

Abstract
Addition of affinity tags to bacteriophage particles facilitates a variety of applications, including vaccine construction and diagnosis of bacterial infections. Addition of tags to phage capsids is desirable, as modification of the tails can lead to poor adsorption and loss of infectivity. Although tags can readily be included as fusions to head decoration proteins, many phages do not have decoration proteins as virion components. The addition of a small (10-amino-acid) Strep-tag II (STAG II) to the mycobacteriophage TM4 capsid subunit, gp9, was not tolerated as a genetically homogenous recombinant phage but could be incorporated into the head by growth of wild-type phage on a host expressing the capsid-STAG fusion. Particles with capsids composed of wild-type and STAG-tagged subunit mixtures could be grown to high titers, showed good infectivities, and could be used to isolate phage-bacterium complexes. Preparation of a STAG-labeled fluoromycobacteriophage enabled capture of bacterial complexes and identification of infected bacteria by fluorescence.
INTRODUCTION
Mycobacteriophages—viruses that infect mycobacterial hosts, including Mycobacterium tuberculosis—represent powerful toolboxes for mycobacterial genetics and for clinical applications to control human tuberculosis (1–3). Over 220 mycobacteriophage genomes have been sequenced, and the sequences have revealed these phages to be highly diverse and organizationally mosaic (4, 5). They have been exploited for the development of genetic tools, such as for delivery of transposons and allelic exchange substrates (6, 7) and for the construction of integration-proficient plasmid vectors (8–12), non-antibiotic-selectable markers (13), and expression systems (14, 15). They have also been proposed for efficient delivery of reporter genes to facilitate simple and rapid determination of drug susceptibilities of M. tuberculosis in clinical specimens (15–19).
TM4 is perhaps one of the best-studied mycobacteriophages. It was isolated in 1984 (20) and was used to construct the first shuttle phasmids (21). It has a genome 52,797 bp in length, contains 89 protein-coding genes and no tRNA genes (22, 23), and is one of several phages with nucleotide sequence similarity to cluster K phage sequences (22). All of the cluster K phages infect both Mycobacterium smegmatis mc2155 and M. tuberculosis H37Rv, and TM4 infects some strains of Mycobacterium avium, as well as Mycobacterium ulcerans (22, 24). TM4 is not temperate and does not form stable lysogens in any known host. However, its cluster K relatives are temperate, as they contain integrase genes and form lysogens in M. smegmatis and M. tuberculosis (22). Comparative genomic analysis suggests that TM4 is a recent virulent derivative of a temperate parent. Temperature-sensitive nonreplicating TM4 mutants have been isolated for use as phage delivery vehicles (7), and the mutations have been mapped (22).
Morphologically, TM4 is siphoviral and contains an isometric icosahedral capsid that is joined to a long flexible noncontractile tail (23). The 305-amino-acid (aa) capsid subunit is encoded by gene 9, is covalently cross-linked in mature particles (23), and is predicted to fold similarly to phage HK97 (Fig. 1). The gene is approximately 100 residues shorter than HK97 gp5, because it lacks the N-terminal 104-aa delta region, which provides an essential assembly function in HK97 (25). The gene upstream of TM4, gene 8, encodes a putative scaffold protein, but genome and virion protein analyses suggest that there are no decoration proteins (22, 23), such as Hoc and Soc, which are components of the phage T4 capsid (26). Hoc and Soc are dispensable for T4 assembly but can be modified, such as to display foreign peptides on the capsid surface, with the potential for vaccine construction and antigen delivery (27). As there are no equivalent proteins in TM4, any modification to the capsid surface would likely require modifications of the capsid subunit itself.
Fig 1.

HHPred alignment of TM4 gp9 with the HK97 gp5 major capsid subunit. TM4 gp9 was used as a query sequence (Q) in an HHPred search using standard parameters (45), and the top two hits (probability of 100%) were matches to the cleaved mature capsid subunit and to the uncleaved procapsid (T), the second of which is shown here. Consensus sequences and secondary structure predictions (ss) are shown. The positions of the lysine and asparagine residues (positions 169 and 356, respectively, in HK97 gp5) that participate in covalent cross-linking in HK97 gp5 are shown by the arrows. The position corresponding to the latter is an aspartic acid residue in TM4 gp9.
Here we describe an approach for addition of a short affinity tag to TM4 particles. This enables the easy purification of phage particles and the capture of phage-bacterium complexes with the potential to simplify diagnostic applications. The approach should be generally applicable to other bacteriophage systems.
MATERIALS AND METHODS
Bacterial strains and reagents.
M. smegmatis mc2155 has been described previously (28) and was grown at 37°C in Middlebrook 7H9 broth (Difco), containing ADC (2 g liter−1 dextrose, 5 g liter−1 albumin, 0.85 g liter−1 NaCl) and 0.05% (vol/vol) Tween 80 or in 7H10 (Difco) containing ADC. Middlebrook Top Agar (MBTA) was prepared using 4.7 g liter−1 Middlebrook 7H9, 7 g liter−1 Bacto agar. Tween was omitted when cultures were used for phage infection. When appropriate, kanamycin 25 μg ml−1 was used. Escherichia coli strains were grown in L-broth. Phage buffer contained 10 mM Tris-HCl (pH 7.5), 68.5 NaCl, 10 mM MgSO4, 1 mM CaCl2.
BRED experiments.
Insertion of a Strep-tag II (STAG II) addition to the C terminus of gp9 of phAE87::hsp60-EGFP (abbreviated gfpϕ here) (18) was achieved by using the bacteriophage recombineering of electroporated DNA (BRED) strategy as already described (29). Briefly, a 230-bp target substrate with 100 bp of homology to each side of the 3′ end of gp9 of TM4 was designed using a 100-nucleotide oligonucleotide (5′-ACAAGACGCCGGTCGCCGTCGTGGCACCGGCTGCCagcgcttggagccacccgcagttcgaaaaaTAGTGCGCTATCGCCACGCCTTGACGGGGGCGGTT-3′) that contained the coding sequence for STAG II plus 2 amino acids used as a linker (shown in lowercase letters) and two 85-mer external primers, one forward (5′-CGCCCTGCGGCTCAAGGCGCGGTTCGCCTACGTGCTGGGTGTGAGCGCGACCGCTCAGGGCGCCAACAAGACGCCGGTCGCCGTC-3′) and one reverse (5′-TACCGGGTCCAGTTGTCGTCGCCCTCGACGAGGGCGGCCAGCAGGGTGCCCTCACGCACCCCGATAACCGCCCCCGTCAAGGCGT-3′), with 20-base overlaps on either side of the 100-mer. Combination PCR was carried out by mixing 10 ng of the 100-mer and 25 pmol of the 85-mers in 100 μl of PCR mix and using cloned Pfu DNA polymerase (Agilent Technologies, Santa Clara, CA). The resulting PCR product, a 230-bp targeting substrate, was extracted from the gel by using the QIAquick gel extraction kit (Qiagen) and reconstituted in sterile water.
M. smegmatis mc2155(pJV53) was grown and induced for recombineering functions as described previously (30). phAE87::hsp60-EGFP DNA and the targeting substrate were coelectroporated in the induced electrocompetent cells, and cells were recovered for 2 h at 30°C and plated in an infectious center assay. Screening of plaques for the presence of the insertion was done by PCR using a forward primer that anneals to the STAG II sequence (5′-GCTTGGAGCCACCCGCAGTTC-3′) and a reverse primer (5′-GACCGCAACCATGCCGGTGCGG-3′) that anneals to the sequence of gp10 (downstream of gp9 in the TM4 genome), giving an amplicon of 500 bp.
Construction of a plasmid expressing STAG-gp9 of TM4.
The major capsid protein gene (gp9) of TM4 containing Strep-tag II plus 2 amino acids used as a linker was cloned in the pNIT plasmid, a nitrile-inducible gene expression vector (31). DNA present in primary positive plaques from the BRED experiment described above was used as a template in a PCR with the following primers: for gp9STAGF, 5′-AATTCCATATGGCTGACATTTCACGCGCC-3′ (containing an NdeI site [underlined]); for gp9STAGR, 5′-TTACAAAGCTTCTATTTTTCGAACTGCGGGTGGCT-3′ (containing a HindIII site [underlined]). The amplicon was digested with NdeI-HindIII and cloned in pNIT digested with the same restriction enzymes.
The resultant plasmid (pNIT gp9-StrepTag) was used to transform M. smegmatis mc2155 electrocompetent cells, and kanamycin-resistant (Kanr) colonies were obtained after incubation for 3 days at 37°C.
Resistant clones were grown to an optical density at 600 nm (OD600) of 0.3 in 7H9-ADC medium, and protein expression was induced by addition of 28 mM ε-caprolactam (Sigma, St. Louis, MO). Expression of STAG-gp9 at different time points was checked by Western blotting with a monoclonal anti-STAG antibody that was horseradish peroxidase (HRP) conjugated (StrepMAB classic-HRP; catalog number 2-1509-001; IBA GmbH, Goettingen, Germany) and the Pierce ECL chemiluminescent substrate (Thermo Fisher Scientific Inc., Rockford, IL).
Preparation of phAE87::hsp60-EGFP stocks containing STAG-gp9 particles.
Phage stocks were prepared in a plaque assay. Briefly, cultures of M. smegmatis mc2155(pNIT gp9-STAG) were grown to an OD600 of 0.3 in 7H9-ADC medium, and then ε-caprolactam (28 mM) was added. When cells reached an OD600 of 1, they were infected with phAE87::hsp60-EGFP and allowed to adsorb for about 15 min at room temperature. After that, bacterium-phage mixes were included in MBTA Top Agar containing ε-caprolactam and poured on top of TH10 plates containing kanamycin. Infections were performed in order to obtain about 5,000 to 10,000 plaques per plate (150-mm plates), with a total of 20 plates prepared. Plaques were visualized after incubation for about 48 h at 30°C. Plates were flooded with phage buffer (10 ml) and left standing for 2 h at 37°C or overnight at 4°C. The buffer was collected and centrifuged to remove cells and debris, and the supernatant was filtered using a Stericup filter unit (EMD Millipore, Billerica, MA). Filtered supernatants were subjected to ultracentrifugation at 31,000 × g by using a Ti45 rotor in a Beckman Coulter Optima L90K apparatus for phage concentration and removal of unincorporated STAG-gp9 capsid proteins. Phage pellets were resuspended in approximately 1 ml of phage buffer, and the PFU ml−1 was calculated via a plaque assay. Titers of 1012 PFU ml−1 were obtained. The resulting phage preparation was designated STAG-gfpϕ.
Western analysis.
Forty microliters of a concentrated phage stock was mixed with 25 μl of water and frozen in dry ice. The frozen mixture was rapidly thawed and mixed by vortexing. This process was repeated twice, and the mixture was then heated to 75°C for 3 to 4 min. Samples were boiled for 3 min in sample buffer (62.5 mM Tris-HCl [pH 6.8], 2% SDS, 20% glycerol, 5% β-mercaptoethanol, 0.1% bromophenol blue) and loaded onto 7% SDS-polyacrylamide gels. Proteins were electrotransferred to a polyvinylidene difluoride membrane for 3 h and visualized using a monoclonal anti-STAG–HRP–conjugated antibody (StrepMAB Classic-HRP; catalog number 2-1509-001; IBA GmbH, Goettingen, Germany) and Pierce ECL chemiluminescent substrate (Thermo Fisher Scientific Inc., Rockford, IL). A cell extract from induced M. smegmatis mc2155(pNIT gp9-STAG) cells was used as a control.
Enzyme-linked immunosorbent assays (ELISA).
StrepMAB-Immo-coated microplates (catalog number 2-1521-001; IBA GmbH, Goettingen, Germany) that bind STAG II were used. Serial dilutions of the phage stock were performed in binding buffer (25 mM Tris-HCl, 2 mM EDTA, 140 mM NaCl; pH 7.6) and incubated for 1 h at room temperature. After that, three washes with washing buffer (binding buffer supplemented with 0.05% Tween 20) were performed to remove unbound phage. Two hundred microliters of a 1:5,000 dilution of StrepMAB Classic-HRP was added and incubated for 1 h at room temperature. After four washes with washing buffer, 100 μl of SureBlue Reserve tetramethylbenzidine (TMB) microwell peroxidase substrate (KPL Inc., Gaithersburg, Md) was added and left at room temperature until blue color developed. Reactions were stopped by addition of an equal volume of TMB Stop solution to the microwell plates, and absorbance was read at 450 nm.
Inmunoelectron microscopy.
Phage preparations were absorbed to a 400-mesh, support film, carbon-coated electron microscopy (EM) grid (01814-F; Ted Pella) previously glow discharged and rinsed with phage buffer. Grids were blocked with bovine serum albumin (BSA) at 50 mg ml−1 for 5 min, rinsed with washing buffer (50 mM Tris-HCl [pH 7.5], 0.1% Tween 20), and incubated for 5 min with the primary antibody (rabbit anti-Strep-tag II polyclonal antibody; Genscript, Piscataway, NJ) diluted 1:500 in the same buffer supplemented with 0.5% BSA. After rinsing with washing buffer, grids were incubated with the secondary antibody (gold-labeled anti-rabbit IgG; KPL Inc., Gaithersburg, MD) diluted 1:1,000. The grid was rinsed with washing buffer and stained with a 1% uranyl acetate solution before examination of several fields under the electron microscope.
Efficiency of capture of free phage particles using Strep-Tactin-coated magnetic beads.
Twenty-five microliters of MagStrep type 2HC beads (catalog number 2-1612-002; IBA GmbH, Goettingen, Germany) was mixed with different dilutions of STAG-gfpϕ, gfpϕ, or D29. Beads and phage were incubated at 4°C for 30 min with occasional mixing. Beads were separated by using a magnetic separator and washed six times with 200 μl of buffer W (100 mM Tris-HCl [pH 8], 150 mM NaCl, 1 mM EDTA). Supernatant (the fraction obtained after magnetic removal of the beads) and washes were combined to calculate the amount of unbound phage. The PFU ml−1 was calculated from a plaque assay. Different amounts of beads were tested to optimize the bead/phage ratio.
Recovery of phage-bacterium complexes using Strep-Tactin-coated magnetic beads.
M. smegmatis cells were grown to an OD600 of 1 in 7H9-ADC in the absence of Tween. Approximately 250 μl of cells (about 2.5 × 107 cells) was infected with STAG-gfpϕ or gfpϕ at an MOI (multiplicity of infection) of 100. Incubation was performed with standing for 10 min at room temperature and shaking for 3.5 h at 37°C. After that, cells were fixed with an equal volume of 4% paraformaldehyde (catalog number HT5011; Sigma, St. Louis, MO) for 30 min at room temperature. Fifty microliters of the fixed cell suspension (approximately 106 cells) was incubated with 50 μl of MagStrep type 2HC beads (catalog number 2-1611-002/006; IBA GmbH, Goettingen, Germany) for 30 min at room temperature on a rocking platform. Beads were separated using a magnetic separator and washed three times using buffer W (100 mM Tris-HCl [pH 8], 150 mM NaCl, 1 mM EDTA) according to the manufacturer's instructions. At this step, either 5 μl of beads was directly spotted on top of a slide for examination or phage-bacterium complexes were eluted from beads by using buffer BE (100 mM Tris-HCl [pH 8], 150 mM NaCl, 1 mM EDTA, 2 mM d-biotin) and eluted cells were observed by epifluorescence microscopy.
Microscopy and settings.
A fluorescence microscope (Axiostar Plus; Carl Zeiss) with a 40× objective, a 100× objective with oil immersion, and phase contrast was used. Fluorescent images were acquired using an AxioCam MRc5 camera (Carl Zeiss) and Carl Zeiss AxioVision release 4.6 software. In all experiments, the same exposure time was used. For detection of enhanced green fluorescent protein (EGFP), the filter CLON ZsGreen1 (42002- HQ 470/30×, HQ 520/40m, Q495LP) from Chroma Technology Corporation was used. Image processing was done by using Adobe Photoshop CS2 (Adobe Systems Inc.), with brightness and contrast modified to the exact same settings to obtain comparable images.
RESULTS
Modification of the TM4 capsid gene is deleterious to phage growth.
Initially, we attempted to construct a recombinant derivative of TM4 in which a Strep II tag consisting of 8 amino acids and a 2-residue linker were added to the extreme C terminus of the TM4 capsid protein (gp9). By extrapolating from HK97 structural studies (25, 32), we reasoned that a short addition to the C terminus might be tolerated for capsid assembly and would be exposed on the capsid surface. Our approach was to use the previously described BRED strategy (29, 33) to engineer a modification to TM4 gene 9 (Fig. 2A). Following coelectroporation of TM4 DNA and a 200-bp mutagenic substrate into recombineering M. smegmatis cells, plaques were recovered in an infectious center assay and screened for the presence of the mutant addition. We were successful in detecting the presence of the mutant in pools of plaques (Fig. 2B), although it was present at a low frequency (<1/100 plaques; in other BRED experiments, typically ∼10% of individual plaques are mixed, and they contain both wild-type and mutant alleles [29]). However, even after multiple rounds of plaque purification and screening, we were unable to purify a homogenous mutant derivative. A simple interpretation is that the mutant can be constructed but cannot be purified to homogeneity, because the recombinant protein is not tolerated if it represents all of the 415 subunits in the assembled capsid.
Fig 2.
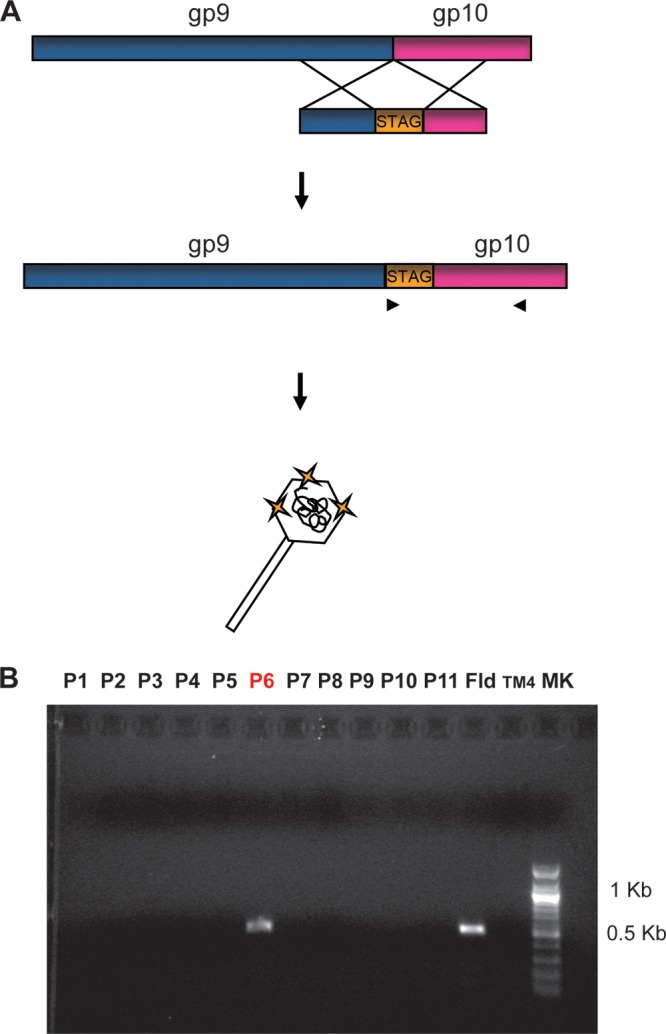
Addition of a STAG to the capsid protein, based on the BRED strategy. (A) Schematic representation of the strategy used to add a STAG to the C terminus of gp9 of TM4 using BRED (see Materials and Methods for details). (B) Screening by PCR for the insertion of the STAG in gp9. Pools of 10 primary plaques (lanes P1 to P11) were screened by PCR, using a forward oligonucleotide that anneals to the STAG II sequence and a reverse oligonucleotide that anneals to the gp10 sequence. In the presence of a positive plaque, the expected product size is 500 bp. Fld, a flooded plate from a BRED experiment, used as a positive control to demonstrate the presence of the phage mutant in the population; TM4, DNA of TM4, used as a negative control for the PCR.
Addition of an affinity tag to the TM4 capsid through mixed assembly.
Because we were able to identify the mutant allele in at least some phage pools, we reasoned that the C-terminal addition could be tolerated if it was present in only a subset of subunits within a particle. We therefore tested the possibility of a mixed assembly, where the recombinant form of the protein is expressed from a plasmid during the process of phage infection and growth and can potentially coassemble with wild-type capsid protein. We constructed a plasmid expression system in which a TM4 gp9-STAG fusion protein (expected size, 33 kDa) was expressed from the inducible pNit promoter (Fig. 3A) (31), and we demonstrated by using anti-STAG antiserum that the protein was well-expressed following induction with ε-caprolactam (Fig. 3B). Interestingly, only an ∼33-kDa protein was observed via SDS-PAGE (Fig. 3C), indicating that while procapsids may assemble, they do not progress to form mature capsids that are covalently cross-linked, as seen in mature TM4 particles (23).
Fig 3.
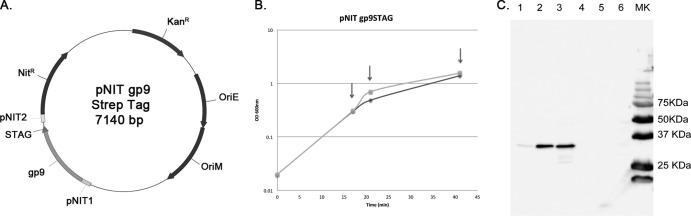
Expression of STAG-tagged TM4 capsid subunit, gp9. (A) Schematic representation of pNIT gp9-STAG. (B) Growth curve of M. smegmatis mc2155(pNIT gp9-STAG). Cells were grown to an OD600 of 0.3 when ε-caprolactam was added. Arrows indicate the different time points when gp9-STAG expression was checked by Western blotting. Diamonds, induced cells; squares, noninduced cells. (C) Expression of gp9-STAG was checked by Western blotting. Lanes 1 to 3, induced cells; lanes 4 to 6, noninduced cells. Samples were removed 1 h (lanes 1 and 4), 3 h (lanes 2 and 5), or 24 h (lanes 3 and 6) after addition or not of the inducer.
Previously, we described construction of a recombinant TM4 derivative carrying a GFP reporter (phAE87::hsp60-EGFP, abbreviated gfpϕ) that yields fluorescent M. smegmatis or M. tuberculosis cells following infection (18). We prepared lysates of gfpϕ on the induced gp9-STAG-expressing strain, and the phage particles were purified and concentrated; the resulting phage preparation is referred to as STAG-gfpϕ. Phage stock titers from gfpϕ (prepared in the control strain) or STAG-gfpϕ (prepared in the gp9-STAG-expressing strain) were comparable, showing that phage production was not compromised in the STAG-gp9-expressing strain and that particles generated were of equivalent infectivity. To determine if the tag was incorporated into the assembled particles, we used anti-STAG antiserum in a Western blot assay of whole phage particles (Fig. 4A). A clear signal was observed, but only at high molecular masses, which presumably corresponded to cross-linked capsomers (Fig. 4A). No signal was observed at a molecular mass (33 kDa), which corresponds to the unassembled gp9 subunit. Thus, gp9-STAG capsid subunits were incorporated as cross-linked subunits into mature virions. The STAG was also clearly detectable by ELISA analysis, and a signal greater than background was readily detected from 109 particles (Fig. 4B). Finally, we could readily detect the STAG in the phage particles by immunoelectron microscopy (Fig. 4C). Because it is difficult to apply these methods quantitatively to such samples, we did not achieve an accurate determination of the proportion of particles containing at least one tag or the average number of STAGs incorporated into each particle.
Fig 4.

Incorporation of an affinity STAG to the phAE87::hsp60-EGFP capsid. The presence of the tag in the capsid of the phage particles was checked by Western blotting (A), ELISA (B), and electroimunomicroscopy (C). (A) In a Western blotting assay, whole phage particles of phAE87::hsp60-EGFP (gfpϕ) (lane 1), phAE87::hsp60-EGFP amplified in M. smegmatis mc2155(pNIT gp9-STAG) (STAG-gfpϕ) (lane 2), or cell extract of M. smegmatis mc2155(pNIT gp9-STAG) (lane 3) were subjected to SDS-PAGE and revealed by chemiluminescence using an HRP-labeled anti-STAG antibody. (B) In the ELISA experiment, dilutions of gfpϕ (lane 1) or STAG-gfpϕ (lane 2) were applied to plates coated with an anti-STAG antibody. From left to right, the number of phage particles adsorbed to the plate was 1 × 1010, 5 × 109, 2.5 × 109, and 1.25 × 109 phage particles. After washing, the capture of the phage particles was checked using an HRP-labeled anti-STAG antibody in a colorimetric assay. (C) Electroimunomicroscopy. Phage particles were adsorbed to a grid and incubated with a rabbit anti-STAG primary antibody and a gold-labeled anti-rabbit secondary antibody. The grid was stained with uranyl acetate, and several fields were examined under an electron microscope. Bar, 100 nm.
Affinity capture of the tagged phages was assayed using magnetic anti-STAG beads (Table 1). PFU values were calculated for initial samples, for supernatants (after removal of the beads), and for washes. Approximately 95% of STAG-gfpϕ particles were captured. Surprisingly, an unexpectedly large proportion (∼80%) of phage particles propagated without STAG-gp9 were also captured, presumably because of Strep-Tactin cross-reactivity with native TM4 proteins. This appears to be specific to TM4, as this was not observed with the unrelated phage D29 (Table 1).
Table 1.
Capture of bacteriophage using Strep-Tactin-coated magnetic beads
| Bacteriophage | Mean ± SD no. of infectious phage particles (PFU)/mla |
% of particles bound | |
|---|---|---|---|
| Initial | Unbound (supernatant and washes) | ||
| D29 | 6.55 × 107 ± 7.07 × 105 | 6.34 × 107 ± 1.48 × 105 | 3.2 ± 1.2 |
| gfpϕ | 4.25 × 108 ± 3.54 × 107 | 8.5 × 107 ± 3.54 × 107 | 80.2 ± 6.6 |
| STAG-gfpϕ | 6.10 × 109 ± 1.41 × 108 | 4.28 × 108 ± 1.59 × 108 | 92.9 ± 2.77 |
Data are means ± standard deviations for 3 experiments and are based on the number of PFU per ml in the initial sample and in the supernatant (the fraction obtained after magnetic removal of the beads) plus washes.
STAG affinity capture of TM4-M. smegmatis complexes.
A potential utility for STAG-TM4 particles is for capture of phage-bacterium complexes from sputum for diagnostic purposes (Fig. 5A). To test whether STAG-gfpϕ particles could be used to capture mycobacterial cells, we prepared a lysate as described above and used this to infect M. smegmatis cells (Fig. 5B). A control lysate of the same phage grown on wild-type M. smegmatis was also used. The same number of cells and MOI were used for further comparison. The phage-bacterium complexes were then fixed and captured using magnetic anti-STAG beads, the beads were collected, and the complexes were eluted and examined by fluorescence microscopy (Fig. 5B). Because we used a high multiplicity of infection (100), fluorescent cells captured with the STAG-gfpϕ particles were readily observed, and these represented about 10% of initial input cells. We were also able to microscopically observe fluorescence by examining the beads directly without elution (Fig. 5C). We have not been able to substantially increase the proportion of captured cells by altering the multiplicity of infection, incubation times, or other reaction conditions, and this may reflect an inherent limitation resulting from relatively poor adsorption, as observed with other mycobacteriophages (34, 35). We also cannot exclude the possibility, although we consider it unlikely, that the virion protein ghost is displaced from the cell surface once DNA injection is complete.
Fig 5.

STAG affinity capture of phage-bacterium complexes. (A) Schematic representation of the protocol used for infection of M. smegmatis with STAG-gfpϕ and recovery of phage-bacterium complexes with Strep-Tactin-coated magnetic beads. (B) Fluorescence micrograph images after elution of phage-bacterium complexes from Strep-Tactin-coated magnetic beads. Cells were infected with STAG-gfpϕ (left) or gfpϕ (right). Magnification, ×400. (C) Fluorescence micrograph (left) and phase-contrast (right) images from Strep-Tactin-coated magnetic beads after recovery of phage-bacterium complexes. Magnification, ×1,000.
DISCUSSION
Addition of affinity tags to bacteriophage particles is useful for a variety of approaches, including antigen delivery for vaccine development (36, 37), capture of phage-bacterium complexes (38), and mechanistic dissection of the process of phage adsorption and infection (39, 40). Nonspecific labeling of the phage particles, such as with addition of fluorescent quantum dots to biotinylated particles, is effective, but a subset of the modifications are likely to specifically interfere with adsorption and DNA injection (40). Phages such as T4 and λ have head decoration proteins (Hoc and Soc for T4 and D for λ) that can be modified, including addition of a fluorescent tag (39), with little or no reduction in infectivity. Although few phage virions have been structurally defined as well as λ and T4 virions have, genome analyses suggest that most phages with siphoviral morphologies likely do not have head decoration proteins and thus cannot be modified simply by using this approach.
Modification of the capsid subunit itself offers an alternative approach. Comparative genomic studies revealed examples in which closely related capsid subunits differed in their lengths as a result of C-terminal extensions (3, 41), suggesting that a C-terminal fusion—especially a small one—would be well tolerated.
A phage display system based on bacteriophage T7 is available from Novagen (T7Select phage display system). This system has the capacity to display peptides up to 50 amino acids long in high copy number (415 copies per particle) and of peptides or proteins of up to 1,200 amino acids in low copy number (0.1 to 1 per particle). The T7 capsid protein is normally made in two forms, 10A (344 aa) and 10B (397 aa), with 10B produced by a translational frameshift of 10A. Thus, 10B contains most of the sequence of 10A, with 52 extra amino acids from the alternate frame added onto the C terminus. In this phage, functional capsids can be composed entirely of either 10A or 10B, or of various ratios of the proteins. Coding sequences for the peptides or proteins to be displayed are cloned in a vector following amino acid 348 of the 10B protein, and because the natural translational frameshift site within the capsid gene has been removed, only a single form of capsid protein is made.
It is thus somewhat surprising that the 10-aa STAG (plus linker) appears not to be tolerated as a C-terminal addition to the TM4 capsid as a genetically homogenous recombinant phage particle. We note that capsid assembly requires a complex and well-coordinated ballet of conformation changes (42), and presumably even this small tag can interfere with capsid assembly when present in all of the subunits. It is plausible that C-terminal capsid fusions are tolerated in other mycobacteriophage genomes, but to our knowledge this is the first time that attempts to do so have been described.
Growth of phage particles on a strain expressing the recombinant capsid-STAG fusion provided opportunities to form mixed particles containing both native and fusion forms of the capsid protein. Capsid assembly with the fusion protein did not generally appear to inhibit phage growth, and recovered phage titers were similar to those propagated on a wild-type strain. Western analysis and immunoelectron microscopy suggested that a substantial proportion of the particles contained at least some STAG-tagged subunits, although precise quantification has proven difficult. Nonetheless, the approach may have broad applicability for addition of tags to other types of phages and phages of other hosts, especially where it is suspected that no head decoration proteins are available. This approach is also attractive because a variety of alternative recombinant phages (such as those containing reporter genes or specific mutations) can be propagated with STAG-labeled capsids by using a single capsid-STAG-expressing strain.
There are a variety of potential applications for STAG-tagged phage particles. One application is for the capture of phage-bacterium complexes in diagnostic use of reporter phages, in which reporter genes, such as firefly luciferase or the gfp gene, are used (17, 18). This could be of particular use in the diagnosis of tuberculosis, where recovery of bacteria from sputum that are competent for phage infection presents a substantial impediment (19). Infection with STAG-tagged reporter phage particles followed by recovery of the complexes provides a plausible solution, although we noted that the efficiency of recovery in the studies reported here was relatively poor. This may reflect observations reported for several mycobacteriophages, including TM4, that adsorption can be relatively inefficient (34, 35, 43). However, at least for some phages, mutants can be isolated with enhanced adsorption, and this might provide a strategy for improving the efficiency of recovery. Many alternative applications and configurations can be envisaged, including attachment of the particles to a solid surface and monitoring the capture and assessment of captured bacterial hosts (44).
ACKNOWLEDGMENTS
This work was supported by the U.S. National Institutes of Health (FIRCA-BB) R03TW008926 to G.H. and M.P. (LMICC), ANPCyT PICT2009-0095, and CONICET PIP 2011-0222 to M.P. L.R. is a doctoral fellow of CONICET (Consejo Nacional de Investigaciones Científicas y Tecnológicas, Argentina).
We thank Robert Duda and Charles Bowman for their help with the inmunoelectron microscopy experiments.
Footnotes
Published ahead of print 12 July 2013
REFERENCES
- 1.Hatfull GF. 1994. Mycobacteriophage L5: a toolbox for tuberculosis. ASM News 60:255–260 [Google Scholar]
- 2.Hatfull GF. 2010. Mycobacteriophages: genes and genomes. Annu. Rev. Microbiol. 64:331–356 [DOI] [PubMed] [Google Scholar]
- 3.Hatfull GF. 2012. The secret lives of mycobacteriophages. Adv. Virus Res. 82:179–288 [DOI] [PubMed] [Google Scholar]
- 4.Hatfull GF. 2012. Complete genome sequences of 138 mycobacteriophages. J. Virol. 86:2382–2384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pedulla ML, Ford ME, Houtz JM, Karthikeyan T, Wadsworth C, Lewis JA, Jacobs-Sera D, Falbo J, Gross J, Pannunzio NR, Brucker W, Kumar V, Kandasamy J, Keenan L, Bardarov S, Kriakov J, Lawrence JG, Jacobs WR, Hendrix RW, Hatfull GF. 2003. Origins of highly mosaic mycobacteriophage genomes. Cell 113:171–182 [DOI] [PubMed] [Google Scholar]
- 6.Bardarov S, Bardarov S, Jr, Pavelka MS, Jr, Sambandamurthy V, Larsen M, Tufariello J, Chan J, Hatfull G, Jacobs WR., Jr 2002. Specialized transduction: an efficient method for generating marked and unmarked targeted gene disruptions in Mycobacterium tuberculosis, M. bovis BCG and M. smegmatis. Microbiology 148:3007–3017 [DOI] [PubMed] [Google Scholar]
- 7.Bardarov S, Kriakov J, Carriere C, Yu S, Vaamonde C, McAdam RA, Bloom BR, Hatfull GF, Jacobs WR., Jr 1997. Conditionally replicating mycobacteriophages: a system for transposon delivery to Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U. S. A. 94:10961–10966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee MH, Pascopella L, Jacobs WR, Jr, Hatfull GF. 1991. Site-specific integration of mycobacteriophage L5: integration-proficient vectors for Mycobacterium smegmatis, Mycobacterium tuberculosis, and bacille Calmette-Guerin. Proc. Natl. Acad. Sci. U. S. A. 88:3111–3115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Morris P, Marinelli LJ, Jacobs-Sera D, Hendrix RW, Hatfull GF. 2008. Genomic characterization of mycobacteriophage Giles: evidence for phage acquisition of host DNA by illegitimate recombination. J. Bacteriol. 190:2172–2182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pham TT, Jacobs-Sera D, Pedulla ML, Hendrix RW, Hatfull GF. 2007. Comparative genomic analysis of mycobacteriophage Tweety: evolutionary insights and construction of compatible site-specific integration vectors for mycobacteria. Microbiology 153:2711–2723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huff J, Czyz A, Landick R, Niederweis M. 2010. Taking phage integration to the next level as a genetic tool for mycobacteria. Gene 468:8–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Freitas-Vieira A, Anes E, Moniz-Pereira J. 1998. The site-specific recombination locus of mycobacteriophage Ms6 determines DNA integration at the tRNAAla gene of Mycobacterium spp. Microbiology 144:3397–3406 [DOI] [PubMed] [Google Scholar]
- 13.Donnelly-Wu MK, Jacobs WR, Jr, Hatfull GF. 1993. Superinfection immunity of mycobacteriophage L5: applications for genetic transformation of mycobacteria. Mol. Microbiol. 7:407–417 [DOI] [PubMed] [Google Scholar]
- 14.Brown KL, Sarkis GJ, Wadsworth C, Hatfull GF. 1997. Transcriptional silencing by the mycobacteriophage L5 repressor. EMBO J. 16:5914–5921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sarkis GJ, Jacobs WR, Jr, Hatfull GF. 1995. L5 luciferase reporter mycobacteriophages: a sensitive tool for the detection and assay of live mycobacteria. Mol. Microbiol. 15:1055–1067 [DOI] [PubMed] [Google Scholar]
- 16.Pearson RE, Jurgensen S, Sarkis GJ, Hatfull GF, Jacobs WR., Jr 1996. Construction of D29 shuttle phasmids and luciferase reporter phages for detection of mycobacteria. Gene 183:129–136 [DOI] [PubMed] [Google Scholar]
- 17.Jacobs WR, Jr, Barletta RG, Udani R, Chan J, Kalkut G, Sosne G, Kieser T, Sarkis GJ, Hatfull GF, Bloom BR. 1993. Rapid assessment of drug susceptibilities of Mycobacterium tuberculosis by means of luciferase reporter phages. Science 260:819–822 [DOI] [PubMed] [Google Scholar]
- 18.Piuri M, Jacobs WR, Jr, Hatfull GF. 2009. Fluoromycobacteriophages for rapid, specific, and sensitive antibiotic susceptibility testing of Mycobacterium tuberculosis. PLoS One 4(3):e4870. 10.1371/journal.pone.0004870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jain P, Hartman TE, Eisenberg N, O'Donnell MR, Kriakov J, Govender K, Makume M, Thaler DS, Hatfull GF, Sturm AW, Larsen MH, Moodley P, Jacobs WR., Jr 2012. ϕ(2)GFP10, a high-intensity fluorophage, enables detection and rapid drug susceptibility testing of Mycobacterium tuberculosis directly from sputum samples. J. Clin. Microbiol. 50:1362–1369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Timme TL, Brennan PJ. 1984. Induction of bacteriophage from members of the Mycobacterium avium, Mycobacterium intracellulare, Mycobacterium scrofulaceum serocomplex. J. Gen. Microbiol. 130:2059–2066 [DOI] [PubMed] [Google Scholar]
- 21.Jacobs WR, Jr, Tuckman M, Bloom BR. 1987. Introduction of foreign DNA into mycobacteria using a shuttle phasmid. Nature 327:532–535 [DOI] [PubMed] [Google Scholar]
- 22.Pope WH, Ferreira CM, Jacobs-Sera D, Benjamin RC, Davis AJ, DeJong RJ, Elgin SCR, Guilfoile FR, Forsyth MH, Harris AD, Harvey SE, Hughes LE, Hynes PM, Jackson AS, Jalal MD, MacMurray EA, Manley CM, McDonough MJ, Mosier JL, Osterbann LJ, Rabinowitz HS, Rhyan CN, Russell DA, Saha MS, Shaffer CD, Simon SE, Sims EF, Tovar IG, Weisser EG, Wertz JT, Weston-Hafer KA, Williamson KE, Zhang B, Cresawn SG, Jain P, Piuri M, Jacobs WR, Jr, Hendrix RW, Hatfull GF. 2011. Cluster K mycobacteriophages: insights into the evolutionary origins of mycobacteriophage TM4. PLoS One 6(10):e26750. 10.1371/journal.pone.0026750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ford ME, Stenstrom C, Hendrix RW, Hatfull GF. 1998. Mycobacteriophage TM4: genome structure and gene expression. Tuberc. Lung Dis. 79:63–73 [DOI] [PubMed] [Google Scholar]
- 24.Rybniker J, Kramme S, Small PL. 2006. Host range of 14 mycobacteriophages in Mycobacterium ulcerans and seven other mycobacteria including Mycobacterium tuberculosis: application for identification and susceptibility testing. J. Med. Microbiol. 55:37–42 [DOI] [PubMed] [Google Scholar]
- 25.Duda RL, Martincic K, Xie Z, Hendrix RW. 1995. Bacteriophage HK97 head assembly. FEMS Microbiol. Rev. 17:41–46 [DOI] [PubMed] [Google Scholar]
- 26.Rao VB, Black LW. 2010. Structure and assembly of bacteriophage T4 head. Virol. J. 7:356. 10.1186/1743-422X-7-356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li Q, Shivachandra SB, Leppla SH, Rao VB. 2006. Bacteriophage T4 capsid: a unique platform for efficient surface assembly of macromolecular complexes. J. Mol. Biol. 363:577–588 [DOI] [PubMed] [Google Scholar]
- 28.Snapper SB, Melton RE, Mustafa S, Kieser T, Jacobs WR., Jr 1990. Isolation and characterization of efficient plasmid transformation mutants of Mycobacterium smegmatis. Mol. Microbiol. 4:1911–1919 [DOI] [PubMed] [Google Scholar]
- 29.Marinelli LJ, Piuri M, Swigonova Z, Balachandran A, Oldfield LM, van Kessel JC, Hatfull GF. 2008. BRED: a simple and powerful tool for constructing mutant and recombinant bacteriophage genomes. PLoS One 3(12):e3957. 10.1371/journal.pone.0003957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.van Kessel JC, Hatfull GF. 2007. Recombineering in Mycobacterium tuberculosis. Nat. Methods 4:147–152 [DOI] [PubMed] [Google Scholar]
- 31.Pandey AK, Raman S, Proff R, Joshi S, Kang CM, Rubin EJ, Husson RN, Sassetti CM. 2009. Nitrile-inducible gene expression in mycobacteria. Tuberculosis (Edinb.) 89:12–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Conway JF, Wikoff WR, Cheng N, Duda RL, Hendrix RW, Johnson JE, Steven AC. 2001. Virus maturation involving large subunit rotations and local refolding. Science 292:744–748 [DOI] [PubMed] [Google Scholar]
- 33.Marinelli LJ, Hatfull GF, Piuri M. 2012. Recombineering: a powerful tool for modification of bacteriophage genomes. Bacteriophage. 2:5–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Barsom EK, Hatfull GF. 1996. Characterization of Mycobacterium smegmatis gene that confers resistance to phages L5 and D29 when overexpressed. Mol. Microbiol. 21:159–170 [DOI] [PubMed] [Google Scholar]
- 35.Jacobs-Sera D, Marinelli LJ, Bowman C, Broussard GW, Guerrero Bustamante C, Boyle MM, Petrova ZO, Dedrick RM, Pope WH, Science Education Alliance Phage Hunters Advancing Genomic and Evolutionary Science Sea-Phages Program, Modlin RL, Hatfull RW, Hatfull GF. 2012. On the nature of mycobacteriophage diversity and host preference. Virology 434:187–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Peachman KK, Li Q, Matyas GR, Shivachandra SB, Lovchik J, Lyons RC, Alving CR, Rao VB, Rao M. 2012. Anthrax vaccine antigen-adjuvant formulations completely protect New Zealand white rabbits against challenge with Bacillus anthracis Ames strain spores. Clin. Vaccine Immunol. 19:11–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sathaliyawala T, Rao M, Maclean DM, Birx DL, Alving CR, Rao VB. 2006. Assembly of human immunodeficiency virus (HIV) antigens on bacteriophage T4: a novel in vitro approach to construct multicomponent HIV vaccines. J. Virol. 80:7688–7698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tolba M, Minikh O, Brovko LY, Evoy S, Griffiths MW. 2010. Oriented immobilization of bacteriophages for biosensor applications. Appl. Environ. Microbiol. 76:528–535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Alvarez LJ, Thomen P, Makushok T, Chatenay D. 2007. Propagation of fluorescent viruses in growing plaques. Biotechnol. Bioeng. 96:615–621 [DOI] [PubMed] [Google Scholar]
- 40.Edgar R, Rokney A, Feeney M, Semsey S, Kessel M, Goldberg MB, Adhya S, Oppenheim AB. 2008. Bacteriophage infection is targeted to cellular poles. Mol. Microbiol. 68:1107–1116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hatfull GF. 2006. Mycobacteriophages, p 602–620 In Calendar R. (ed), The bacteriophages. Oxford University Press, New York, NY [Google Scholar]
- 42.Hendrix RW, Johnson JE. 2012. Bacteriophage HK97 capsid assembly and maturation. Adv. Exp. Med. Biol. 726:351–363 [DOI] [PubMed] [Google Scholar]
- 43.Piuri M, Hatfull GF. 2006. A peptidoglycan hydrolase motif within the mycobacteriophage TM4 tape measure protein promotes efficient infection of stationary phase cells. Mol. Microbiol. 62:1569–1585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bennett AR, Davids FG, Vlahodimou S, Banks JG, Betts RP. 1997. The use of bacteriophage-based systems for the separation and concentration of Salmonella. J. Appl. Microbiol. 83:259–265 [DOI] [PubMed] [Google Scholar]
- 45.Soding J. 2005. Protein homology detection by HMM-HMM comparison. Bioinformatics 21:951–960 [DOI] [PubMed] [Google Scholar]