequencing. hsp65 codes were assigned according to the nomenclature of Turenne et al. (41), and novel sequences were reported to GenBank.
MIRU-VNTR analysis.
MIRU-VNTR analysis was carried out using primer sequences from Thibault et al. (16). For loci 3, 7, 10, 25, 32, 47, and 292, PCR mixtures consisted of 2.5 μl genomic DNA extraction mix, 1× PCR buffer with MgCl2, 0.2 mM dNTPs, 1 U EconoTaq, 1 M betaine, 2.0 μM each primer, and nanopure water to adjust the final volume to 25 μl. Reaction mixtures for loci 3 and 32 also contained 1.0 μl of dimethyl sulfoxide. For locus X3, the volume of genomic DNA extraction mix used was reduced to 1 μl. Cycling conditions for loci 3, 7, 10, 25, 32, 47, and 292 were 10 min at 95°C, followed by 40 cycles of 95°C for 1 min, 58°C for 1 min, and 72°C for 30 s, and then a final 10 min at 72°C. Cycling conditions for reactions at locus X3 consisted of 5 min at 94°C, followed by 40 cycles of 94°C for 30 s, 58°C for 30 s, and 72°C for 30 s, and then a final 10 min at 72°C. Products were analyzed on 1.0 to 2.0% agarose gels stained with ethidium bromide.
Because four of the eight MIRU loci described by Thibault et al. (16) showed notably greater allelic diversity than the other four, we considered both eight-locus and four-locus MIRU (alone or in combination with other methods) as possible fingerprinting methods for this study. In particular, we evaluated the replacement of the four least-variable MIRU loci with four LSP loci that appeared to be more variable, thereby achieving greater resolution in a total of 8 PCRs. MIRU types were assigned to represent the number of repeats inferred to be present from the product length at that locus based on product sizes and repeat numbers from Thibault et al. (16) and personal correspondence with V. Thibault and F. Biet.
LSP analysis.
LSP analysis was carried out at four genomic loci, HSDR, LSP2 (also known as DA2), LSP7 (also known as DA7), and LSPP5 (also known as DEL11), using published primer sequences (15). For most isolates, LSP analysis was carried out on 1 μl of genomic DNA using the GC-rich PCR system (Roche Diagnostics, Basel, Switzerland) as described previously (15). For a small number of isolates, EconoTaq was used with the following reaction mixture: 1 μl genomic DNA, 1× PCR buffer with MgCl2, 0.2 mM dNTPs, 1 U EconoTaq, 1 M betaine, 0.6 μM each primer, and nanopure water to adjust the final volume to 25 μl. Visualization and nomenclature techniques employed have been previously described (15).
LSP-MVR analysis.
An eight-digit genotype combining information from LSP and MIRU-VNTR analyses was assigned. The first four digits represent results for the four LSP loci, and the last four digits represent results for the MIRU-VNTR loci that showed the highest allelic diversity in Thibault et al.: X3, 25, 32, and 47. We refer to this combined approach as LSP-MVR analysis.
Rep-PCR.
Rep-PCR was carried out using the Bacterial Barcodes mycobacterial kit (Athens, GA) as described previously (14, 15). Results for isolates analyzed in separate PCRs and on separate chips were combined into a single report using the Diversilab software, and letter type designations were assigned to branches of the dendrogram with at least 92% average similarity.
PFGE.
PFGE of M. avium isolates was carried out using previously described methods (11). Patterns considered indistinguishable by the Tenover criteria (29) were assigned numeric type designations.
Calculation of DI.
Assessment of DI was conducted on a 20% random sample of all clinical M. avium isolates for which DNA was stored at Seattle BioMed at the outset of the study. A uniform distribution in Microsoft Excel was used to assign a random number between 0 and 1 to each sample. Samples with random numbers of 0.20 or less (n = 29) were subjected to multiple genotyping methods. One of the selected genomic DNA samples was exhausted before all methods could be applied, so 28 samples were used to evaluate discrimination. The DI of each method was calculated using the approach of Hunter (20) as implemented by BioPHP at http://biophp.org/stats/discriminatory_power/demo.php.
Identification of GDCs.
Genomic DNA from a convenience sample of 127 clinical M. avium isolates was subjected to LSP-MVR analysis. Each clinical isolate was classified as having a unique genotype, sharing its genotype only with other clinical isolates from the same location, or sharing its genotype with at least one clinical isolate from another location. These latter genotypes were termed “geographically dispersed clinical types,” or GDC types.
Environmental comparison.
Genomic DNA from a convenience sample of 52 environmental M. avium isolates was subjected to LSP-MVR analysis.
Ethical review.
This study was determined by the investigators to be exempt from ethical review under 45 CFR 46.101.b.4.
Nucleotide sequence accession numbers.
Novel hsp65 sequences have been deposited in the GenBank database under accession numbers FJ839884.1 and FJ839885.1.
RESULTS
Selection of PCR-based genotyping methods.
In order to assess reproducibility within each method, DNA extracted from isolates HMC02 and 104 was subjected to hsp65 sequencing 5 times, rep-PCR 11 and 8 times, and LSP-MVR more than 7 times. Identical hsp65 sequencing results were seen all five times. Eighty-five percent of all possible pairwise comparisons between rep-PCR runs of HMC02 (47 of 55) were ≥92% similar, and 88% (7 of 8) of replicate rep-PCR runs of 104 were ≥92% similar. LSP-MVR results were identical for HMC02 on every occasion and identical for 104 all but one time.
To compare DIs, all PCR-based methods were applied to the panel of 28 randomly selected clinical isolates (see Table S1 in the supplemental material). hsp65 sequencing alone had a DI of 0.58, LSP of 0.87, four-locus MIRU of 0.84, eight-locus MIRU of 0.87, and rep-PCR of 0.97 (Table 1). The combination of four LSP and four MIRU-VNTR (LSP-MVR) loci improved the DI to 0.95. When combined with LSP, the use of eight MIRU-VNTR loci did not improve resolving power over the use of four MIRU-VNTR loci (Table 1). Thus, LSP-MVR, with four LSP and four MIRU-VNTR loci, exhibited high resolving power in a high-throughput, portable, PCR-based method and was chosen as the first-line approach for further analysis.
Table 1.
Discriminatory indices of genotyping methods for Mycobacterium avium
| Methoda | Discriminatory index of method plus: |
|||||
|---|---|---|---|---|---|---|
| Alone | hsp65 | LSP | 4-Locus MIRU | 8-Locus MIRU | Rep-PCR | |
| hsp65 | 0.58 | 0.91 | 0.89 | 0.91 | 0.98 | |
| LSP | 0.87 | 0.95 | 0.95 | 0.97 | ||
| 4-Locus MIRU | 0.84 | 0.98 | ||||
| 8-Locus MIRU | 0.87 | 0.98 | ||||
| Rep-PCR | 0.97 | |||||
hsp65, sequencing of a 1,059-bp portion of the 3′ end of the gene for heat shock protein 65; LSP, large-sequence polymorphism analysis; 4-locus MIRU, MIRU-VNTR analysis using loci X3, 25, 32 and 47; 8-locus MIRU, MIRU-VNTR analysis using loci 292, X3, 25, 47, 3, 7, 10, and 32; rep-PCR, repetitive-sequence-based PCR.
Identification of geographically dispersed clinical strains.
Clinical M. avium isolates from 127 individuals were typed by LSP-MVR. Of these, 24/127 (19%) had genotypes unique among clinical isolates. The remaining 103 isolates had 1 of 18 nonunique genotypes. Of these 18 nonunique genotypes, 6 genotypes were found only in one geographic collection. Twelve genotypes were found at more than one geographic location. Isolates exhibiting these geographically dispersed clinical (GDC) genotypes accounted for 50% (63/127) of all typed clinical isolates.
GDCs by location are shown in Fig. 1; other LSP-MVR genotypes observed among clinical isolates are shown in Table S5 in the supplemental material. GDC1 includes isolate HMC02 and some genotypically similar isolates previously identified by rep-PCR and IS1245 RFLP (14, 15). GDC6 and GDC7 both contain some isolates previously characterized by rep-PCR as 104-like (14, 15). Strain 104 itself shared its LSP-MVR type only with other clinical isolates from its region of isolation, southern California. The other GDCs have not previously been described.
Fig 1.

Geographic distribution of LSP-MVR genotypes of clinical Mycobacterium avium. The size of a pie chart is proportional to the number of cases from each location included in the analysis. Yellow circles on the map correspond to catchment areas of the clinical laboratory(ies) providing isolates or DNA for analysis. GDCs were defined by the same LSP-MVR type being found for at least one clinical isolate at each of two or more geographical locations. LSP-MVR digits of type represent loci HSDR, DA2/LSP2, DA7/LSP7, DEL11/LSPP5, X3, 25, 32, and 47 (21, 39). “1” is used to indicate the absence of the large-sequence polymorphism, which corresponds to product lengths of 343 bp (for HSDR), 834 bp (for DA2/LSP2), 329 bp (for DA7/LSP7), and 728 bp (for DEL11/LSPP5). “2” is used to indicate the presence of the large-sequence polymorphism, which corresponds to product lengths of 480 bp (for HSDR), 954 bp (for DA2/LSP2), 181 bp (for DA7/LSP7), and 794 bp (for DEL11/LSPP5) (21); digits for MIRU-VNTR type represent the number of repeats inferred from the product length at that locus based on product sizes and repeat numbers from Thibault et al. (16) and personal correspondence with V. Thibault: 196 bp when 2 repeats of 53 bp are present (locus X3), 350 bp when 3 repeats of 58 bp are present (25), 298 bp when 8 repeats of 18 bp are present (32), and 217 bp when 3 repeats of 35 bp are present (47). LSP-MVR types of GDCs are as follows: GDC1, 2112-3383; GDC2, 1111-5282; GDC3, 1121-3282; GDC4, 1211-2483; GDC5, 1211-4282; GDC6, 1221-3282; GDC7, 1221-4282; GDC8, 2211-2283; GDC9, 2212-3383; GDC10, 1121-2282; GDC11, 1121-4282; and GDC12, 2112-5383. Local strains were defined by finding the same LSP-MVR type for two or more clinical isolates at a single geographic location but not being found among clinical isolates from other geographic locations; unique types were found for a single isolate in the entire set of clinical genotypes. Environmental isolates are not included in these numbers and were not considered in GDC designations. Genotypes for local clusters and unique types are shown in Table S5 in the supplemental material. The map is adapted from a Wikimedia Commons map (http://commons.wikimedia.org/wiki/File:A_large_blank_world_map_with_oceans_marked_in_blue-edited.png) published under a Creative Commons agreement.
Although 8-locus MIRU-VNTR had a lower DI than LSP-MVR (Table 1), it is a widely used method that increased in popularity over the course of this study. Therefore, a post hoc analysis was conducted to determine if GDCs identified by LSP-MVR would have been identified if the main analysis had been carried out by using 8-locus MIRU-VNTR instead. Complete 8-locus MIRU-VNTR types were determined for the isolates randomly selected to evaluate the DIs of genotyping methods (see Table S1 in the supplemental material) and for the isolates selected for PFGE analysis (Table 2). Thus, 8-locus MIRU-VNTR results were available for at least two isolates from different geographical locations within 4 GDCs defined by LSP-MVR (GDC1, GDC2, GDC3, and GDC7). Three of the 4 GDCs contained multiple isolates from different locations with the same 8-locus MIRU-VNTR genotype (see Table S6 in the supplemental material). Therefore, these GDCs would have been identified if this widely used approach had been employed in the main analysis.
Table 2.
PFGE results for some clinical isolates sharing GDCa genotypes
| Name and LSP-MVR typeb | Isolate name | Site | PFGE pattern |
|---|---|---|---|
| GDC1 | |||
| 2112-3383 | 4904 | Montreal, Canada | 10 |
| HMC02 | Seattle | Not interpretable | |
| 108 | Southern California | 11 | |
| W355 | Southern California | 10 | |
| 03240 | Montreal | 13 | |
| GDC7 | |||
| 1221-4282 | W214 | Southern California | 7 |
| HMC08 | Seattle | 7 | |
| HMC24 | Seattle | 4 | |
| W216-8 | Southern California | 5 | |
| 105 | Southern California | 8 |
GDCs, geographically dispersed clinical strains. GDCs were defined by the same LSP-MVR type being found for at least one clinical isolate at each of two or more geographical locations.
LSP-MVR and PFGE types are given for all members of LSP-MVR-defined GDCs for which archived cultures from more than one location were available for PFGE analysis; identity by PFGE of isolates from distant geographic locations could be tested only for GDC1 and GDC7.
A subset of clinical isolates was further analyzed by PFGE (Fig. 2). These isolates were chosen for their membership in GDCs defined by LSP-MVR and the availability of permanent cultures (required for PFGE). For two GDCs (GDC1 and GDC7), at least one isolate from more than one geographic location was available for PFGE analysis (Table 2). In both of these GDCs, two isolates from distant sites exhibited indistinguishable PFGE patterns.
Fig 2.
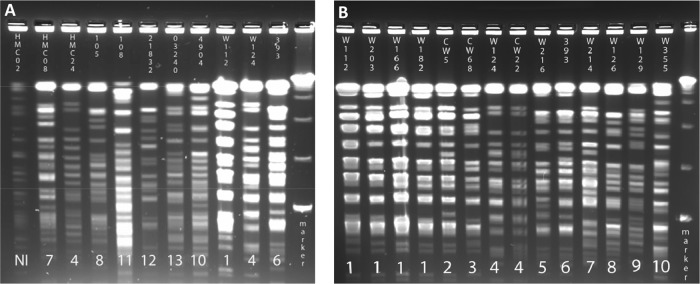
Pulse-field gel electrophoresis of M. avium clinical isolates. Isolates were selected for this analysis as described in the text. (A) Analysis of 11 isolates, including four from GDC1 and three from GDC7. (B) Analysis of 14 isolates, including one from GDC1 and two from GDC7. Data at the top of each lane indicate the isolate identity. Numbers at the bottom of each lane indicate different PFGE patterns (isolates with the same horizontal number have indistinguishable patterns). DNA markers in the far right lane (from bottom to top) are 48.5, 97.0, 145.5, and 194.0 bp. NI, not interpretable.
GDC genotypes among environmental isolates.
The occurrence of GDC genotypes in the environment was investigated. M. avium isolates from 52 water, food, and soil samples were typed by LSP-MVR. Thirty-three isolates (out of 52; 63%) from two distinct western U.S. geographic locations shared a single genotype, GDC1. Five additional GDCs were observed (GDC5, GDC9, GDC10, GDC11, and GDC12) in environmental collections, accounting for 13 additional isolates (out of 52; 25%). Only six environmental isolates (out of 52; 12%) had genotypes not observed in the clinical collections. Genotypes observed among environmental isolates by location are shown in Table S7 in the supplemental material.
DISCUSSION
Studies on the epidemiology and etiology of MAC diseases have often compared the genetic fingerprints of MAC isolated from patients' clinical specimens and the environments to which those patients have been exposed (22, 23, 25–28). Few of these studies were conducted in a comparative fashion, and the underlying distribution of MAC in the environment is only partially understood. To date, we know of only one comparative study, that of Fujita et al. (42), that included MAC isolated from the environments of nondiseased individuals.
Studies of the epidemiology of M. tuberculosis have been aided by the use of PCR-based strain typing methods, namely, MIRU-VNTR and spoligotyping. The portability of these methods is such that results generated by independent investigators around the world can be fed into unified numerical databases (43, 44). At the outset of the present study, no such portable methods had emerged for MAC. Therefore, we combined two previously reported, moderate-resolution methods, LSP analysis and MIRU-VNTR (15, 16), to yield a simple, highly reproducible approach with resolving power that approaches those of well-characterized methods such as PFGE and IS1245-RFLP. LSP-MVR is inexpensive to perform, requires only small amounts of DNA, and yields portable results.
LSP-MVR analysis identified distinct genotypic groups among M. avium isolates from clinical cases separated by distance and without identified epidemiologic links. Forty-two genotypes were observed among 127 clinical isolates. Twelve genotypes (GDC1 to GDC12) were observed at multiple geographic locations, isolated from patients with no apparent connection to each other. This may reflect common exposure to commercial products, such as food or potting soil, which are shipped across long distances and are known to be colonized by MAC (1, 26). It may also reflect the presence of these genotypes in environmental sources at multiple, distant locations. These genotypes may have traits that favor exposure to humans, such as survival in shipped products, built environments, domestic water supplies, or domesticated animals. They may have characteristics that favor colonization, invasion, or pathogenicity in human hosts.
Alternatively, these results could be an artifact of the genotyping approach. These results were obtained using a novel method, but it is likely that other established genotyping methods would yield similar results for several reasons. First, LSP-MVR showed good resolving power. It displayed a DI of 0.95 in a random set of clinical M. avium isolates. This value approached a DI of 0.97, which was observed for rep-PCR using the same isolates, and exceeded that of the widely used 8-locus MIRU-VNTR (0.87).
Second, the existence of GDCs has been partially corroborated by use of other methods: IS1245-RFLP, PFGE, rep-PCR, and MIRU-VNTR. Three GDCs identified in the present study, GDC1, GDC6, and GDC7, include isolates previously reported to share IS1245-RFLP patterns (14, 15). In the present study, GDC1 and GDC7 contained geographically distant members that shared PFGE patterns. Likewise, for the four GDCs identified by LSP-MVR in this study that had sufficient 8-locus MIRU-VNTR data to evaluate (GDC1 to GDC3 and GDC7), 3 of 4 GDCs were confirmed. Thus, the combined high-resolution genotypic data support the conclusion that some genotypes are geographically widespread.
Our observations are consistent with a recent MIRU-VNTR study of 47 clinical isolates collected over 2 decades from a single clinical site in Italy (31). Although the Italian isolates were not separated geographically, the authors concluded that some M. avium genotypes are stable over time and can be associated with epidemiologically unlinked cases. Our study, which applied a higher-resolution method (LSP-MVR) to a larger and more diverse sample set (179 clinical and environmental isolates from geographically distinct sources), confirms and extends these conclusions.
Six GDC genotypes (GDC1, GDC5, GDC9, GDC10, GDC11, and GDC12) were observed in isolates from environmental samples. Therefore, GDC genotypes are not unique to clinical isolates. Genotypic diversity appeared to be lower among the environmental isolates examined than among the clinical isolates examined. Limited genotypic diversity among environmental isolates has been reported elsewhere (23, 24) and must be interpreted with caution. It may be due to selection by methods of sample collection, decontamination, and culture or peculiarities of the environments represented in the convenience sample typed. It also implies that high-resolution genotyping methods that were developed for use with clinical MAC isolates do not have the same ability to distinguish environmental isolates. In other words, the molecular markers that vary among clinical MAC strains may be less variable among environmental strains despite a high level of diversity at other loci. Alternatively, it may reflect truly low genotypic diversity among M. avium strains in any given environment. The greater diversity seen among clinical isolates may reflect the fact that humans are exposed to many different environments; thus, they are able to reflect the diversity across different environments more effectively than purposeful environmental sampling.
Our results suggest that caution should be employed in interpreting genotypic matches between clinical and environmental isolates in studies of MAC lung disease that do not include appropriate control subjects. Matches between rarely observed genotypes of M. avium may provide evidence for an acquisition event, but the observation of common genotypes at distant sites shows that some genotypic matches can occur in the apparent absence of an epidemiologic link. When Tenover et al. presented their criteria for interpreting PFGE data in epidemiologic investigations, they emphasized that high-resolution genotypic data should be applied in the context of an outbreak investigation with a defined time frame, knowledge of the strains of a pathogen that are endemic to the region, and other epidemiologic data (29). The importance of this context was recently noted by investigators of a nationwide Salmonella outbreak associated with fresh produce (45).
This context is infrequently present in studies of human disease caused by MAC. The timing of etiologically relevant exposures is unknown, and the underlying distribution of strains in the environment is poorly characterized. In genotypic studies to date, other epidemiologic data are often limited and comparator groups are rarely included. Comparison to representative control subjects or development of an expanded worldwide genotypic database will facilitate the interpretation of molecular epidemiological data in studies of MAC disease. Use of PCR-based genotyping technologies with portable, numerical results, such as LSP-MVR, will further assist in large-scale, multisite studies and development of shared databases.
Supplementary Material
ACKNOWLEDGMENTS
This publication was developed under a STAR Research Assistance Agreement (no. FP-91695601) and a Collaborative Agreement (no. 833030010) awarded by the U.S. Environmental Protection Agency. Additional funding was provided to M.A.D. by the Medical Scientist Training Program (T32 GM07266, a National Research Service Award from the National Institute of General Medical Sciences).
We thank Joseph Falkinham, Marcel Behr, Sylvia Leao, Richard van Soolingen, and Carolyn Wallis for providing strains and DNA samples.
This work was performed at Seattle Biomedical Research Institute, Seattle, WA, and the Centers for Disease Control and Prevention, Atlanta, GA.
The views expressed in this report are those of the individual authors and do not necessarily reflect the views and policies of the U.S. Environmental Protection Agency or the Centers for Disease Control and Prevention. Mention of trade names or commercial products does not constitute endorsement or recommendation for use.
Footnotes
Published ahead of print 12 July 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.01443-13.
REFERENCES
- 1.Argueta C, Yoder S, Holtzman AE, Aronson TW, Glover N, Berlin OG, Stelma GN, Jr, Froman S, Tomasek P. 2000. Isolation and identification of nontuberculous mycobacteria from foods as possible exposure sources. J. Food Prot. 63:930–933 [DOI] [PubMed] [Google Scholar]
- 2.Falkinham JO., III 2002. Nontuberculous mycobacteria in the environment. Clin. Chest Med. 23:529–551 [DOI] [PubMed] [Google Scholar]
- 3.Griffith DE, Aksamit T, Brown-Elliott BA, Catanzaro A, Daley C, Gordin F, Holland SM, Horsburgh R, Huitt G, Iademarco MF, Iseman M, Olivier K, Ruoss S, von Reyn CF, Wallace RJ, Jr, Winthrop K, ATS Mycobacterial Disease Subcommittee, American Thoracic Society, Infectious Disease Society of America 2007. An official ATS/IDSA statement: diagnosis, treatment, and prevention of nontuberculous mycobacterial diseases. Am. J. Resp. Crit. Care Med. 175:367–416 [DOI] [PubMed] [Google Scholar]
- 4.Bang D, Herlin T, Stegger M, Andersen AB, Torkko P, Tortoli E, Thomsen VO. 2008. Mycobacterium arosiense sp. nov., a slowly growing, scotochromogenic species causing osteomyelitis in an immunocompromised child. Int. J. Syst. Evol. Microbiol. 58:2398–2402 [DOI] [PubMed] [Google Scholar]
- 5.Ben Salah I, Cayrou C, Raoult D, Drancourt M. 2009. Mycobacterium marseillense sp. nov., Mycobacterium timonense sp. nov., and Mycobacterium bouchedurhonense sp. nov., members of the Mycobacterium avium complex. Int. J. Syst. Evol. Microbiol. 59:2803–2808 [DOI] [PubMed] [Google Scholar]
- 6.Murcia MI, Tortoli E, Menendez MC, Palenque E, Garcia MJ. 2006. Mycobacterium colombiense sp. nov., a novel member of the Mycobacterium avium complex, and description of MAC-X as a new ITS genetic variant. Int. J. Syst. Evol. Microbiol. 56:2049–2054 [DOI] [PubMed] [Google Scholar]
- 7.Tortoli E, Rindi L, Garcia MJ, Chiaradonna P, Dei R, Garzelli C, Kroppenstedt RM, Lari N, Mattei R, Mariottini A, Mazzarelli G, Murcia MI, Nanetti A, Piccoli P, Scarparo C. 2004. Proposal to elevate the genetic variant MAC-A, included in the Mycobacterium avium complex, to species rank as Mycobacterium chimaera sp. nov. Int. J. Syst. Evol. Microbiol. 54:1277–1285 [DOI] [PubMed] [Google Scholar]
- 8.van Ingen J, Boeree MJ, Kosters K, Wieland A, Tortoli E, Dekhuijzen PN, van Soolingen D. 2009. Proposal to elevate Mycobacterium avium complex ITS sequevar MAC-Q to Mycobacterium vulneris sp. nov. Int. J. Syst. Evol. Microbiol. 59:2277–2282 [DOI] [PubMed] [Google Scholar]
- 9.Mijs W, de Haas P, Rossau R, Van der Laan T, Rigouts L, Portaels F, van Soolingen D. 2002. Molecular evidence to support a proposal to reserve the designation Mycobacterium avium subsp. avium for bird-type isolates and “M. avium subsp. hominissuis” for the human/porcine type of M. avium. Int. J. Syst. Evol. Microbiol. 52:1505–1518 [DOI] [PubMed] [Google Scholar]
- 10.Behr MA. 2008. Mycobacterium du jour: what's on tomorrow's menu? Microbes Infect. 10:968–972 [DOI] [PubMed] [Google Scholar]
- 11.Mazurek GH, Hartman S, Zhang Y, Brown BA, Hector JS, Murphy D, Wallace RJ., Jr 1993. Large DNA restriction fragment polymorphism in the Mycobacterium avium-M. intracellulare complex: a potential epidemiologic tool. J. Clin. Microbiol. 31:390–394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ritacco V, Kremer K, van der Laan T, Pijnenburg JE, de Haas PE, van Soolingen D. 1998. Use of IS901 and IS1245 in RFLP typing of Mycobacterium avium complex: relatedness among serovar reference strains, human and animal isolates. Int. J. Tuberc. Lung Dis. 2:242–251 [PubMed] [Google Scholar]
- 13.van Soolingen D, Bauer J, Ritacco V, Leao SC, Pavlik I, Vincent V, Rastogi N, Gori A, Bodmer T, Garzelli C, Garcia MJ. 1998. IS1245 restriction fragment length polymorphism typing of Mycobacterium avium isolates: proposal for standardization. J. Clin. Microbiol. 36:3051–3054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cangelosi GA, Freeman RJ, Lewis KN, Livingston-Rosanoff D, Shah KS, Milan SJ, Goldberg SV. 2004. Evaluation of a high-throughput repetitive-sequence-based PCR system for DNA fingerprinting of Mycobacterium tuberculosis and Mycobacterium avium complex strains. J. Clin. Microbiol. 42:2685–2693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Horan KL, Freeman R, Weigel K, Semret M, Pfaller S, Covert TC, van Soolingen D, Leao SC, Behr MA, Cangelosi GA. 2006. Isolation of the genome sequence strain Mycobacterium avium 104 from multiple patients over a 17-year period. J. Clin. Microbiol. 44:783–789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thibault VC, Grayon M, Boschiroli ML, Hubbans C, Overduin P, Stevenson K, Gutierrez MC, Supply P, Biet F. 2007. New variable-number tandem-repeat markers for typing Mycobacterium avium subsp. paratuberculosis and M. avium strains: comparison with IS900 and IS1245 restriction fragment length polymorphism typing. J. Clin. Microbiol. 45:2404–2410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pfaller SL, Aronson TW, Holtzman AE, Covert TC. 2007. Amplified fragment length polymorphism analysis of Mycobacterium avium complex isolates recovered from southern California. J. Med. Microbiol. 56:1152–1160 [DOI] [PubMed] [Google Scholar]
- 18.Wojtasik A, Kubiak AB, Krzyzanowska A, Majchrzak M, Augustynowicz-Kopec E, Parniewski P. 2012. Comparison of the (CCG)4-based PCR and MIRU-VNTR for molecular typing of Mycobacterium avium strains. Mol. Biol. Rep. 39:7681–7686 [DOI] [PubMed] [Google Scholar]
- 19.Cayrou C, Turenne C, Behr MA, Drancourt M. 2010. Genotyping of Mycobacterium avium complex organisms using multispacer sequence typing. Microbiology 156:687–694 [DOI] [PubMed] [Google Scholar]
- 20.Hunter PR. 1990. Reproducibility and indices of discriminatory power of microbial typing methods. J. Clin. Microbiol. 28:1903–1905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Telles MA, Yates MD, Curcio M, Ueki SY, Palaci M, Hadad DJ, Drobniewski FA, Pozniak AL. 1999. Molecular epidemiology of Mycobacterium avium complex isolated from patients with and without AIDS in Brazil and England. Epidemiol. Infect. 122:435–440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.von Reyn CF, Maslow JN, Barber TW, Falkinham JO, III, Arbeit RD. 1994. Persistent colonisation of potable water as a source of Mycobacterium avium infection in AIDS. Lancet 343:1137–1141 [DOI] [PubMed] [Google Scholar]
- 23.Aronson T, Holtzman A, Glover N, Boian M, Froman S, Berlin OG, Hill H, Stelma G., Jr 1999. Comparison of large restriction fragments of Mycobacterium avium isolates recovered from AIDS and non-AIDS patients with those of isolates from potable water. J. Clin. Microbiol. 37:1008–1012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hilborn ED, Covert TC, Yakrus MA, Harris SI, Donnelly SF, Rice EW, Toney S, Bailey SA, Stelma GN., Jr 2006. Persistence of nontuberculous mycobacteria in a drinking water system after addition of filtration treatment. Appl. Environ. Microbiol. 72:5864–5869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.von Reyn CF, Arbeit RD, Horsburgh CR, Ristola MA, Waddell RD, Tvaroha SM, Samore M, Hirschhorn LR, Lumio J, Lein AD, Grove MR, Tosteson AN. 2002. Sources of disseminated Mycobacterium avium infection in AIDS. J. Infect. 44:166–170 [DOI] [PubMed] [Google Scholar]
- 26.De Groote MA, Pace NR, Fulton K, Falkinham JO., III 2006. Relationships between Mycobacterium isolates from patients with pulmonary mycobacterial infection and potting soils. Appl. Environ. Microbiol. 72:7602–7606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Falkinham JO., III 2011. Nontuberculous mycobacteria from household plumbing of patients with nontuberculous mycobacteria disease. Emerg. Infect. Dis. 17:419–424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Falkinham JO, III, Iseman MD, de Haas P, van Soolingen D. 2008. Mycobacterium avium in a shower linked to pulmonary disease. J. Water Health 6:209–213 [DOI] [PubMed] [Google Scholar]
- 29.Tenover FC, Arbeit RD, Goering RV, Mickelsen PA, Murray BE, Persing DH, Swaminathan B. 1995. Interpreting chromosomal DNA restriction patterns produced by pulsed-field gel electrophoresis: criteria for bacterial strain typing. J. Clin. Microbiol. 33:2233–2239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bauer J, Andersen AB, Askgaard D, Giese SB, Larsen B. 1999. Typing of clinical Mycobacterium avium complex strains cultured during a 2-year period in Denmark by using IS1245. J. Clin. Microbiol. 37:600–605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rindi L, Buzzigoli A, Medici C, Garzelli C. 2013. High phylogenetic proximity of isolates of Mycobacterium avium subsp. hominissuis over a two decades-period. Infect. Genet. Evol. 16:99–102 [DOI] [PubMed] [Google Scholar]
- 32.Cangelosi GA, Palermo CO, Laurent JP, Hamlin AM, Brabant WH. 1999. Colony morphotypes on Congo red agar segregate along species and drug susceptibility lines in the Mycobacterium avium-intracellulare complex. Microbiology 145(Part 6):1317–1324 [DOI] [PubMed] [Google Scholar]
- 33.Komijn RE, de Haas PE, Schneider MM, Eger T, Nieuwenhuijs JH, van den Hoek RJ, Bakker D, van Zijderveld FG, van Soolingen D. 1999. Prevalence of Mycobacterium avium in slaughter pigs in The Netherlands and comparison of IS1245 restriction fragment length polymorphism patterns of porcine and human isolates. J. Clin. Microbiol. 37:1254–1259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mayer BK, Falkinham JO., III 1986. Superoxide dismutase activity of Mycobacterium avium, M. intracellulare, and M. scrofulaceum. Infect. Immun. 53:631–635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mukherjee S, Petrofsky M, Yaraei K, Bermudez LE, Cangelosi GA. 2001. The white morphotype of Mycobacterium avium-intracellulare is common in infected humans and virulent in infection models. J. Infect. Dis. 184:1480–1484 [DOI] [PubMed] [Google Scholar]
- 36.Oliveira RS, Sircili MP, Oliveira EM, Balian SC, Ferreira-Neto JS, Leao SC. 2003. Identification of Mycobacterium avium genotypes with distinctive traits by combination of IS1245-based restriction fragment length polymorphism and restriction analysis of hsp65. J. Clin. Microbiol. 41:44–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Semret M, Zhai G, Mostowy S, Cleto C, Alexander D, Cangelosi G, Cousins D, Collins DM, van Soolingen D, Behr MA. 2004. Extensive genomic polymorphism within Mycobacterium avium. J. Bacteriol. 186:6332–6334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stormer RS, Falkinham JO., III 1989. Differences in antimicrobial susceptibility of pigmented and unpigmented colonial variants of Mycobacterium avium. J. Clin. Microbiol. 27:2459–2465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.von Reyn CF, Waddell RD, Eaton T, Arbeit RD, Maslow JN, Barber TW, Brindle RJ, Gilks CF, Lumio J, Lahdevirta J, Ranki A, Dawson D, Falkinham JO., III 1993. Isolation of Mycobacterium avium complex from water in the United States, Finland, Zaire, and Kenya. J. Clin. Microbiol. 31:3227–3230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ashworth M, Horan KL, Freeman R, Oren E, Narita M, Cangelosi GA. 2008. Use of PCR-based Mycobacterium tuberculosis genotyping to prioritize tuberculosis outbreak control activities. J. Clin. Microbiol. 46:856–862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Turenne CY, Semret M, Cousins DV, Collins DM, Behr MA. 2006. Sequencing of hsp65 distinguishes among subsets of the Mycobacterium avium complex. J. Clin. Microbiol. 44:433–440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fujita K, Ito Y, Hirai T, Maekawa K, Imai S, Tatsumi S, Ichiyama S, Mishima M. 2012. Comparison of genotype distribution of Mycobacterium avium-intracellulare complex (MAC) isolates from clinical samples and residential soils in patients with or without pulmonary MAC disease, abstr. A4029. American Thoracic Society International Meeting, San Francisco, CA, 1 May 2012. DOI: 10.1164/ajrccm-conference.2012.185.1_MeetingAbstracts.A4028 [DOI] [Google Scholar]
- 43.Allix-Beguec C, Harmsen D, Weniger T, Supply P, Niemann S. 2008. Evaluation and strategy for use of MIRU-VNTRplus, a multifunctional database for online analysis of genotyping data and phylogenetic identification of Mycobacterium tuberculosis complex isolates. J. Clin. Microbiol. 46:2692–2699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brudey K, Driscoll JR, Rigouts L, Prodinger WM, Gori A, Al-Hajoj SA, Allix C, Aristimuno L, Arora J, Baumanis V, Binder L, Cafrune P, Cataldi A, Cheong S, Diel R, Ellermeier C, Evans JT, Fauville-Dufaux M, Ferdinand S, Garcia de Viedma D, Garzelli C, Gazzola L, Gomes HM, Guttierez MC, Hawkey PM, van Helden PD, Kadival GV, Kreiswirth BN, Kremer K, Kubin M, Kulkarni SP, Liens B, Lillebaek T, Ho ML, Martin C, Martin C, Mokrousov I, Narvskaia O, Ngeow YF, Naumann L, Niemann S, Parwati I, Rahim Z, Rasolofo-Razanamparany V, Rasolonavalona T, Rossetti ML, Rusch-Gerdes S, Sajduda A, Samper S, Shemyakin IG, Singh UB, Somoskovi A, Skuce RA, van Soolingen D, Streicher EM, Suffys PN, Tortoli E, Tracevska T, Vincent V, Victor TC, Warren RM, Yap SF, Zaman K, Portaels F, Rastogi N, Sola C. 2006. Mycobacterium tuberculosis complex genetic diversity: mining the fourth international spoligotyping database (SpolDB4) for classification, population genetics and epidemiology. BMC Microbiol. 6:23. 10.1186/1471-2180-6-23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mody RK, Greene SA, Gaul L, Sever A, Pichette S, Zambrana I, Dang T, Gass A, Wood R, Herman K, Cantwell LB, Falkenhorst G, Wannemuehler K, Hoekstra RM, McCullum I, Cone A, Franklin L, Austin J, Delea K, Behravesh CB, Sodha SV, Yee JC, Emanuel B, Al-Khaldi SF, Jefferson V, Williams IT, Griffin PM, Swerdlow DL. 2011. National outbreak of Salmonella serotype saintpaul infections: importance of Texas restaurant investigations in implicating jalapeno peppers. PLoS One 6:e16579. 10.1371/journal.pone.0016579 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.