

Abstract
Hydrophobins are small fungal proteins that are amphiphilic and have a strong tendency to assemble at interfaces. By taking advantage of this property, hydrophobins have been used for a number of applications: as affinity tags in protein purification, for protein immobilization, such as in foam stabilizers, and as dispersion agents for insoluble drug molecules. Here, we used site-directed mutagenesis to gain an understanding of the molecular basis of their properties. We especially focused on the role of charged amino acids in the structure of hydrophobins. For this purpose, fusion proteins consisting of Trichoderma reesei hydrophobin I (HFBI) and the green fluorescent protein (GFP) that contained various combinations of substitutions of charged amino acids (D30, K32, D40, D43, R45, K50) in the HFBI structure were produced. The effects of the introduced mutations on binding, oligomerization, and partitioning were characterized in an aqueous two-phase system. It was found that some substitutions caused better surface binding and reduced oligomerization, while some showed the opposite effects. However, all mutations decreased partitioning in surfactant systems, indicating that the different functions are not directly correlated and that partitioning is dependent on finely tuned properties of hydrophobins. This work shows that not all functions in self-assembly are connected in a predictable way and that a simple surfactant model for hydrophobin function is insufficient.
INTRODUCTION
Hydrophobins are surface-active proteins produced by filamentous fungi. In some cases they are secreted in large amounts into the liquid environment outside the organism, and in other cases they are assembled on structures such as spores or fruiting bodies (1). When secreted they fulfill functions such as forming coatings to allow adhesion on substrates or lowering the surface tension of the surrounding liquid to allow aerial growth modes (2). When assembled on fungal structures, they can also have different roles. In some cases they form protective coatings that for certain pathogenic fungi can act to mask immune recognition (3). It has become clear that hydrophobins can have a multitude of functions but that all of the functions are related in some way to their unique properties as surface-active compounds (4).
The surface adhesive properties and exceptional behavior of hydrophobins at interfaces have inspired a number of different biotechnological applications. For example, the extraordinary stability of hydrophobin foams has led to uses as novel food ingredients (5), and the unique surfactant properties of hydrophobins have led to new solutions for dispersing insoluble compounds in pharmaceutical applications (6). In these applications, the interfacial activity of hydrophobins is a unifying function.
Significantly, many hydrophobins function very well as parts of fusion proteins, where functionally active components are linked by recombinant DNA techniques to the hydrophobins. Examples of such fusion proteins include glucose oxidase (7), laccase (8), growth factors (9), and parts of cellulose enzymes (10), which have led to applications in colloidal materials, sensors, enhancement of enzyme function, and production of cell growth-promoting layers and as tags to purify recombinant fusion proteins by surfactant extraction.
Structural analyses of hydrophobins (11–14) have given significant insight into how hydrophobins function as surface-active and surface-adhering proteins and what confers their special properties. They have a clearly distinguishable hydrophobic patch on the surface that suggests how the hydrophobin can act as an amphiphilic particle-like structure. This type of rigid macromolecular amphiphile can be expected to have interesting properties (15). For example, the hydrophobic patch has a relatively large size (diameter, about 2 nm), which can lead to a very high surface energy compared to that of smaller surfactants (16). The hydrophobic patch consists entirely of aliphatic side chains and comprises about half of all the aliphatic side chains of the hydrophobin molecules.
However, it seems that many of the observed properties of hydrophobins are not easily explained by a simple amphiphile model. Especially noteworthy with the hydrophobins is the very high modulus observed in surface-shear rheological measurements of the surface films that they form (17). Some hydrophobins also show a distinct oligomerization behavior in solution that makes them highly soluble in water (18). At high protein concentrations, tetramers are formed, while at lower concentrations, dimers are formed. It is also clear from atomic force microscopy (AFM) studies that assembled layers of hydrophobins have a very regular structure (19). It is also clear that both structural and functional differences exist between the two main classes of hydrophobins, class I and class II (20). Especially for class II hydrophobins, very little is known about the molecular basis of their function, apart from the above-mentioned observations. In this work, we focus on hydrophobin I (HFBI) from Trichoderma reesei, which belongs to class II. It is also clear that there are significant differences in how individual hydrophobins function.
The importance of hydrophobins for the growth and development of fungi as well as their application potential motivates pursuit of a deeper understanding of the relationship between molecular structure and function within this protein family (21). We address the role of possible intermolecular interactions by investigating features of the hydrophilic region of the protein. The most prominent features of the hydrophilic region are the charged residues that are located in two different areas of the hydrophilic region of the protein (Fig. 1). Accordingly, the molecular basis of hydrophobin function was investigated through mutation of these regions. The proteins were produced as fusion proteins with green fluorescent protein (GFP) in order to facilitate production and analysis of the behavior of these proteins.
Fig 1.
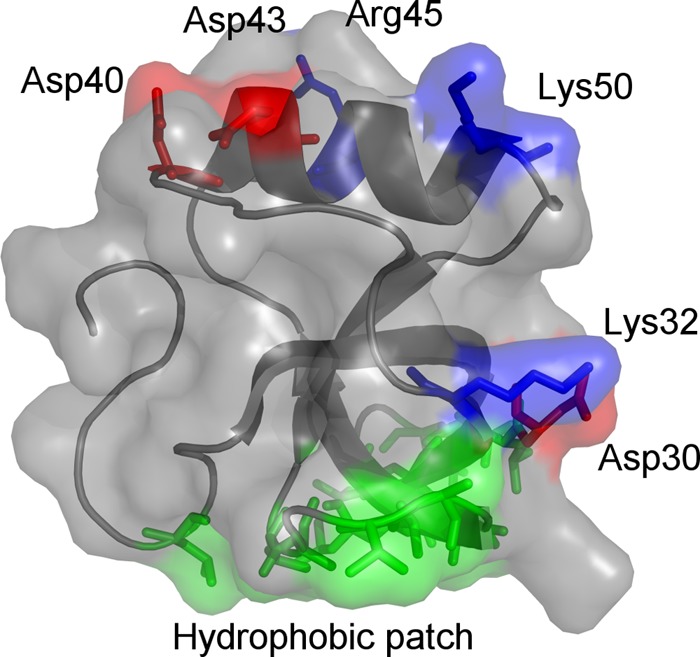
Three-dimensional structure of Trichoderma reesei HHBI (Protein Data Bank accession number 2FZ6). Basic and acidic residues are annotated and colored blue and red, respectively. The protein binds to hydrophobic substrates through the hydrophobic patch (shown in green).
MATERIALS AND METHODS
Site-directed mutations and construction of expression vectors for recombinant protein production.
Plasmids coding for GFP-HFBI variants harboring one to four substitutions of the charged HFBI amino acids Asp30, Lys32, Asp40, Asp43, Arg45, and Lys50 were produced using site-directed mutagenesis. The substitutions were performed with structurally related amino acids with polar side chains; e.g., Arg and Lys were replaced with Gln, while Asp was replaced with Asn, except if a change to Asn would create a glycosylation site, in which case Gln was used instead. Nine variants with the following point mutations were made: D40Q, D43N, R45Q, K50Q, D30N/K32Q, D40Q/D43N, R45Q/K50Q, D40Q/D43N/R45Q/K50Q, and D40Q/D43N/K50Q.
Mutations were introduced into the binary transfer DNA vector pJJJ161 (7) using a QuikChange II XL site-directed mutagenesis kit (Agilent, CA). This vector contains the GFP-HFBI gene, which encodes a 348-amino-acid-long fusion protein, and its expression is controlled by the dual enhancer cauliflower mosaic virus (CaMV) 35S promoter (22), the soybean vegetative storage protein B gene terminator (23), and the tCUP translational enhancer (24). For production as intracellular protein bodies, the Pr1b secretory signal (25) and the KDEL endoplasmic reticulum (ER) retention sequence were included at the 5′ and 3′ termini of the coding region, respectively. All fusion protein variants contained a 12-amino-acid-long Gly-Ser linker and a tobacco etch virus (TEV) protease recognition site joining the GFP and HFBI sequences (molecular mass of fusion protein, 37.8 kDa). In addition to these features, a StrepII tag (26) was included in the fusion protein carboxy terminus as an alternative purification strategy. Introduction of the desired mutations was confirmed by sequencing. The sequence of the fusion protein with the wild-type HFBI is shown in Fig. 2, with the sites for mutations indicated.
Fig 2.

Amino acid sequence of GFP-HFBI fusion protein. From residue 1, enhanced GFP; from residue 241, Gly-Ser linker; from residue 253, TEV protease recognition site; from residue 261, T. reesei HFBI; from residue 335, StrepII affinity tag; and from residue 344, ER retention signal. The sites where mutations were introduced are shown in red.
Protein production.
The transformation of Agrobacterium tumefaciens and infiltration of Nicotiana benthamiana were done essentially as described previously (7). Briefly, the expression constructs were agroinfiltrated into the leaves of 7-week-old N. benthamiana plants in the presence of the gene-silencing suppressor vector p19. The infiltrated leaves were left to absorb the suspension liquid and then moved to a cultivation room for production of the recombinant protein for 7 days. GFP-HFBI production was monitored by fluorescence, and at the end of the cultivation period, fluorescent plant leaves were harvested, ground in liquid nitrogen, and stored frozen as a powder until purification of the contained recombinant protein.
Purification of GFP-HFBI variants by ATPS.
The purification of GFP-HFBI variants by aqueous two-phase separation (ATPS) was performed essentially as described previously (27). Soluble protein was extracted from the leaf cells by adding chilled (4°C) phosphate-buffered saline (PBS) buffer (15 mM sodium phosphate, 150 mM NaCl, pH 7.3) to the frozen leaves (6 ml per 1 g of leaf powder) and ground for about 10 min in a chilled mortar. The insoluble cell debris was removed by centrifugation (20,800 × g, 10 min at 4°C). This step was performed twice, the supernatant was equilibrated to 22°C, and nonionic alkyl polyoxyethylene ether (C12EO5) surfactant (Agrimul 1205 NRE; Henkel, Germany) was added at 4% (wt/vol). The solution was gently mixed for 10 min, incubated at 22°C for 1 h, and then centrifuged for 10 min at 20,800 × g and 22°C for phase separation. The surfactant phase was isolated, and a small amount of PBS buffer (typically, 1:4.5 [vol/vol]) was added. Then, the surfactant was separated from the aqueous components by adding a volume of isobutanol that corresponded to about 10 times the amount of added surfactant. The bottom phase was isolated and applied to a desalting column (Econo-Pac 10DG; Bio-Rad, CA) for exchange of buffer for 50 mM HEPES (pH 7.0). The protein concentration in the elution fractions was determined using a bicinchoninic acid assay (Thermo, MA) and checked for purity by SDS-PAGE. Fusion protein-rich fractions were pooled, and protease inhibitor (cOmplete EDTA-free cocktail; Roche, Switzerland) and EDTA (5 mM) were added for sample stabilization. For control experiments, a GFP fragment was prepared from GFP-HFBI through proteolysis with TEV protease (Eton Bioscience Inc., CA), followed by TEV protease removal by incubation with Ni-nitrilotriacetic acid resin (Qiagen, Germany) at 4°C in 100 mM Tris buffer (pH 8.0) containing 20 mM imidazole. The resin was removed by centrifugation, the supernatant was applied to 1 ml equilibrated Strep-Tactin MacroPrep resin (IBA GmbH, Germany), unbound protein was washed off, and purified GFP fragment was eluted using 2.5 mM desthiobiotin. The GFP fragment was transferred into 50 mM HEPES buffer (pH 7.0) containing 5 mM EDTA and protease inhibitor (cOmplete EDTA-free cocktail; Roche, Switzerland) using a desalting column, as described above.
QCM-D measurements.
The effect of the amino acid substitutions in the HFBI domain of GFP-HFBI on the ability of the fusion protein to bind to hydrophobic surfaces was studied by quartz crystal microbalance (QCM) analysis (D4-QCM system; Biolin Scientific AB, Sweden). The method is a robust and sensitive way to obtain the mass of a layer adsorbed to a surface. In addition, the dissipation monitoring that forms part of the measurements provides information on the viscoelastic properties of the adsorbed layers (28). QCM sensors with a hydrophobic sensor surface were prepared by first cleaning gold-coated QCM sensor disks (model QSX301; Biolin Scientific AB, Sweden) in a UV/ozone chamber for 10 min and immersing them for another 10 min in a 5:1:1 mixture of water, H2O2, and aqueous ammonia solution (25%, wt/wt) at ∼75°C. This was followed by thorough rinsing with water, drying under a stream of N2, and incubation in ethanol (94%, wt/vol) for 2 min. The prewetted sensor disks were immersed in a 50 mM 1-hexanethiol solution in ethanol and left to react at room temperature overnight. As a final step, unreacted 1-hexanethiol was washed off with ethanol, and water and the sensors were dried under a flow of N2. The QCM dissipation (QCM-D) measurements were performed at 23°C, and buffer/sample injection was performed at a rate of 0.1 ml/min. All buffers were degassed by vacuum filtration before use. The adsorption of GFP-HFBI and variants and sensor equilibration were performed at pH 7.0 using 50 mM HEPES buffer by repeated injection of 300 μl protein sample solution containing the different GFP-HFBI variants (0.33 mg/ml).
Partitioning analysis with surfactant ATPS.
For determining the partitioning coefficients of the variants in ATPS, 580 μl protein sample was prepared by dilution in PBS (7.5 mM NaPO4, 75 mM NaCl, pH 7.3) to a final concentration of 0.2 mg/ml. The sample was transferred into a 1.5-ml reaction vial, an 80-μl sample was withdrawn for later analysis, and 20 mg Agrimul surfactant (4% [wt/vol]) was added. The solution was gently mixed for 10 min at room temperature, incubated at 22°C for 140 min, and centrifuged at 3,220 × g for 5 min at 22°C. After centrifugation, a sample (80 μl) was taken from the aqueous phase. From these samples and GFP-HFBI reference samples of known concentration, a series of 1:10, 1:20, 1:40, 1:80, 1:160, 1:320, and 1:640 dilutions was prepared in black microtiter plates (Microfluor 2; Thermo Fisher Scientific, MA) by addition of PBS containing 1% (wt/vol) bovine serum albumin (BSA). The fluorescence of the diluted samples was determined at 485/527 nm using a Victor2 plate reader (PerkinElmer, MA) at a 12-nm bandwidth and with a 100-ms measurement time. The volumes of the aqueous and surfactant phases were determined, and the concentration of the GFP-HFBI variant in the surfactant phase was calculated from the measured fraction volumes and fluorescence intensities in 1:20 sample dilutions. Partitioning coefficients were calculated as the ratio of the protein concentration in the surfactant phase to the protein concentration in the aqueous phase.
AF4.
The solution assembly and size distribution of the GFP-HFBI variants were measured by asymmetric flow field flow fractionation (AF4) using an AF2000 MT instrument (Postnova Analytics GmbH, Germany) equipped with a Fluoroscan Ascent fluorescence spectrophotometer (Thermo Fisher Scientific, MA). With this method, proteins of different sizes were first focused in a flow channel with a flow across the flow channel (perpendicular to the separation flow from the sample inlet to the detector outlet) (29). Then, proteins were separated according to their molecular masses by a velocity gradient, resulting in particle elution in the order of increasing molecular mass. The AF4 analysis was performed using a 1-kDa-cutoff AF2000 focus polyethersulfone membrane (Postnova Analytics GmbH, Germany) at 23°C in 50 mM HEPES buffer (pH 7.0) containing 200 mM NaCl. For sample separation, 21.1 μl sample containing 5.2 μg protein (determined by measurement of the absorption at 280 nm) was focused for 5 min at a flow rate of injection of 0.2 ml/min, a flow rate when focused in the flow channel (ffocus) of 3 ml/min, a flow rate across the channel (fcross) of 3.2 ml/min, and a flow rate for the detector (fdetector) of 0 ml/min and then eluted for 30 min at a flow rate at the tip of 3.09 ml/min, an ffocus of 0 ml/min, an fcross of 3 ml/min, and an fdetector of 0.09 ml/min. GFP or GFP-HFBI variant elution was detected through determination of the fluorescence at 485/538 nm. For calibration, 12.5 μl of a protein standard mix containing 5 μg lysozyme (14.3 kDa), 2.5 μg ovalbumin (44.3 kDa), 2.5 μg bovine serum albumin (66.5 kDa), and 2.5 μg ferritin (440 kDa) was injected. Lysozyme was purchased from AppliChem GmbH (Germany), and the remaining protein standards were obtained from Sigma-Aldrich Chemie GmbH (Germany).
RESULTS AND DISCUSSION
In this work, we wanted to investigate how the hydrophilic region of the HFBI hydrophobin affects its function. The most prominent features of the hydrophilic part are the charged residues Asp30, Lys32, Asp40, Asp43, Arg45, and Lys50 (Fig. 1 and 2). Structurally, these residues are grouped into two regions: Asp30 and Lys32 are closest to the hydrophobic patch, while the remaining four residues are grouped together as a patch on the opposite side of the hydrophobic patch. In this discussion, we refer to the latter region as the charged patch. To probe the function of the residues on the changed patch, we made four variants, with each having one of the mutations D40Q, D43N, R45Q, or K50Q. That is, in these variants each residue was changed to a similarly sized but charge-neutral one. In D43N, N instead of Q was chosen to avoid glycosylation of the site. In addition, we made two variants in which all negative side residues were mutated (variant D40Q/D43N) or all positive ones were mutated (variant R45Q/K50Q). Also, we made one variant in which all the residues in this patch were simultaneously mutated (D40Q/D43N/R45Q/K50Q) and one variant in which residues all except R45 were mutated (D40Q/D43N/K50Q). To probe the role of the charged residues close to the hydrophobic patch, we made one variant in which these were mutated (D30N/K32Q). All proteins were produced as fusion proteins with GFP as a tag in N. benthamiana and purified using the two-phase extraction method.
AF4 was used to characterize the oligomerization of the wild-type and variant HFBI proteins. Oligomerization was chosen as a parameter for understanding hydrophobins because structural (12) and functional (18) data show that solution interactions are characteristic properties, but the exact mechanisms are not completely understood. The AF4 method is a good alternative to size exclusion chromatography analysis, as the absence of a matrix minimizes shear forces, allowing the observation of weak interactions, and also minimizes interaction of the sample with the stationary phase. For assigning molecular masses to retention times, a set of protein standards with known molecular masses was used to make a calibration curve. For the wild type, elution maxima were recorded at 15 and 28 min (Fig. 3). The peak at 15 min corresponded to monomeric GFP-HFBI. This was verified using a control consisting of pure, isolated GFP, prepared by cleaving the fusion protein with TEV. It eluted as a single sharp peak at the same position as the monomeric GFP-HFBI. The variants showed different distributions of oligomers that were in the range of 120 to 300 kDa. To evaluate the effects of the mutations, we analyzed the data by integrating the monomer and the oligomer peaks and comparing their relative sizes. In this way, we obtained a value for the ratio of oligomer to monomer. This value should be seen as a characteristic number and not a true equilibrium constant. That is, during the analysis run time, the equilibrium can potentially change as monomers and lower-molecular-mass oligomers move in the system. Therefore, the equilibrium is constantly changing and the observed ratio value is not necessarily the same as that at equilibrium in solution. These ratios varied widely (x axes in Fig. 4A and B), with the variant D40Q/D43N/K50Q being almost exclusively in the monomeric state and the variant R45Q mostly being in the oligomeric form. We noted that even single mutations greatly affected oligomerization, but it is not clear what the structural background of this effect was. Mutations at the charged patch opposite the hydrophobic patch in some cases increased and in some cases decreased oligomerization. In contrast, mutations near the hydrophobic patch increased oligomerization. As an example of the complex interactions involved, we note the set of single mutation variants R45Q and K50Q and the variant R45Q/K50Q, in which both point mutations were introduced simultaneously. Both single mutations resulted in an increased oligomer-monomer ratio, while combining both mutations in the same molecule resulted in a slightly decreased oligomerization compared to that for the wild type.
Fig 3.
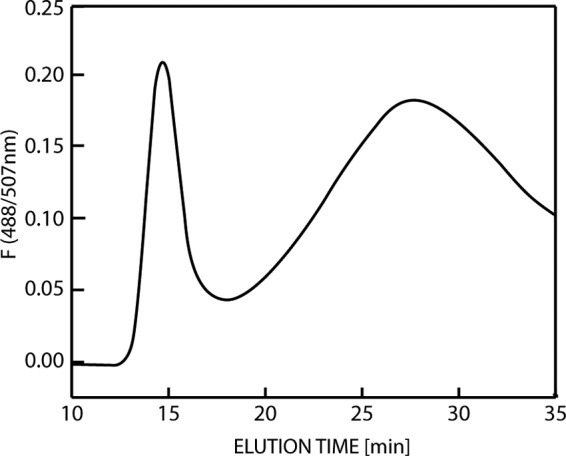
Asymmetric flow field flow fractionation elution profile of GFP-HFBI detected by elution of fluorescent protein (F; 488/507 nm). The first maximum at about 15 min is the monomer, and the second maximum at 28 min corresponds to the oligomeric state.
Fig 4.
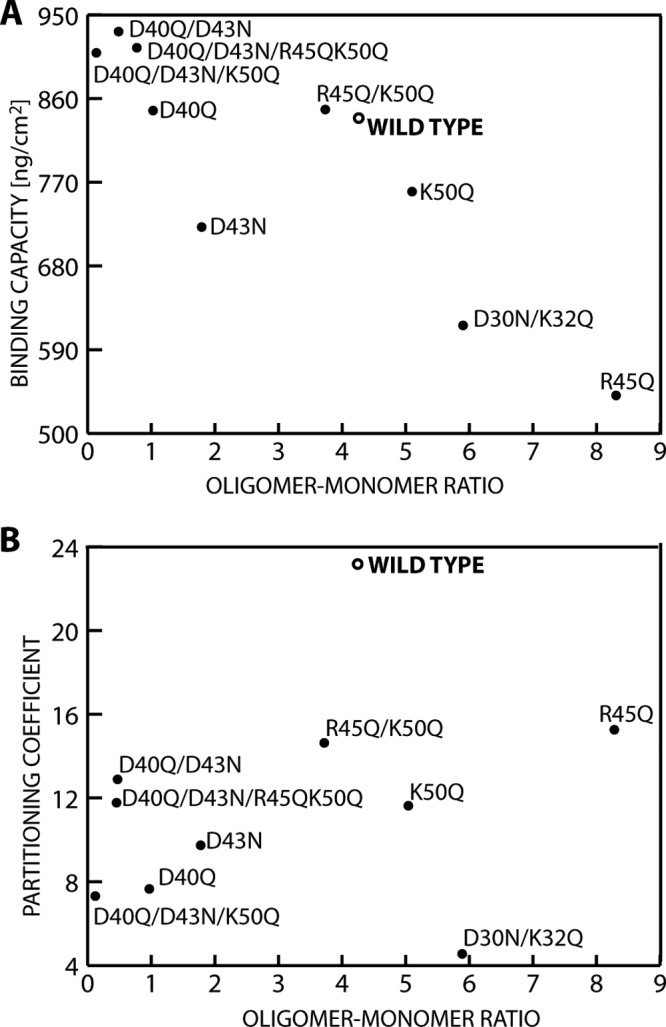
Correlation of GFP-HFBI variant oligomerization to adsorption to a 1-hexanethiol-coated hydrophobic substrate (A) and variant association with a nonionic surfactant in an ATPS (B).
To then evaluate how oligomerization affects other functional characteristics of hydrophobins, we compared the ratio of oligomerization of the variants with two reliably quantifiable properties, surface binding (Fig. 4A) and partitioning in two-phase aqueous surfactant systems (ATPSs) (Fig. 4B). Surface binding was measured as the maximum bound amount of each variant, as measured by QCM using hydrophobic hexanethiol surfaces. Adsorption is a key characteristic of hydrophobins because this property relates strongly to their biological function. A particularly interesting correlation was observed when plotting the oligomer-monomer ratio of wild-type HFBI and variant HFBI versus the density of the adsorbed mass. HFBI variants that bound equally well or somewhat better than the wild-type protein also showed comparatively low oligomer-monomer ratios. However, variants having comparatively higher oligomer-monomer ratios showed progressively lower binding densities. This suggests that even though we do not understand exactly how charged residues affect oligomerization, we can see that the degree of oligomerization clearly correlated with the adhesiveness of hydrophobins. Intuitively, this correlation is understandable if we interpret increased stabilization of oligomers as leading to a better shielding of the hydrophobic patch of the hydrophobin. On the other hand, an impaired ability to form oligomers leads to an increased fraction of monomers that is energetically not favored because it leads to exposure of the hydrophobic patches. This drives the equilibrium toward the bound state, which would increase the tendency of HFBI to bind surfaces (Fig. 5).
Fig 5.

Mutations affecting oligomerization also affect the adhesion of hydrophobins to a hydrophobic surface. The results show that a greater stability of oligomers in solution leads to less binding to surfaces and a lower stability of oligomers in solution leads to more binding to surfaces.
The ATPS method was used to quantify how strongly the proteins interacted with a nonionic surfactant. This characterization is motivated by the fact that hydrophobins partition with uniquely high coefficients in ATPS (10). The uniquely high partitioning makes ATPS separation a distinct property for hydrophobins, even though it remains unclear exactly how hydrophobins interact with the surfactant. It is known that membrane proteins also partition very well in similar systems (30), and this is generally ascribed to the high hydrophobicity of their membrane-spanning domains. The experimental conditions were such that the nonionic surfactant (Agrimul) formed a separate phase. The upper phase mainly consisted of surfactant, and the lower phase was mainly aqueous. Between the phases was a clear border. The protein concentration could be determined in each phase. The partitioning coefficient was then calculated as the ratio of these concentrations (31). Typical soluble proteins partition very poorly into the surfactant phase and therefore have values below 1. We noted that partitioning of all variants to the nonionic surfactant decreased compared to that for the wild type (Fig. 4B). Accordingly, there was no observable correlation between the oligomer-monomer distribution and partitioning. This is interesting, because it implies that there is not a clear correlation between surface adhesion and partitioning in ATPS and, additionally, that the overall increased hydrophobicity of variants in which charged residues had been removed did not improve partitioning. Notably, the variant D30N/K32Q had the lowest partitioning coefficient. Since corresponding side chains are located close to the hydrophobic patch, we hypothesize that in addition to the neighboring hydrophobic amino acids, residues D30 and K32 might contribute to interactions that lead to the characteristic two-dimensional crystalline lattice that hydrophobins show. It is also noteworthy that among the HFBI variants, R45Q had the highest oligomer-to-monomer ratio and the lowest binding to surfaces but the highest partitioning in ATPS. We conclude that the mechanism behind partitioning (i.e., affinity to a hydrophobic fluid interface) is in some way different from the mechanism behind interactions with a solid hydrophobic interface. The fact that all variants were clearly inferior to the wild type in ATPS partitioning indicates that a finely tuned mechanism drives this interaction, whereas general impacts on solubility affect HFBI binding to solid surfaces.
The fact that the HFBI was produced and characterized as a fusion protein naturally has an effect on both the absolute values obtained and the interpretation of the results. The ATPS partitioning coefficient has been shown to be very high with the wild-type HFBI without fusion partners, even over several hundred (32). The values drop markedly when hydrophobins are fused with other proteins, such as to below 25 here. It was shown that this reduction in partitioning most likely is due to size exclusion effects and the increased hydrophilicity of the fusion proteins (10). Similar factors can also probably result in differences for the HFBI oligomerization and surface adhesion. However, in this work all mutations were made in the HFBI molecule itself, and the changes in behavior were therefore based on only the functional differences in the hydrophobins, while the contribution of the GFP fusion was constant. Therefore, we expect that the behavior of the fusion protein reflected the functional differences in HFBI, but in a qualitative way and not in absolute values.
In conclusion, the HFBI function is sensitive to mutations outside the hydrophobic patch. The protein functions are easily affected by mutations but are not affected in a predictable way. Partitioning in ATPS was not related to oligomerization, and it was clear that this characteristic function was very easily disrupted, which implies that finely tuned molecular mechanisms are involved. The data show that it is insufficient to explain the functional properties of hydrophobins only through the hydrophobic patch, as several residues around the hydrophilic regions are clearly involved in the function of the hydrophobin as well.
ACKNOWLEDGMENTS
We thank Päivi Tuomainen and Leena Pitkänen at the University of Helsinki for assistance with AF4 and performing measurements. Mathias Grunér is thanked for helpful discussions. We are grateful to Eero Mustalahti for TEV protease-digested GFP-HFBI. Riitta Suihkonen is thanked for excellent technical assistance.
Financial support from the Academy of Finland (grant 131055) is acknowledged.
Footnotes
Published ahead of print 8 July 2013
REFERENCES
- 1.Linder MB, Szilvay GR, Nakari-Setälä T, Penttilä ME. 2005. Hydrophobins: the protein-amphiphiles of filamentous fungi. FEMS Microbiol. Rev. 29:877–896 [DOI] [PubMed] [Google Scholar]
- 2.Wösten HAB, Willey JM. 2000. Surface-active proteins enable microbial aerial hyphae to grow into the air. Microbiology 146:767–773 [DOI] [PubMed] [Google Scholar]
- 3.Bayry J, Aimanianda V, Guijarro JI, Sunde M, Latgé J-P. 2012. Hydrophobins—unique fungal proteins. PLoS Pathog. 8:e1002700. 10.1371/journal.ppat.1002700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cox PW, Hooley P. 2009. Hydrophobins: new prospects for biotechnology. Fungal Biol. Rev. 23:40–47 [Google Scholar]
- 5.Cox AR, Aldred DL, Russell AB. 2009. Exceptional stability of food foams using class II hydrophobin HFBII. Food Hydrocolloids 23:366–376 [Google Scholar]
- 6.Valo HK, Laaksonen PH, Peltonen LJ, Linder MB, Hirvonen JT, Laaksonen TJ. 2010. Multifunctional hydrophobin: toward functional coatings for drug nanoparticles. ACS Nano 4:1750–1758 [DOI] [PubMed] [Google Scholar]
- 7.Joensuu JJ, Conley AJ, Lienemann M, Brandle JE, Linder MB, Menassa R. 2010. Hydrophobin fusions for high-level transient protein expression and purification in Nicotiana benthamiana. Plant Physiol. 152:622–633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Paananen A, Ercili-Cura D, Saloheimo M, Lantto R, Linder MB. 2013. Directing enzymatic cross-linking activity to the air-water interface by a fusion protein approach. Soft Matter 9:1612. 10.1039/c2sm27234b [DOI] [Google Scholar]
- 9.Scholtmeijer K, Janssen MI, Gerssen B, de Vocht ML, van Leeuwen BM, van Kooten TG, Wösten HAB, Wessels JGH. 2002. Surface modifications created by using engineered hydrophobins. Appl. Environ. Microbiol. 68:1367–1373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Linder MB, Qiao M, Laumen F, Selber K, Hyytiä T, Nakari-Setälä T, Penttilä ME. 2004. Efficient purification of recombinant proteins using hydrophobins as tags in surfactant-based two-phase systems. Biochemistry 43:11873–11882 [DOI] [PubMed] [Google Scholar]
- 11.Hakanpää J, Linder M, Popov A, Schmidt A, Rouvinen J. 2006. Hydrophobin HFBII in detail: ultrahigh-resolution structure at 0.75Å. Acta Crystallogr. D Biol. Crystallogr. 62:356–367 [DOI] [PubMed] [Google Scholar]
- 12.Kallio JM, Linder MB, Rouvinen J. 2007. Crystal structures of hydrophobin HFBII in the presence of detergent implicate the formation of fibrils and monolayer films. J. Biol. Chem. 282:28733–28739 [DOI] [PubMed] [Google Scholar]
- 13.Hakanpaa J, Paananen A, Askolin S, Nakari-Setälä T, Parkkinen T, Penttilä M, Linder MB, Rouvinen J. 2004. Atomic resolution structure of the HFBII hydrophobin, a self-assembling amphiphile. J. Biol. Chem. 279:534–539 [DOI] [PubMed] [Google Scholar]
- 14.Kwan A, Winefield RD, Sunde M, Matthews JM, Haverkamp RG, Templeton MD, Mackay JP. 2006. Structural basis for rodlet assembly in fungal hydrophobins. Proc. Natl. Acad. Sci. U. S. A. 103:3621–3626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Walther A, Müller AHE. 2013. Janus particles: synthesis, self-assembly, physical properties, and applications. Chem. Rev. 113:5194–5261 [DOI] [PubMed] [Google Scholar]
- 16.Chandler D. 2005. Interfaces and the driving force of hydrophobic assembly. Nature 437:640–647 [DOI] [PubMed] [Google Scholar]
- 17.Blijdenstein TBJ, de Groot PWN, Stoyanov SD. 2010. On the link between foam coarsening and surface rheology: why hydrophobins are so different. Soft Matter 6:1799. 10.1039/b925648b [DOI] [Google Scholar]
- 18.Szilvay GR, Kisko K, Serimaa R, Linder MB. 2007. The relation between solution association and surface activity of the hydrophobin HFBI from Trichoderma reesei. FEBS Lett. 581:2721–2726 [DOI] [PubMed] [Google Scholar]
- 19.Szilvay GR, Paananen A, Laurikainen K, Vuorimaa E, Lemmetyinen H, Peltonen J, Linder MB. 2007. Self-assembled hydrophobin protein films at the air-water interface: structural analysis and molecular engineering. Biochemistry 46:2345–2354 [DOI] [PubMed] [Google Scholar]
- 20.Wessels JG. 1997. Hydrophobins: proteins that change the nature of the fungal surface. Adv. Microb. Physiol. 38:1–45 [DOI] [PubMed] [Google Scholar]
- 21.Sunde M, Kwan AHY, Templeton MD, Beever RE, Mackay JP. 2008. Structural analysis of hydrophobins. Micron 39:773–784 [DOI] [PubMed] [Google Scholar]
- 22.Kay R, Chan A, Daly M, McPherson J. 1987. Duplication of CaMV 35S promoter sequences creates a strong enhancer for plant genes. Science 236:1299–1302 [DOI] [PubMed] [Google Scholar]
- 23.Mason HS, Guerrero FD, Boyer JS, Mullet JE. 1988. Proteins homologous to leaf glycoproteins are abundant in stems of dark-grown soybean seedlings. Analysis of proteins and cDNAs. Plant Mol. Biol. 11:845–856 [DOI] [PubMed] [Google Scholar]
- 24.Wu K, Malik K, Tian L, Hu M, Martin T, Foster E, Brown D, Miki B. 2001. Enhancers and core promoter elements are essential for the activity of a cryptic gene activation sequence from tobacco, tCUP. Mol. Genet. Genomics 265:763–770 [DOI] [PubMed] [Google Scholar]
- 25.Cutt J, Dixon D, Carr J, Klessig D. 1988. Isolation and nucleotide-sequence of cDNA clones for the pathogenesis-related proteins Pr1a, Pr1b and Pr1c of Nicotiana tabacum cv. Xanthi nc induced by TMV infection. Nucleic Acids Res. 16:9861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schmidt TG, Koepke J, Frank R, Skerra A. 1996. Molecular interaction between the Strep-tag affinity peptide and its cognate target, streptavidin. J. Mol. Biol. 255:753–766 [DOI] [PubMed] [Google Scholar]
- 27.Joensuu JJ, Conley AJ, Linder MB, Menassa R. 2012. Bioseparation of recombinant proteins from plant extract with hydrophobin fusion technology. Methods Mol. Biol. 824:527–534 [DOI] [PubMed] [Google Scholar]
- 28.Marx KA. 2003. Quartz crystal microbalance: a useful tool for studying thin polymer films and complex biomolecular systems at the solution-surface interface. Biomacromolecules 4:1099–1120 [DOI] [PubMed] [Google Scholar]
- 29.Roda B, Zattoni A, Reschiglian P, Moon MH, Mirasoli M, Michelini E, Roda A. 2009. Field-flow fractionation in bioanalysis: a review of recent trends. Anal. Chim. Acta 635:132–143 [DOI] [PubMed] [Google Scholar]
- 30.Bordier C. 1981. Phase separation of integral membrane proteins in Triton X-114 solution. J. Biol. Chem. 256:1604–1607 [PubMed] [Google Scholar]
- 31.Sivars U, Tjerneld F. 2000. Mechanisms of phase behaviour and protein partitioning in detergent/polymer aqueous two-phase systems for purification of integral membrane proteins. Biochim. Biophys. Acta 1474:133–146 [DOI] [PubMed] [Google Scholar]
- 32.Linder M, Selber K, Nakari-Setälä T, Qiao M, Kula M-R, Penttilä M. 2001. The hydrophobins HFBI and HFBII from Trichoderma reesei showing efficient interactions with nonionic surfactants in aqueous two-phase systems. Biomacromolecules 2:511–517 [DOI] [PubMed] [Google Scholar]