

Abstract
Two-day-old (P2), but not 9-day-old (P9), rat pups are susceptible to systemic infection following gastrointestinal colonization by Escherichia coli K1. Age dependency reflects the capacity of colonizing K1 to translocate from gastrointestinal (GI) tract to blood. A complex GI microbiota developed by P2, showed little variation over P2 to P9, and did not prevent stable K1 colonization. Substantial developmental expression was observed over P2 to P9, including upregulation of genes encoding components of the small intestinal (α-defensins Defa24 and Defa-rs1) and colonic (trefoil factor Tff2) mucus barrier. K1 colonization modulated expression of these peptides: developmental expression of Tff2 was dysregulated in P2 tissues and was accompanied by a decrease in mucin Muc2. Conversely, α-defensin genes were upregulated in P9 tissues. We propose that incomplete development of the mucus barrier during early neonatal life and the capacity of colonizing K1 to interfere with mucus barrier maturation provide opportunities for neuropathogen translocation into the bloodstream.
INTRODUCTION
The newborn infant is particularly vulnerable to systemic bacterial infection during the first 4 weeks of life, and mortality and morbidity associated with neonatal bacterial meningitis (NBM) and accompanying sepsis remain significant despite advances in antibacterial chemotherapy and supportive care (1, 2). In the developed world, Escherichia coli and group B streptococci are responsible for the majority of cases of NBM, and bacteria isolated from the cerebrospinal fluid of infected neonates invariably elaborate a protective polysaccharide capsule. Of neuroinvasive E. coli isolates, 80 to 85% express the K1 capsule (3, 4), a homopolymer of α-2,8-linked polysialic acid (polySia) that mimics the molecular structure of the polySia modulator of neuronal plasticity in mammalian hosts (5) and enables these strains to evade detection by a neonatal innate immune system undergoing a process of age-dependent maturation (6).
Risk factors for NBM include obstetric and perinatal complications, premature birth, and low birth weight, particularly in low socioeconomic groups (7), but predisposition to infection is critically dependent on vertical transmission of the causative agent from mother to infant at or soon after birth (8). Although many aspects of the pathogenesis of E. coli K1 in NBM are unclear, maternally derived E. coli K1 bacteria are known to colonize the neonatal gastrointestinal (GI) tract (8, 9, 10), which is sterile at birth but rapidly acquires a complex microbiota that eventually converges toward a profile characteristic of the adult GI tract (11). E. coli K1 bacteria then translocate from the lumen of the small intestine or colon into the systemic circulation before entering the central nervous system (CNS) across the blood-brain barrier at the cerebral microvascular endothelium of the arachnoid membrane (12) or the blood-cerebrospinal fluid (CSF) barrier at the choroid plexus epithelium (13).
Many of the temporal and spatial aspects of NBM can be reproduced in a rodent model of E. coli K1 infection initially developed by Glode et al. (14) and subsequently refined by others (15, 16). Thus, oral (15, 16, 17) or intragastric (14, 18) administration of E. coli K1 results in stable and persistent GI colonization of adults and neonates. E. coli K1-colonized neonatal rat pups, but not adult animals, subsequently develop lethal systemic infection, with E. coli K1 present in the blood circulation and brain tissue (15, 19, 20). Persistence of bacteria in the blood is dependent on the continued expression of the polySia capsule, as evidenced by the inability of capsule-defective mutants to cause systemic infection (21) and by the capacity of intraperitoneally delivered capsule-selective depolymerase to abrogate infection (16). Bacteria enter the CSF compartment of infected rat pups predominantly at the choroid plexus and penetrate superficial brain tissue (19), where they induce inflammation via proinflammatory cytokine-induced pathways involving interleukin-1β (IL-1β), IL-6, and tumor necrosis factor α (TNF-α) (20).
The experimental rodent model of infection has yielded fresh insights into the transit of the E. coli K1 neuropathogen from the blood circulation into the CNS; in particular, the age dependency of experimental NBM in rodents is striking, with clear evidence of systemic infection at 2 days of age. We now employ this model to shed light on the mechanism of bacterial translocation from the GI tract to the blood compartment. As it is unlikely that conventional prophylactic measures such as vaccination can be readily implemented to prevent infection in the at-risk neonatal cohort—the poor immunogenicity of polySia, the relative unresponsiveness of the neonatal immune system, lack of IgA-mediated protection of mucosal surfaces, and the age profile of the target patient population mitigate against the successful development of vaccination as a preventative strategy for the control of E. coli NBM (22, 23)—a more detailed understanding of the early stages of NBM will underpin alternative approaches to the prevention of E. coli K1 systemic neonatal infection, such as bacteriophage-mediated selective elimination of the E. coli K1 bacterial cohort from maternal or neonatal reservoirs, and forms the basis of the study reported here.
The preterm rodent GI tract is sterile but becomes colonized immediately after birth (24). Little is known of the dynamics of colonization of the rat GI tract by maternally derived microbes, but any lag in the development of a mature microbiota may allow colonizing E. coli K1 to achieve sufficient density in the colon or small intestine to permit entry into the blood circulation during the first few days of life. K1 bacteria transit to the blood from the GI tract via the mesenteric lymph nodes, and there is compelling evidence that they do so with a very low frequency (15). In the study reported here, we found no evidence that the development of a mature microbiota in neonatal rat pups influenced the size or location within the GI tract of the colonizing E. coli K1 population, and it is unlikely to be responsible for the strong age dependency of experimental NBM in rodents. There were, however, differences in the appearance of the GI tract between susceptible and nonsusceptible animals; tissues of younger, susceptible pups contained fewer goblet cells, and the intestinal mucus layer was less developed in comparison to those of older, resistant animals. We also identified major differences in the capacity of susceptible and nonsusceptible neonatal pups to respond to GI tract colonization by E. coli K1, potentially enabling a closer association of E. coli K1 with the epithelial surface in susceptible compared to nonsusceptible pups. Furthermore, susceptible rat pups, in contrast to nonsusceptible animals, were unable to respond to E. coli K1 colonization by increasing the concentrations of the defensin peptides Defa-rs1 and Defa24 in the lumen of the small intestine. These changes were not evident following GI tract colonization by nonpathogenic E. coli K-12. We propose a model based on these observations to account for the strong age dependency of E. coli K1 NBM.
MATERIALS AND METHODS
Bacterial strains and bacterial colonization of neonatal rat pups.
E. coli O18:K1 strain A192PP was derived from E. coli A192, an isolate from a patient with septicemia (25), by serial passage through neonatal rat pups as described previously (16, 19); the passaged strain was significantly more virulent for rat pups than E. coli A192 (17). E. coli K-12 was obtained from the Coli Genetic Stock Center, Yale University as CGSC5073 (K-12 wild type). Wistar rat litters (9 to 14 individuals), obtained from Harlan UK, were retained in a single cage with their natural mothers after birth. For GI tract colonization, all members of a litter were fed 20 μl of mid-exponential-phase Mueller-Hinton (MH) broth culture of E. coli K1 (2 × 106 to 6 × 106 CFU, warmed to 37°C) from an Eppendorf micropipette. Colonization was determined by MacConkey agar culture of perianal swabs as described earlier (16). Bacteremia was detected by culture on MacConkey agar of daily blood samples taken from superficial veins in the footpad. Animal experiments were approved by the Ethical Committee of the UCL School of Pharmacy and the UK Home Office (HO) and were conducted under HO Project license PPL 80/2243.
Processing of animal material.
Stools were collected from adult animals, and neonates were killed by decapitation; postmortem, the neonatal GI tract, from stomach to colon, was excised aseptically. For E. coli K1 enumeration and DNA extraction, samples were transferred to 2 ml ice-cold phosphate-buffered saline (PBS) and homogenized. For enumeration, the homogenate was serially diluted in PBS and plated onto MacConkey agar. Coliform colonies were restreaked and assayed for sensitivity to the K1-specific lytic phage K1E (26). DNA was extracted from homogenates using the QIAamp stool DNA minikit (Qiagen). For RNA extraction, tissues were transferred to RNAlater (Ambion) and stored overnight at 4°C. RNA was then extracted using the RNeasy Midi kit (Qiagen). Nucleic acid extractions were quantified and assessed for purity using a NanoDrop 1000 spectrophotometer (Thermo Scientific) and native DNA/RNA agarose gel electrophoresis. For protein extraction, tissues were homogenized in 2 ml tissue lysis buffer containing 1% (vol/vol) NP-40, 1% (vol/vol) Tween 20, and Tris-EDTA (10 mM Tris-HCl, 1 mM EDTA [pH 7.4]) in PBS supplemented with 1× Complete mini-EDTA-free protease inhibitor cocktail (Roche). Protein extracts were quantified using Bio-Rad protein assay reagent in a cuvette format. All homogenizations were performed on ice using an Ultra-Turrax T-10 homogenizer (IKA Werke); extracts were stored at −80°C prior to analysis.
GI microbiota profiles.
Analysis of the composition of complex microbial communities has been facilitated by an expanding database of small subunit (SSU) rRNA gene sequences. Palmer et al. (11, 27) designed a microarray-based platform incorporating probes which target the more variable SSU rRNA gene regions; the customized microarray (Agilent Technologies) used in the present study consisted of 10,266 unique oligonucleotide sequences covering 1,590 bacterial and 39 archeal species as well as 649 higher-order taxonomic groups, which together ensure broad coverage of known SSU rRNA gene sequences within the Prokaryotic Multiple Sequence (prokMSA) database (http://greengenes.lbl.gov/cgi-bin/nph-index.cgi). The platform can be used to quantify different SSU rRNA gene populations from mixed samples using standard microarray technology. DNA was extracted from three stool samples for each adult and three GI tissue samples per litter for each neonatal age group examined using the QIAamp stool DNA minikit. The SSU rRNA gene was amplified from 1 μg extracted DNA using the GoTaq Green PCR kit (Promega) and the broad-range bacterium-specific primers Bact-8F and T7-1391R described by Palmer et al. (11). Amplified DNA was quantified using a NanoDrop 1000 spectrophotometer, and the products were confirmed by agarose gel electrophoresis. A reference pool was constructed from an equimolar mixture of all samples for use as a template for in vitro transcription. Reference pool amplicons were labeled with Cy3, and test samples comprising amplified DNA from animals in individual age groups were labeled with Cy5 using the genomic DNA (gDNA) labeling kit Plus (Agilent Technologies); combined Cy3/5-labeled samples were purified using MinElute columns (Qiagen). For hybridization, 4.5 μl blocking agent and 22.5 μl hybridization buffer from the Hyb kit (Agilent Technologies) were added to 18 μl of eluted DNA. After 3 min at 95°C and 30 min at 37°C, the 45-μl sample was loaded into a hybridization chamber (Agilent Technologies), and the arrays were hybridized at 65°C in a rotating oven for 24 h. Slides were washed in Oligo aCGH wash buffers (Agilent Technologies) according to the manufacturer's instructions. Microarrays were scanned using an Agilent high-resolution C scanner at a 5-μm resolution with the extended dynamic range settings at 100 and 10. Microarray images were processed using feature extraction software v9.5.1.1 (Agilent Technologies) utilizing linear normalization and rank-consistent probe dye normalization methods. Data were processed using GeneSpring GX v7.3.1 (Agilent Technologies) to combine data from replicate spots and multiple node/species reporters. Test samples were normalized to adult control samples to give an estimate of relative abundance.
Histology and immunohistochemistry.
Segments of proximal, mid-, and distal small intestine were removed from P2 and P9 pups without washing; segments of whole colon were removed from P2 animals, and the colons from P9 pups were divided into proximal and distal portions, again without washing. Tissues were placed in methanol-Carnoy's fixative and maintained at room temperature for at least 3 h. Paraffin-embedded sections were dewaxed and hydrated; mucin was visualized using anti-MUC2C3 antiserum with Alexa 546-conjugated rabbit immunoglobulins (Life Technologies). Immunostained sections were mounted in ProLong antifade (Life Technologies). Images were obtained at room temperature using a Nikon Eclipse E1000 fluorescence microscope with a Plan-Fluor 20×/0.50 differential interference contrast (DIC) M or 10×/0.30 DIC L objective (Nikon). Images were acquired with a Nikon digital sight DS5M camera and the Nikon Eclipse E1000 control software. Further processing was performed with Adobe Photoshop software. All image adjustments were performed equally for all comparably stained images.
qPCR.
Quantitative PCR (qPCR) was used to amplify and quantify DNA from the bacterial components of the GI tract microbiota; a qPCR assay was developed to enumerate E. coli K1 in the GI tract. The bacterial load was quantified by amplification of 16S rRNA genes essentially according to Palmer et al. (11). Briefly, universal forward primer 8FM (0.9 μM) and Bifidobacterium longum forward primer 8FB (0.09 μM) were used in conjunction with universal reverse primer Bact515R (0.9 μM). The thermal cycling program comprised 95°C for 3 min followed by 40 cycles of 95°C for 20 s, 55°C for 20 s, 60°C for 35 s, 65°C for 15 s, and 72°C for 15 s. E. coli K1 was quantified using a method validated in this study (see the supplemental material), by amplification of the K1-specific gene neuS (28). Forward primer neuSF3 (0.625 μM) was used in conjunction with reverse primer neuSR3 (0.625 μM). The E. coli K1 thermal cycling program comprised 95°C for 3 min followed by 40 cycles of 95°C for 20 s and 61°C for 20 s. In both assays, each 20-μl reaction mixture comprised 1× Brilliant III SYBR green Ultra-Fast qPCR master mix (Agilent), the assay-specific primers, ROX reference dye (30 nM), and 5 μl extracted DNA. Reactions were performed using an Mx3000P instrument (Stratagene), and fluorescence data for SYBR1 and ROX were acquired at the annealing step of each cycle. Tenfold serial dilutions of triplicate genomic DNA extractions from known quantities of E. coli strains A192PP (for the E. coli K1 assay) and CGSC5073 (for assay of total bacteria) were used to generate standard curves for each qPCR plate. The abundance of neuS or 16S rRNA genes was calculated based on these standard curves using Mx3000P v2.0 software (Stratagene) to normalize SYBR1 to ROX fluorescence and to determine cycle threshold values using adaptive baseline and amplification-based threshold algorithm enhancements. qPCR plates were run in duplicate and contained duplicate standard, sample, and no-template control reaction mixtures.
Semiquantitative RT-PCR.
cDNA was amplified from RNA extracts by one-step reverse transcriptase (RT)-PCR and amplicons resolved by agarose gel electrophoresis. RT-PCRs were prepared in a class II biological safety cabinet to reduce the risk of contamination. RNA (50 ng) was mixed with Brilliant II RT-PCR master mix (Agilent), gene-specific forward and reverse primer pairs (0.5 μM each), and AffinityScript RT-RNase block enzyme mixture (Agilent) to a final reaction volume of 25 μl. Control reaction mixtures without RNA template or RT-RNase block enzyme were also prepared. RT-PCR was performed using a thermocycling program of 30 min at 50°C and 10 min at 95°C followed by 35 cycles of 30s at 95°C, 1 min at 60°C, and 30 s at 72°C. Reaction mixtures were mixed with 5 μl of 6× gel loading buffer (0.05% [wt/vol] bromophenol blue, 40%, [wt/vol] sucrose, 0.1 M [pH 8] EDTA, 0.5% [wt/vol] SDS) and resolved by agarose gel electrophoresis. Gels were visualized using a Molecular Imager FX system (Bio-Rad) set to detect UV fluorescence.
Gene expression analysis.
RNA was extracted from GI tissues using the Qiagen RNeasy animal tissue Midi kit (Qiagen). RNA integrity and concentration were determined with a NanoDrop 1000 spectrophotometer (Thermo Scientific). Labeled cDNA was hybridized to Affymetrix GeneChip rat genome 230 2.0 arrays. The arrays comprised over 31,000 probe sets representing variants from greater than 28,000 rat genes; there are 11 probe sets represented for each coding sequence. GeneChip expression analysis was performed using samples extracted from uninfected rat pups of age comparable to that of the reference control. Labeled cDNA synthesis, fragmentation, hybridization, washing, and scanning of rat genome arrays were performed according to the Affymetrix GeneChip expression analysis technical manual (702232; revision 2). Hybridizations were incubated for 16 h at 45°C. GeneChips were scanned using Affymetrix Gene Array Scanner 3000. Hybridization data were analyzed on GeneChip Operating Software (GCOS v1.4). DAT, CEL, and CHP files were removed using the Data Transfer Tool (v1.1.1); CEL files were imported into the third-party data analysis software GeneSpring v7.3 (Agilent Technologies). GC robust multiarray averaging (GC-RMA) normalization was performed on all data generated using a chip from a comparable uninfected rat pup as the reference.
Competitive ELISA for Tff2.
For the enzyme-linked immunosorbent assay (ELISA) for Tff2, goat polyclonal anti-Tff2 primary antibody (sc-23558; Santa Cruz Biotechnology) was biotinylated, diluted 1:1,000 in blocking buffer (1%, [wt/vol] casein in Tris-buffered saline), and dispensed in 100-μl aliquots, and 10 μg of tissue protein extract was added. Serial 2-fold dilutions of a 15-ng/ml PBS solution of recombinant human Tff2 (rhTff2; Sigma-Aldrich) were prepared, and 100 μl of each dilution was added to individual tubes to give a standard range of 1,500 to 23.4 pg rhTff2. Control tubes containing only anti-Tff2 antibody were also prepared. Standard-antibody, sample-antibody, and control tubes were incubated with rotation for 6 h at 20°C. Separately, aliquots (0.1 μg in 100 μl bicarbonate-carbonate buffer [pH 9.6]) of rhTff2 were transferred to each well of 96-well Maxisorp Immunoplates (Nunc) and incubated with rotation at 20°C for 2 h, the buffer was aspirated, and the wells were washed twice with 0.05% (vol/vol) Tween 20 in PBS. Wells were blocked with 350 μl of blocking buffer and incubated for 2 h, the buffer was aspirated, and the wells were washed twice.
Standard-antibody, sample-antibody, and control solutions were applied to individual wells, and the plates were incubated for 16 h at 20°C. Antibody solutions were aspirated, and the wells were washed four times. Horseradish peroxidase (HRP)-streptavidin conjugate (0.5 μg in 100 μl PBS; Vector Laboratories) was applied to each well, the plates were incubated for 1 h, supernatants were removed, and the wells were washed four times. The plates were developed by addition of 100 μl of 3,3′,5,5′-tetramethylbenzidine liquid substrate and incubated in the dark for 15 to 30 min. Color development was terminated by the addition of 100 μl 0.4 M sulfuric acid. The optical density at 450 nm (OD450) was measured using a SPECTROstar Omega plate reader (BMG Labtech), and Tff2 was quantified by comparison to standard curves obtained by plotting the OD450 values against the amount of rhTff2 incubated with anti-Tff2 IgG. All standard, sample, and control wells were run in triplicate on each plate, and each plate was duplicated.
Serum cytokine assays.
Serum was obtained by centrifugation (1,500 × g, 10 min) of blood collected from culled neonatal rats. Levels of IL-1β and IL-6 were determined by sandwich ELISAs, utilizing, respectively, rabbit or goat polyclonal cytokine-specific capture and biotinylated detection antibodies and recombinant rat cytokine standards from an appropriate ELISA development kit (Peprotech). Reactions were developed by incubation with streptavidin-HRP conjugate (Vector Laboratories) for 30 min followed by incubation with 3,3′,5,5′-tetramethylbenzidine. Reactions were terminated after 15 min by addition of 0.5 M H2SO4, and A450 was measured using a SPECTROstar plate reader (BMG Labtech). All assays were performed in triplicate in 96-well MaxiSorp immunoplates (Nunc).
Extraction of tissue cell nuclear proteins.
Prior to extraction of nuclear proteins, single-cell suspensions were obtained from whole tissue samples. Tissues were washed in 2 ml ice-cold PBS and then transferred to 4.7 ml HEPES buffer (10 mM HEPES, 150 mM NaCl, 5 mM KCl, 1 mM MgCl2, 1 mM CaCl2) supplemented with collagenase (2 mg/ml) and DNase I (80 U/ml). Samples were homogenized for 10 s on ice, incubated at 37°C for 30 min, and homogenized for 10 s. Homogenates were filtered (100-μm cell strainer; BD Falcon), and tissue cells were recovered by centrifugation (500 × g, 10 min). Cell pellets were suspended in 5 ml nuclear extraction buffer (0.32 M sucrose, 10 mM Tris-HCl, 3 mM CaCl2, 2 mM MgOAc, 0.1 mM EDTA, 1 mM dithiothreitol [DTT]) supplemented with 0.5% (vol/vol) NP-40 to lyse the plasma membrane, followed by centrifugation (500 × g, 5 min) to obtain nuclei. After aspiration of the cytoplasmic protein fraction, nuclei were washed twice in nuclear extraction buffer and recovered by centrifugation (500 × g, 5 min). Nuclei were suspended in 1.5 ml low-salt buffer (20 mM HEPES, 1.5 mM MgCl2, 20 mM KCl, 0.2 mM EDTA, 25% [vol/vol] glycerol, 0.5 mM DTT) and lysed by the gradual addition of an equal volume of high-salt buffer (20 mM HEPES, 1.5 mM MgCl2, 800 mM KCl, 0.2 mM EDTA, 25% [vol/vol] glycerol, 0.5 mM DTT, 1% [vol/vol] NP-40). Samples were incubated for 45 min at 4°C on a rotator and then centrifuged (14,000 × g, 15 min). The nuclear protein fraction was aspirated and stored at −80°C prior to analysis. All nuclear protein extraction buffers were supplemented with 1× Complete mini-EDTA-free protease inhibitor cocktail (Roche).
NF-κB electrophoretic mobility shift assay.
A fluorophore-labeled double-stranded oligonucleotide probe containing the NF-κB consensus binding site was prepared by annealing equimolar volumes of 5′-Cy5-conjugated sense and antisense single-stranded oligonucleotides at room temperature for 10 min. Binding reaction mixtures were prepared from 1 μl poly(dIdC) (1 mg/ml), 3 μl 5× binding buffer (50 mM Tris-HCl, 750 mM KCl, 2.5 mM EDTA, 0.5% [vol/vol] Triton X-100, 62.5% [vol/vol] glycerol, 1 mM DTT), 5 μg nuclear protein extract, 1 μl labeled probe (10 ng/μl), and double-distilled water (ddH2O) in a total volume of 15 μl. Reaction mixtures were incubated at room temperature for 30 min prior to electrophoresis on 5% (vol/vol) polyacrylamide gels. Cy5 fluorescence was detected using a Molecular Imager FX scanner (Bio-Rad).
RESULTS
Translocation of E. coli K1 to the blood compartment and capacity to cause lethal infection are dependent on the age of the colonized host.
The septicemia isolate E. coli A192 efficiently colonized the GI tract of neonatal rat pups (frequency of 100%), but only 23% (17) to 35% (15) of colonized pups progressed to the bacteremic state. We therefore enhanced the capacity of the A192 strain to translocate to the blood compartment by two rounds of serial passage in neonatal rats. Bacteria recovered from the blood following passage produced bacteremia at higher frequency (100% of colonized pups); one colony from the blood culture was propagated for further experimentation and designated strain A192PP. Feeding of E. coli A192PP to 2-day-old (P2) pups resulted in 100% colonization of the GI tract within 24 to 48 h; 48 h after dosing, E. coli K1 began to appear in the bloodstream, and by day 7 all animals had succumbed to a lethal bacteremia (Fig. 1A). P5 pups were less susceptible to infection, with, typically, around 50% of animals displaying bacteremia following GI tract colonization rates of 100%. P9 animals were efficiently colonized by A192PP but were refractory to bacteremia and lethal infection (Fig. 1A). Patterns of colonization and infection were highly reproducible in Wistar rat pup litters. For practical reasons, fecal material could only be sampled from the perianal area, and the colonization lag displayed in Fig. 1A is likely to be due to increasing contamination of this anatomical region (engendering an increasingly large coliform population) over the first week of life. There was an age-dependent decrease in susceptibility to lethal infection over the period from P2 to P9 (Fig. 1B); pups began to acquire resistance from day 3 and were completely resistant by day 9. Reductions in susceptibility to infection following colonization will be accompanied by structural, physiological, and microbiological changes of GI tract tissues (29, 30), and we therefore undertook parallel histological and immunohistochemical investigations of GI tissue in order to ensure that processes that affect susceptibility to infection can be correlated with the dynamic process of postnatal development of the rat intestine. Over the P2-to-P9 period, the length of the digestive system (stomach to colon) increased in an incremental fashion from a mean of 25.6 cm at P2 to 47.6 cm at P9 and was characterized by rapid postnatal physiological and anatomical development due to expansion of the small intestine and increased differentiation of the cecum (Fig. 1C). As is evident from Alcian blue–periodic acid-Schiff stain (PAS)-stained sections of intestinal tissue from pups over the P2-to-P9 period (Fig. 2), organ maturation was accompanied by increases in the thickness of the submucosal layer and the number of mucin-containing cells. In accord with others (31), we found no histological evidence for any lack of structural integrity during the early phases of neonatal development. No discernible histological changes occurred 24 h and 48 h after colonization of P2 and P9 pups with A192PP.
Fig 1.

Age-dependent susceptibility of the neonatal rat to systemic E. coli K1 infection. (A) GI tract colonization, bacteremia, and accumulated deaths of neonatal rat pups fed E. coli A192PP two (P2), five (P5), and nine (P9) days after birth. All pups in a litter received the oral dose on day 0. Colonies expressing the K1 capsule were detected using K1-specific bacteriophage from perianal swab cultures and from blood samples. The data shown are from representative experiments involving single litters of 9 to 14 pups. (B) Survival of P2-to-P9 rat pups fed E. coli A192PP. Two litters (12 pups per litter) of 2- to 9-day-old pups were used for each time point, and the animals were monitored for a period of 1 week following oral dosing of bacterial cultures; n = 24. Values are means ± standard deviations (SD). (C) Gross morphological appearance of GI tract tissue (stomach to colon) from P2-to-P9 rat pups.
Fig 2.
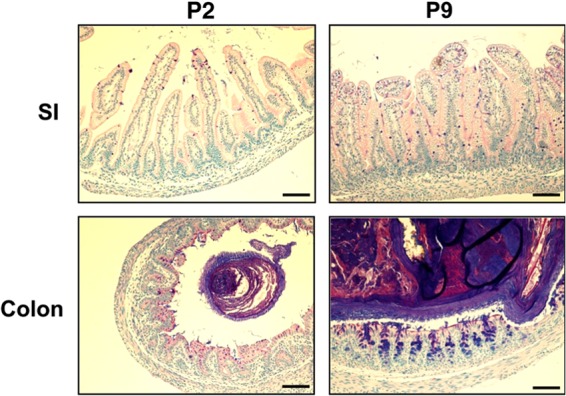
Rapid development of GI tract tissue morphology during the early postnatal period. Small intestinal (SI) and colonic tissues from P2 and P9 animals were fixed, sectioned, and stained with Alcian blue-PAS. The scale bar is 100 μm.
Systemic infection is unrelated to either the size of the colonizing E. coli K1 cohort or the composition of the GI tract microbiota.
Translocation of E. coli K1 from the neonatal rat GI tract to the blood occurs via the mesenteric lymph nodes, and each bacterial cell has a low probability of overcoming the mechanical and immunological barriers to gain access to the blood compartment (15), even though neonatal intestinal mucosal barriers may be incompletely developed at birth (32). As selective elimination of the resident GI tract microbiota may facilitate the translocation of bacteria to systemic organs and tissues (33) and as E. coli K1 replicates in the neonatal rat intestine concomitant with translocation to the blood (15), we hypothesized that K1 intestinal colonization prior to the acquisition of a fully developed microbiota would permit sufficient replication of the neuropathogen to enable translocation to take place. We therefore determined the qualitative and quantitative bacterial composition of GI tract tissues from P2, P5, and P9 animals using qPCR of conserved 16S small subunit (SSU) rRNA genes and a customized SSU rRNA gene microarray designed to detect most currently recognized species and taxonomic groups of bacteria; we also quantified E. coli K1 from GI tract whole-tissue homogenates using qPCR of the K1-specific neuS gene.
There was a high degree of similarity, both quantitatively and qualitatively, between the GI tract microbiota of rat litters (Fig. 3; see Fig. S1 in the supplemental material); by P2, uninfected pups had acquired large numbers of resident bacteria and the numbers, expressed as CFU/g tissue, did not increase significantly over the period from P2 to P9 (Fig. 3A). Even at P2, the range of phyla representative of mammalian GI tract bacterial populations (34, 35) was generally present with an abundance comparable to that found in adult feces, although Bacteriodetes comprised a smaller proportion of the GI tract microbiota in P2-to-P9 rats compared to adult feces (Fig. 3B). The Firmicutes/Actinobacteria and Proteobacteria dominated bacterial populations over the period from P2 to P9, and there were no significant (P ≥ 0.57) differences in the abundance of these phyla in samples from these pups. In accord with observations in human infants (11), Bacteriodetes comprised a smaller proportion of the GI tract microbiota in P2-to-P9 rats compared to adult feces (Fig. 3B) as a result of underrepresentation of Bacteroides (see Fig. S1C). There were no significant differences between P2, P5, and P9 pups with regard to the three dominant phyla (see Fig. S1A to C). At the species level within these phyla, there were significant differences between pups of the three age groups for only eight species, and these are described in Fig. S1D. Less abundant phyla included Acidobacteria, Nitrospina, and Fusobacteria. It is noteworthy that members of the Acidobacteria, a recently established (36) phylum of metabolically versatile bacteria associated in the main with diverse environmental habitats that encompass soil, freshwater habitats, and metal-contaminated subsurface sediments (37, 38) and are occasionally found in gastrointestinal material (39, 40), were present in all GI tract samples we examined. Similarly, the chemolithoautotrophic bacteria of the phylum Nitrospina (Nitrospora) are generally associated with aquatic environments (41) but were encountered in all groups of neonatal rats. Thus, we found a high degree of comparability in the composition of the neonatal rat gut microbiota over the period from P2 to P9 and also good qualitative and quantitative accord between neonatal and adult GI tract bacterial populations.
Fig 3.

Comparable rates of GI tract colonization by E. coli A192PP over the P2-to-P9 neonatal period complements rapid development of the GI tract microbiota in neonatal rats. (A) Bacterial load, determined by qPCR of conserved 16S SSU rRNA genes, of GI tract tissues and fecal content from P2, P5, and P9 rat pups. At each time point, four animals from each of three litters were sampled. (B) Relative abundance of bacterial phyla of the GI microbiota by SSU rRNA gene microarray. Data were normalized to adult data, as indicated by the dashed line at x = 1. Four litters were employed. Four animals were removed from each litter at P2, P5, and P9; bacterial DNA extracted from tissue samples at each time point hybridized with DNA from the appropriate maternal fecal sample. Results from each of four litters were combined. Probes were ranked according to average Cy5 and Cy3 fluorescence, with the highest shown at the top. (C) Temporal aspects of E. coli K1 GI tract colonization of P2, P5, and P9 animals. K1 bacteria were quantified by qPCR of the neuS gene. DNA was extracted from GI tissues and their contents, and a calibration curve was constructed to convert data to CFU. Two litters of neonatal rats were employed. Four animals were removed from each litter at each time point, the GI tissue was removed and processed, and DNA representing each time point was pooled. LOD, limit of detection. (D) Colonization of the proximal small intestine, distal small intestine, and cecum/colon over the 48-h period following feeding of E. coli A192PP to P2 and P9 pups. E. coli K1 bacteria were quantified by qPCR with the neuS probe. Data were normalized to tissue mass following conversion to CFU. Error bars represent the standard errors of the mean (SEM) from n = 4 animals. Significant differences, as determined by 2-tailed Mann-Whitney test: *, P < 0.05; **, P < 0.01; and ***, P < 0.001.
No significant differences were found in the capacity of E. coli A192PP to colonize and persist in the P2, P5, and P9 GI tract for at least 5 days following oral administration of a bacterial suspension (Fig. 3C). Immediately after feeding the E. coli K1 strain to both P2 and P9 animals, a major portion of the inoculum distributed to the lower intestine and cecum/colon over the following 48-h period (Fig. 3D). Within 24 h, the largest E. coli K1 population was found in cecum/colon tissue samples. Significantly larger numbers of E. coli K1 cells were detected in the upper GI tissues of P2-inoculated compared to P9-inoculated neonates at all time points sampled, but the numbers were comparable in cecum/colon tissues (Fig. 3D). There was clear evidence of extensive E. coli K1 replication in the GI tract after introduction of strain A192PP (see Fig. S2 in the supplemental material).
GI tissue responses to E. coli A192PP colonization are notably different in P2 and P9 rat pups.
In the first hours and days following birth, there is a distinct expression pattern of innate immune molecules by mucosal epithelia aimed at preventing infection while avoiding excessive inflammatory responses to bacteria and their products (6). During early postnatal development, this bias against TH1 cell-polarizing cytokine-mediated effects alters following exposure to microbes, diminishing TH2 cell polarization and enhancing TH1 cell polarization, and these effects are likely to be pronounced in gut tissue due to the rapid accumulation of a complex microbiota. We therefore examined the response of P2 pups to colonization by E. coli K1 to determine if any differences in response compared to P9 animals could shed light on the basis of the age dependency of systemic infection. P2 and P9 pups were fed strain A192PP, GI tract tissues were removed after 12 h, and duodenum to colon samples were prepared for microarray analysis. Arrays were performed by pooling equimolar amounts of RNA extracted from three infected rat pups. Responses were standardized against tissues removed at the same time from noncolonized animals of identical age and processed in an identical fashion.
Major differences were found in the responses of P2 and P9 pups to colonization (Fig. 4; see Tables S1 to S5 in the supplemental material); A total of 241 genes were upregulated 2-fold or more in the P2 GI tract compared to 354 in P9 animals, but only 22 genes were common to both data sets. More genes (n = 240) were downregulated at least 2-fold in P9 samples than in P2 samples (n = 36), with eight genes common to both (Fig. 4A; see Table S6 in the supplemental material). With both pooled P2 and P9 tissue arrays, up- and downregulated genes belonged to a wide range of functional categories, with genes involved in growth, differentiation, and development and cell metabolism and transcriptional regulation strongly represented (see Table S1). In both P2 and P9 tissues, genes encoding major histocompatibility complex (MHC) class I and class II proteins were differentially expressed. For example, RT1-Aw2, encoding RT1 class Ib, was the most strongly upregulated (17.7-fold) gene in P2-colonized GI tissues (see Table S2) and RT1-A3 and RT1-Db1, encoding RT1 class I and class II proteins, respectively, were highly downregulated in P2-colonized tissues (see Table S3). In pups colonized at P9, the most prominently upregulated (11.8-fold) gene was the MHC class II protein-encoding RT1-Bb gene, and expression of RT1-Aw2 was also significantly increased (see Table S4); RT1-A3 and RT1-Db1 were also downregulated in P9 pups (see Table S5). These and other MHC genes feature in the relatively short list of genes whose regulation was modulated by colonization in both P2 and P9 tissues, along with genes involved in mRNA splicing, DNA damage repair, and protein modification (see Table S6). A number of genes involved in initiation of apoptosis, such as pcdc4, stk17b, casp2, and casp3, were upregulated in P9- but not P2-colonized tissues (see Table S4), and genes encoding antiapoptotic factors such as Tgfb2 and Hspa5 were concomitantly downregulated (see Table S5) in P9 only. Also noteworthy was the 2.5-fold upregulation in P9 but not P2 of DDX3X, encoding a helicase that together with TBK1 is central to the induction of type 1 interferons in response to pathogens (42).
Fig 4.

P2 and P9 host responses to E. coli K1 intestinal colonization. (A) P2 and P9 intestinal tissues show differential transcriptomic responses to E. coli K1 colonization, as demonstrated by comparison of microarray analyses of RNA extracted from colonized and noncolonized P2 and P9 tissues 12 h postinoculation with E. coli K1 or broth. n = 4. (B) Expression of tff2, defa24, defa-rs1, and the reference gene rps23 was analyzed by semiquantitative RT-PCR followed by resolution of pooled cDNA amplicons on agarose gels and by qRT-PCR analysis (C, D, and E) of RNA extracted from GI tract tissues from colonized and noncolonized P2 and P9 animals 6, 12, 24, and 48 h postinoculation with E. coli K1 or broth. Data from colonized animals were normalized to data from an equal number of noncolonized (broth-fed) animals. Tff2 protein was quantified by competitive ELISA of protein extractions from colonized and noncolonized animals (F). The normal developmental expression of tff2, defa24, and defa-rs1 over the period from P1 to P11 was examined by normalizing qRT-PCR data from noncolonized animals to data obtained from RNA extracted from noncolonized P1 neonatal intestinal tissues (G). Error bars for all figures represent the SEM of results from either n = 12 (C, D, and E) or n = 6 (F) animals. Statistically significant differences, as determined by 2-tailed t test, between colonized and noncolonized animals are indicated: *, P < 0.05; **, P < 0.01; ***, P < 0.001.
A very limited number of genes whose products are likely to be directly linked to the susceptibility of P2 pups to postcolonization infection were differently regulated by colonizing E. coli K1 in P2 compared to P9 pups. The genes encoding the α-defensins Defa24 and Defa-rs1 were upregulated 3.1- and 5.4-fold, respectively, in colonized P9 but not P2 pups (see Table S4 in the supplemental material). This class of bactericidal peptides, produced by Paneth cells in the small intestine (43, 44), regulate the composition of the intestinal bacterial microbiome (45), control intestinal barrier penetration by commensal and pathogenic bacteria (46), and are necessary for antibacterial defense in the neonate (47). The failure of P2 animals to respond to E. coli K1 colonization in similar fashion to P9 pups may enhance the capacity of this neuropathogen to reach the small intestinal epithelium, as α-defensins are thought to limit the bacterial load at the epithelial surface (48). We therefore investigated temporal aspects of α-defensin gene expression in relation to colonization by E. coli A192PP. The most highly downregulated gene (26.4-fold) in P2 animals was tff2, encoding trefoil factor 2. This small peptide plays a role in GI mucosal protection, stabilizes the mucus layer, and enhances intestinal epithelial repair (49). As the mucus layer continues to develop postpartum in the rat (50) and the goblet cell complement of the P2 gut appears to be low in comparison to those in P9 and adult animals (Fig. 2), we investigated the impact of tff2 downregulation on the development of the mucus layer between P2 and P9.
Microarray data were validated by qRT-PCR analysis of 11 differentially regulated genes from the P2 (RT1-Aw2, Btg2, Cald1, tff2, and Ins2) and P9 (defa-rs1, Pdcd4, Clic4, Cav, Afp, and Amy2) data sets. A highly significant (P < 0.0001) Pearson correlation was obtained when microarray and qRT-PCR data were compared.
GI tract colonization by E. coli A192PP modulates defa24 and defa-rs1 expression over the P2-to-P9 period of neonatal development.
No information is available regarding temporal aspects of α-defensin gene expression in the rat over the neonatal period. In order to provide context for the modulation of defa-rs1 and defa24 by colonizing E. coli K1, we determined the expression of these genes in duodenum to colon GI tract tissues from noncolonized pups over the P1-to-P11 postpartum period. The level of expression of both genes progressively increased over this period, peaking at P10; over the 11 days, defa-rs1 expression increased ∼12-fold and defa24 expression decreased ∼4-fold (Fig. 4G). To determine the effects of colonization on these background levels of gene expression, P2 and P9 pups were fed E. coli A192PP and gene expression was compared to that of same-age noncolonized animals 6, 12, 24, and 48 h postcolonization by semiquantitative PCR (Fig. 4B). The data suggested that, in P2 animals, the expression of both genes increased 24 to 48 h after E. coli K1 dosing; in P9 pups, the increases in defa24 expression peaked 24 h after dosing, whereas defa-rs1 expression continued to increase over the 48-h period in comparison to the level in animals fed broth rather than bacteria. As the levels of defa24 and defa-rs1 continue to rise in control pups over this period, providing a changing background upon which to assess the degree of gene modulation resulting from colonization, we quantified levels of gene expression for both genes by qPCR and normalized the data against the normal developmental levels (Fig. 4G).
Expression of defa24 in P2 gut tissues was similar to background levels following E. coli K1 colonization. In contrast, defa24 expression in the P9 gut was significantly increased up to 12 h postcolonization but returned to levels comparable to those found in the control group within 24 to 48 h (Fig. 4D). Levels of defa-rs1 expression in colonized P2 pups remained comparable to those found in control animals over the 48-h period, whereas expression was increased at all time points in colonized P9 pups; differences in defa-rs1 expression between P2 and P9 animals were significant at all time points over this period (Fig. 4E). We conclude that P2 pups, in contrast to P9 rats, are unable to respond to E. coli A192PP colonization by upregulation of expression of α-defensin peptide genes.
Suppression of Tff2 in the P2 GI tract by E. coli K1 colonization.
We established that tff2 expression in non-E. coli K1-colonized pups increased incrementally from P1 up to P9 but substantially declined over P9 to P11 (Fig. 4G). Semiquantitative PCR indicated that tff2 expression was downregulated over the 48-h postcolonization period in P2 but not P9 pups (Fig. 4B); qPCR confirmed that whereas GI tissue levels of tff2 in P9-colonized pups did not differ significantly over this period from the normal developmental levels shown in Fig. 4G, tff2 levels were markedly reduced by colonization at P2 (Fig. 4C). Suppression of gene expression in P2 tissues was evident at 24 to 48 h; the degree of downregulation compared to expression in P9 pups was highly significant and was tightly correlated with the reduced levels of Tff2 protein found in P2 and P9 tissue homogenates (Fig. 4F).
Transcription of tff genes is repressed by IL-1β and IL-6 (51); cell stimulation by these proinflammatory cytokines leads to release of NF-κB and C/EBPβ from resting-state complexes in the cytoplasm and their translocation to the nucleus (52), where they inhibit transcription of trefoil factor genes. We therefore assessed the systemic levels of these cytokines by cytokine-specific ELISA of serum samples obtained from P2 and P9 neonates colonized with E. coli K1 and compared these to the levels in noncolonized animals. A 2- to 3-fold elevation in systemic IL-1β from levels found in noncolonized controls was observed 6 to 24 h after feeding E. coli K1 in P2-colonized but not in P9-colonized neonates (see Fig. S2A in the supplemental material). No IL-6 was detected in either colonized or noncolonized P2 neonates; although IL-6 was detected in both colonized and noncolonized P9 cohorts, no significant differences were observed between the experimental groups (see Fig. S2B). In order to provide evidence for the role of NF-κB in tff2 transcriptional repression, we assessed the extent of NF-κB localization to intestinal cell nuclei by electrophoretic mobility shift assay using a Cy5-labeled double-stranded DNA (dsDNA) probe containing the NF-κB consensus binding motif incubated with nuclear protein extracts obtained from intestinal tissues of neonates colonized with E. coli K1 and broth-fed, noncolonized animals (see Fig. S2C). Although a degree of band-shift was observed in all samples, the intensity of the shifted band was greater in colonized compared to noncolonized P2 animals. A control assay utilizing nonlabeled wild-type and mutant dsDNA competitors yielded reduced band shift in the wild-type but not the mutant competition reactions, indicating that the band shift was specific for the NF-κB binding motif sequence. These results suggest that E. coli K1 intestinal colonization provokes a significant increase in the release of IL-1β in P2-colonized pups, leading to increased nuclear localization of NF-κB.
The observation that P2 pups possess fewer goblet cells than P9 rats (Fig. 2) is reflected in less small intestine-associated mucin and a thinner Muc2-immunostaining layer in the colon compared to the case in infection-resistant P9 neonates (Fig. 5). Less mucin was evident 48 h after oral feeding of E. coli K1 (Fig. 5C) compared to the level in broth-fed pups (Fig. 5B), although much of this can be accounted for by a large decrease in Muc2 stored in colonic goblet cells, presumably expelled into the gut lumen in response to E. coli K1 colonization. At P4, animals are relatively susceptible to E. coli K1 infection (Fig. 1B), so the colonic mucin content of animals sham fed at P2 and culled at P4 (Fig. 5B) may not be sufficient to afford protection against systemic infection.
Fig 5.
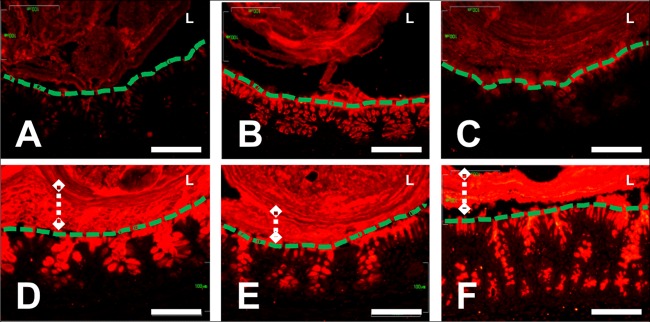
E. coli K1 colonization of P2 pups results in depletion of Muc2 mucin from colonic epithelial cells. Anti-Muc2 immunostaining of sections of colonic tissues from P2 (A to C) and P9 (D to F) neonatal rats. Tissues were obtained at P2 (A) or P9 (D) and at 48 h after feeding neonates a control dose (B and E) or E. coli A192PP (C and F). The epithelial surface (green dashed lines), lumen (L), and stratified mucus layer (white dashed lines with diamonds) are indicated where appropriate. The scale bars are 100 μm.
DISCUSSION
The data presented here and by other workers (14, 17) provide compelling evidence that the capacity of E. coli K1 to translocate from the GI tract to the bloodstream constitutes the basis of susceptibility to systemic infection in the neonatal rat model. It is generally accepted that the gut microbiota confers colonization resistance, prohibiting both potentially harmful (53, 54) and potentially beneficial (55) microbes from establishing a foothold in the GI tract. This protective role is complemented by the capacity of members of the microbiota to modulate the virulence of some opportunistic pathogens (56, 57). We therefore hypothesized that the gut microbiota of susceptible P2 pups was likely to be less well developed than that of resistant P9 rats and would permit colonizing E. coli K1 cells to reach sufficient numbers in the GI tract to allow transit to the blood; colonial expansion would be denied to E. coli K1 in the P9 GI tract.
In broad terms, this hypothesis is not supported by our data. Although the intestinal microbiota of P2, P5, and P9 pups were quantitatively and qualitatively different from the adult microbiota, they were remarkably similar to each other. Even 2 days postpartum, a highly complex bacterial consortium had developed in the GI tract that was barely distinguishable from that found in P9 animals. However, the methods used in this investigation do not take into account spatial aspects of the distribution of the microbial population. The individual GI compartments have distinct biochemical and physical properties and represent diverse ecological niches. Accordingly, the gastric, small intestinal, and colonic compartments play host to a partially distinct subset of the GI microbiota (58, 59, 60), and it is possible that significant changes to the microbiota of these regions occur over the period from P2 to P9 in a way that could influence susceptibility to infection.
The microbiota appeared to exert little influence on the capacity of E. coli A192PP to stably colonize the GI tract of P2-to-P9 pups, notwithstanding that the strain was selected for this study on the basis of its systemic virulence following GI tract colonization. No differences in the E. coli K1 burden after colonization at P2, P5, or P9 could be demonstrated after 120 h, and the temporal development of the K1 population was very similar in these age groups. We did, however, note some significant differences in the temporal and spatial distributions of E. coli A192PP along the GI tract that almost certainly relate to differences in innate competence between P2 and P9 pups, lending support to the contention that the bacteria are more likely to translocate from the small intestine than the colon. The stabilization of the E. coli K1 intestinal load 24 h after colonization indicates that there may be an upper limit to the size or growth capacity of the bacterium in the neonatal intestine. The population climaxed immediately prior to the bacterial translocation window, suggesting that a critical E. coli K1 population density must be reached before translocation to the blood compartment can occur.
The intestinal tissues are subject to significant postpartum development in response to exposure to the extrauterine environment and the initiation of enteral feeding. Changes include the proliferation of two secretory epithelial cell lineages which play a key role in maintaining intestinal barrier function in the small intestinal and colonic compartments. The colonic goblet cell population continues to expand postpartum and is accompanied by increased production of Muc2 and trefoil peptides (61, 62). The small intestinal Paneth cell population also proliferates rapidly in the postnatal period (63), as does their secretion of antimicrobial peptides (64). The proteins secreted by these cells are vital for maintaining the microbiota away from the enteric epithelial surface (65, 66). The fact that they are developmentally regulated indicates that the intestinal barrier function in younger neonates is immature and partly informed our view that the development of the neonatal intestine over the P2-to-P9 period modulates susceptibility to E. coli K1 systemic infection.
The neonatal intestinal tract grew substantially over the P2-to-P9 period and was accompanied by a significant degree of developmental gene regulation. Based on our observations and the current state of knowledge regarding the developmental regulation of Paneth (40) and goblet cells (67), we propose a model to account for the development of innate defense barriers in the neonatal rat intestine over the P2-to-P9 period (Fig. 6). Thus, at P2 the ileum secretes lower quantities of α-defensins and the colon lower quantities of Muc2 and trefoil factor than at P9, resulting in an underdeveloped stratified inner mucus layer in comparison to that in P9 pups which permits closer association between the intestinal microbiota (which inhabit the outer mucus layer) and the intestinal epithelium. The comparative lack of α-defensin expression in P2 tissues indicates that the antimicrobial peptide-dependent barrier function of the small intestine is weaker at P2 than at P9 and is likely to be a factor that determines the differences in E. coli A192PP numbers along the GI tract. In future work, we will examine the possibility that defects in α-defensin-mediated barrier function allow K1 colonizers to penetrate enterocytes in P2 pups. The development of the colonic mucus barrier may be related to the transient increase in Tff2 expression from P2 to P9. Expression of Tff3 in the rat colon does not start to increase toward adult levels until P12 to P17 (58, 68); given the apparent role of Tff3 as a structural component of the colonic mucus barrier (69), this pattern of developmental expression seems unusual. Tff2 may play a similar role to Tff3 in the early neonatal intestine, stabilizing the developing colonic mucus barrier prior to the developmental increase in Tff3 expression, or it may be localized to the small intestine rather than the colon.
Fig 6.

Proposed model of GI barrier development in rats from P2 (A) to P9 (B). (C and D) Colonization of the P2 (C) and P9 (D) GI tract by E. coli K1. The numbered steps in panels C and D are expanded in the text.
The transcriptional responses of P2 and P9 intestinal tissues to colonization by E. coli K1 were highly divergent. Several genes encoding products involved in host defense were differentially expressed, including developmentally regulated α-defensins and Tff2. There were comparatively few differentially expressed genes shared between colonized P2 and P9 neonates, indicating that the intestinal tissue of the refractive neonate responds very differently from that of the susceptible neonate and may in part determine susceptibility to systemic infection. Suppression of Tff2 with concomitant loss of Muc2 in P2-colonized tissues may allow E. coli K1 to gain access to the intestinal epithelium. Conversely, the upregulation of α-defensins by P9-colonized tissues may prevent this interaction. Thus, we propose (Fig. 6) the following. In P2 animals, the relative deficiency of α-defensins allows the pathogen to access and invade the tissues of the small intestine (step 1). Bacterial colonization is detected by intestinal leukocytes (step 2). Activated leukocytes secrete IL-1β, which activates NF-κB transcription factor (step 3). Finally activated NF-κB suppresses trefoil factor in goblet cells with concomitant breakdown of the inner mucus layer structure (step 4), allowing bacteria to access and invade colonic tissue (step 5). In P9 pups, upregulation of α-defensins denies the bacteria access to tissues of the small intestine (step 1) and prevents IL-1β secretion by leukocytes (step 2); a thick and intact inner mucus layer prevents bacterial access to colonic tissue (step 3). In all likelihood, the mucus layer keeps the colonizing bacteria away from the gut wall, preventing interactions with the epithelial surface. Evidence that E. coli K1 cells gain access to the enterocytes in susceptible pups will provide strong support for this model, and such interactions will be sought using immunohistochemical procedures. The model would be further strengthened by demonstration that E. coli K1 colonization of α-defensin knockout rats at P9 induces the bacteremic state, and we are actively pursuing this line of investigation.
E. coli K1 colonization of the P2 intestine invariably results in translocation of the pathogen from the intestine into the systemic circulation. The site or sites of translocation have yet to be resolved, and the proximal small intestine is a likely candidate. However, developmental deficits appear to be present in the P2 small intestinal and colonic barriers, and consequently, both regions represent a potential route of invasion. Lack of secreted α-defensins could allow E. coli K1 to access the small intestinal epithelium. Alternatively, dysregulation of Tff2 expression may provide access to the colonic epithelium. We have provided some indication that tff2 transcriptional repression involves IL-1β and NF-κB. Colonization of the P2 intestine induces the secretion of IL-1β into the systemic circulation, but this could be due to the early presence of bacteria in the blood; we therefore plan to examine cytokine levels in tissues in more detail in order to provide a mechanistic basis for susceptibility to infection. This is most likely due to the detection of pathogen-associated molecular patterns (PAMPs) by the intestinal leukocyte population. Adult intestinal macrophages lack the CD14 receptor, which systemic macrophages use to detect bacterial lipopolysaccharide (70). Lipopolysaccharide tolerance prevents intestinal macrophages from inducing potentially damaging inflammatory reactions in response to the intestinal microbiota; tolerance does not develop until the perinatal period (71, 72) and may explain why IL-1β secretion is not induced in P9 neonates. Furthermore, α-defensins represent another potential inhibitor of IL-1β secretion from macrophages in the P9 intestine (73).
E. coli K1 colonization at P2 may suppress Muc2 synthesis or induce goblet cells to expel stored Muc2 into the intestinal lumen in an attempt to clear the pathogen from the intestine, an effect that has been observed following colonization by the rodent intestinal pathogen Citrobacter rodentium (74, 75). The loss of stored Muc2 at such an early stage in the development of the colonic mucus barrier could compromise developmental processes and provide a possible route of infection for E. coli K1.
Supplementary Material
ACKNOWLEDGMENTS
This study was supported by research grant G0400268 from the Medical Research Council, Swedish Research Council grant 7461, and the Knut and Alice Wallenberg Foundation. This research was supported by the National Institute for Health Research University College London Hospitals Biomedical Research Centre.
We thank Ozan Gundogdu and Melissa Martin for advice on microarray procedures.
The authors have no conflicting financial interests.
Footnotes
Published ahead of print 24 June 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.00268-13.
REFERENCES
- 1.Harvey D, Holt DE, Bedford H. 1999. Bacterial meningitis in the newborn: a prospective study of mortality and morbidity. Semin. Perinatol. 23:218–225 [DOI] [PubMed] [Google Scholar]
- 2.Brouwer MC, Tunkel AR, van de Beek D. 2010. Epidemiology, diagnosis, and antimicrobial treatment of acute bacterial meningitis. Clin. Microbiol. Rev. 23:467–492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Robbins JB, McCracken GH, Gotschlich EC, Ørskov F, Ørskov I, Hanson LA. 1974. Escherichia coli K1 polysaccharide associated with neonatal meningitis. N. Engl. J. Med. 290:1216–1220 [DOI] [PubMed] [Google Scholar]
- 4.Korhonen TK, Valtonen MV, Parkkinen J, Väisänen-Rhen V, Finne J, Ørskov F, Ørskov I, Svenson SB, Mäkelä PH. 1985. Serotypes, hemolysin production, and receptor recognition of Escherichia coli strains associated with neonatal sepsis and meningitis. Infect. Immun. 48:486–491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rutishauser U. 2008. Polysialic acid in the plasticity of the developing and adult vertebrate nervous system. Nat. Rev. Neurosci. 9:26–35 [DOI] [PubMed] [Google Scholar]
- 6.Levy O. 2007. Innate immunity of the newborn: basic mechanisms and clinical correlates. Nat. Rev. Immunol. 7:379–390 [DOI] [PubMed] [Google Scholar]
- 7.de Louvois J. 1994. Acute bacterial meningitis in the newborn. J. Antimicrob. Chemother. 34(Suppl A):61–73 [DOI] [PubMed] [Google Scholar]
- 8.Sarff LD, McCracken GH, Schiffer MS, Glode MP, Robbins JB, Ørskov I, Ørskov F. 1975. Epidemiology of Escherichia coli K1 in healthy and diseased newborns. Lancet i:1099–1104 [DOI] [PubMed] [Google Scholar]
- 9.Schiffer MS, Oliveira E, Glode MP, McCracken G, Sarff LM, Robbins JB. 1976. A review: relation between invasiveness and the K1 capsular polysaccharide of Escherichia coli. Pediatr. Res. 10:82–87 [DOI] [PubMed] [Google Scholar]
- 10.Obata-Yasuoka M, Ba-Thein W, Tsukamoto T, Yoshikawa H, Hayashi H. 2002. Vaginal Escherichia coli share common virulence factor profile, serotypes and phylogeny with other extraintestinal E. coli. Microbiology 148:2745–2752 [DOI] [PubMed] [Google Scholar]
- 11.Palmer C, Bik EM, DiGuilio DB, Relman DA, Brown PO. 2007. Development of the human infant intestinal microbiota. PLoS Biol. 5:1556–1573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nassif X, Bourdoulous S, Eugène E, Couraud P-E. 2002. How do extracellular pathogens cross the blood-brain barrier? Trends Microbiol. 10:227–232 [DOI] [PubMed] [Google Scholar]
- 13.Tunkel AR, Scheld WM. 1993. Pathogenesis and pathophysiology of bacterial meningitis. Clin. Microbiol. Rev. 6:118–136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Glode MP, Sutton A, Moxon ER, Robbins JB. 1977. Pathogenesis of neonatal Escherichia coli meningitis: induction of bacteremia and meningitis in infant rats fed E. coli K1. Infect. Immun. 16:75–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pluschke G, Mercer A, Kusećek B, Pohl A, Achtman M. 1983. Induction of bacteremia in newborn rats by Escherichia coli K1 is correlated with only certain O (lipopolysaccharide) antigen types. Infect. Immun. 39:599–608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mushtaq N, Redpath MB, Luzio JP, Taylor PW. 2004. Prevention and cure of systemic Escherichia coli K1 infection by modification of the bacterial phenotype. Antimicrob. Agents Chemother. 48:1503–1508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mushtaq N, Redpath MB, Luzio JP, Taylor PW. 2005. Treatment of experimental Escherichia coli infection with recombinant bacteriophage-derived capsule depolymerase. J. Antimicrob. Chemother. 56:160–165 [DOI] [PubMed] [Google Scholar]
- 18.Martindale J, Stroud D, Moxon ER, Tang CM. 2000. Genetic analysis of Escherichia coli K1 gastrointestinal colonization. Mol. Microbiol. 37:1293–1305 [DOI] [PubMed] [Google Scholar]
- 19.Zelmer A, Bowen M, Jokilammi A, Finne J, Luzio JP, Taylor PW. 2008. Differential expression of the polysialyl capsule during blood-to-brain transit of neuropathogenic Escherichia coli K1. Microbiology 154:2522–2532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zelmer A, Martin MJ, Gundogdu O, Birchenough G, Lever R, Wren BW, Luzio JP, Taylor PW. 2010. Administration of capsule-selective endosialidase E minimizes upregulation of organ gene expression induced by experimental systemic infection with Escherichia coli K1. Microbiology 156:2205–2215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pluschke G, Pelkonen S. 1988. Host factors in the resistance of newborn mice to K1 Escherichia coli infection. Microb. Pathog. 4:93–102 [DOI] [PubMed] [Google Scholar]
- 22.Polin RA, Harris MC. 2001. Neonatal bacterial meningitis. Semin. Neonatol. 6:157–172 [DOI] [PubMed] [Google Scholar]
- 23.Jennings HJ, Brisson J-R, Kulakowska M, Michon F. 1993. Polysialic acid vaccines against meningitis caused by Neisseria meningitidis B and Escherichia coli K1, p 25–38 In Roth J, Rutishauser U, Troy FA. (ed), Polysialic acid. Birkhäuser Verlag, Basel, Switzerland [Google Scholar]
- 24.Lee A, Gordon J, Lee C-J, Dubos R. 1971. The mouse intestinal microflora with emphasis on the strict anaerobes. J. Exp. Med. 133:339–352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Achtman M, Mercer A, Kusećek B, Pohl A, Heuzenroeder M, Aaronson W, Sutton A, Silver RP. 1983. Six widespread bacterial clones among Escherichia coli K1 isolates. Infect. Immun. 39:315–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gross RJ, Cheasty T, Rowe B. 1977. Isolation of bacteriophages specific for the K1 polysaccharide antigen of Escherichia coli. J. Clin. Microbiol. 6:548–550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Palmer C, Bik EM, Eisen MB, Echberg PB, Sana TR, Wolber PK, Relman DA, Brown PO. 2006. Rapid quantitative profiling of complex microbial populations. Nucleic Acids Res. 34:e5. 10.1093/nar/gnj007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Steenbergen SM, Vimr ER. 2008. Biosynthesis of the Escherichia coli K1 group 2 polysialic acid capsule occurs within a protected cytoplasmic compartment. Mol. Microbiol. 68:1252–1267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cummins AG, Jones BJ, Thompson FM. 2006. Postnatal epithelial growth of the small intestine in the rat occurs by both crypt fission and crypt hyperplasia. Dig. Dis. Sci. 51:718–723 [DOI] [PubMed] [Google Scholar]
- 30.Hansson J, Panchaud A, Favre L, Bosco N, Mansourian R, Benyacoub J, Blum S, Jensen ON, Kussmann M. 2011. Time-resolved quantitative proteome analysis of in vivo intestinal development. Mol. Cell Proteom. 10:M110.005231. 10.1074/mcp.M110.005231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pácha J. 2000. Development of intestinal transport function in mammals. Physiol. Rev. 80:1633–1667 [DOI] [PubMed] [Google Scholar]
- 32.Udall JM, Pang K, Fritze L, Kleinman R, Walker WA. 1981. Development of gastrointestinal mucosal barrier. I. The effect of age on intestinal permeability to macromolecules. Pediatr. Res. 15:241–244 [DOI] [PubMed] [Google Scholar]
- 33.Wells CL, Maddaus MA, Reynolds CM, Jechorek RP, Simmons RL. 1987. Role of anaerobic flora in the translocation of aerobic and facultatively anaerobic intestinal bacteria. Infect. Immun. 55:2689–2694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Andersson AF, Lindberg M, Jakobsson H, Bäckhed F, Nyrén P, Engstrand L. 2008. Comparative analysis of human gut microbiota by barcoded pyrosequencing. PLoS One 3:e2836. 10.1371/journal.pone.0002836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ley RE, Hamady M, Lozupone C, Turnbaugh PJ, Ramey RR, Bircher JS, Schlegel ML, Tucker TA, Schrenzel MD, Knight R, Gordon JI. 2008. Evolution of mammals and their gut microbes. Science 320:1647–1651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ludwig W, Bauer SH, Bauer M, Held I, Kirchhof G, Schulze R, Haber I, Spring S, Hartmann A, Schleifer KH. 1997. Detection and in situ identification of representatives of a widely distributed new bacterial phylum. FEMS Microbiol. Lett. 153:181–190 [DOI] [PubMed] [Google Scholar]
- 37.Quaiser A, Ochsenreiter T, Lanz C, Schuster SC, Treusch AH, Eck J, Schleper C. 2003. Acidobacteria form a coherent but highly diverse group within the bacterial domain: evidence from environmental genomics. Mol. Microbiol. 50:563–575 [DOI] [PubMed] [Google Scholar]
- 38.Barns SM, Cain EC, Sommerville L, Kuske CR. 2007. Acidobacteria phylum sequences in uranium-contaminated subsurface sediments greatly expand the known diversity within the phylum. Appl. Environ. Microbiol. 73:3113–3116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ley RE, Lozupone CA, Hamady M, Knight R, Gordon JI. 2008. Worlds within worlds: evolution of the vertebrate gut microbiota. Nat. Rev. Microbiol. 6:776–788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stearns JC, Lynch MDJ, Senadheera DB, Tenenbaum HC, Goldberg MB, Cvitkovitch DG, Croitoru K, Moreno-Hagelsieb G, Neufeld JD. 2011. Bacterial biogeography of the human digestive tract. Sci. Rep. 1:170. 10.1038/srep00170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Spieck E, Bock E. 2005. The lithoautotrophic nitrogen-oxidizing bacteria, p 149–153 In Garrity G. (ed), Bergey's manual of systematic bacteriology, 2nd ed, vol 2 Springer, Berlin, Germany [Google Scholar]
- 42.Soulat D, Bürckstümmer T, Westermayer S, Goncalves A, Bauch A, Stefanovic A, Hantschel O, Bennett KL, Decker T, Superti-Furga G. 2008. The DEAD-box helicase DDX3X is a critical component of the TANK-binding kinase 1-dependent innate immune response. EMBO J. 27:2135–2146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Karlsson J, Pütsep K, Chu H, Kays RJ, Bevins CL, Andersson M. 2008. Regional variations in Paneth cell antimicrobial peptide expression along the mouse intestinal tract. BMC Immunol. 9:37. 10.1186/1471-2172-9-37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bevins CL, Salzman NH. 2011. Paneth cells, antimicrobial peptides and maintenance of intestinal homeostasis. Nat. Rev. Microbiol. 9:356–368 [DOI] [PubMed] [Google Scholar]
- 45.Salzman NH, Hung K, Haribhai D, Chu H, Karlsson-Sjöberg J, Amir E, Teggatz P, Barman M, Hayward M, Eastwood D, Stoel M, Zhou Y, Sodergren E, Weinstock GM, Bevins CL, Williams CB, Bos NA. 2010. Enteric defensins are essential regulators of intestinal microbial ecology. Nat. Immunol. 11:76–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vaishnava S, Behrendt CL, Ismail AS, Eckmann L, Hooper LV. 2008. Paneth cells directly sense gut commensals and maintain homeostasis at the intestinal host-microbial interface. Proc. Natl. Acad. Sci. U. S. A. 105:20858–20863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sherman MP, Bennett SH, Hwang FFY, Sherman J, Bevins CL. 2005. Paneth cells and antibacterial host defense in neonatal small intestine. Infect. Immun. 73:6143–6146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Johansson MEV, Hansson GC. 2011. Keeping bacteria at a distance. Science 334:182–183 [DOI] [PubMed] [Google Scholar]
- 49.Kjellev S. 2009. The trefoil factor family—small peptides with multiple functionalities. Cell. Mol. Life Sci. 66:1350–1369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shub MD, Pang KY, Swann DA, Walker WA. 1983. Age-related changes in chemical composition and physical properties of mucus glycoproteins from rat small intestine. Biochem. J. 215:405–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dossinger V, Kayademir T, Blin N, Gött P. 2002. Down-regulation of TFF expression in gastrointestinal cell lines by cytokines and nuclear factors. Cell Physiol. Biochem. 12:197–206 [DOI] [PubMed] [Google Scholar]
- 52.Oeckinghaus A, Hayden MS, Ghosh S. 2011. Crosstalk in NF-κB signalling pathways. Nat. Immunol. 12:695–708 [DOI] [PubMed] [Google Scholar]
- 53.Van der Waaij D, Burghuis JM, Lekkerkerk JE. 1972. Colonization resistance of the digestive tract of mice during systemic antibiotic treatment. J. Hyg. (Lond.) 70:605–610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fukuda S, Toh H, Hase K, Oshima K, Nakanishi Y, Yoshimura K, Tobe T, Clarke JM, Topping DL, Suzuki T, Taylor TD, Itoh K, Kikuchi J, Morita H, Hattori M, Ohno H. 2011. Bifidobacteria can protect from enteropathogenic infection through production of acetate. Nature 469:543–547 [DOI] [PubMed] [Google Scholar]
- 55.McNulty NP, Yatsunenko T, Hsiao A, Faith JJ, Muegge BD, Goodman AL, Henrissat B, Oozeer R, Cools-Portier S, Gobert G, Chervaux C, Knights D, Lozupone CA, Knight R, Duncan AE, Bain JR, Muehlbauer MJ, Newgard CB, Heath AC, Gordon JI. 2011. The impact of a consortium of fermented milk strains on the gut microbiome of gnotobiotic mice and monozygotic twins. Sci. Transl. Med. 3:106ra106. 10.1126/scitranslmed.3002701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Coconnier MH, Lievin V, Lorrot M, Servin AL. 2000. Antagonistic activity of Lactobacillus acidophilus LB against intracellular Salmonella enterica serovar Typhimurium infecting human enterocyte-like Caco-2/TC-7 cells. Appl. Environ. Microbiol. 66:1152–1157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Altenhoefer A, Oswald S, Sonnenborn U, Enders C, Schulze J, Hacker J, Oelschlaeger TA. 2004. The probiotic Escherichia coli strain Nissle 1917 interferes with invasion of human intestinal epithelial cells by different enteroinvasive bacterial pathogens. FEMS Immunol. Med. Microbiol. 40:223–229 [DOI] [PubMed] [Google Scholar]
- 58.Eckburg PB, Bik EM, Bernstein CN, Purdom E, Dethlefsen L, Sargent M, Gill SR, Nelson KE, Relman DA. 2005. Diversity of the human intestinal microbial flora. Science 308:1635–1638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hayashi H, Takahashi R, Nishi T, Sakamoto M, Benno Y. 2005. Molecular analysis of jejunal, ileal, caecal and recto-sigmoidal human colonic microbiota using 16S rRNA gene libraries and terminal restriction fragment length polymorphism. J. Med. Microbiol. 54:1093–1101 [DOI] [PubMed] [Google Scholar]
- 60.Bik EM, Eckburg PB, Gill SR, Nelson KE, Purdom EA, Francois F, Perez-Perez G, Blaser MJ, Relman DA. 2006. Molecular analysis of the bacterial microbiota in the human stomach. Proc. Natl. Acad. Sci. U. S. A. 103:732–737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chambers JA, Hollingsworth MA, Trezise AE, Harris A. 1994. Developmental expression of mucin genes MUC1 and MUC2. J. Cell Sci. 107:413–424 [DOI] [PubMed] [Google Scholar]
- 62.Fança-Berthon P, Michel C, Pagniez A, Rival M, Van Seuningen I, Darmaun D, Hoebler C. 2009. Intrauterine growth restriction alters postnatal colonic barrier maturation in rats. Pediatr. Res. 66:47–52 [DOI] [PubMed] [Google Scholar]
- 63.Bry L, Falk P, Huttner K, Ouellette A, Midtvedt T, Gordon JI. 1994. Paneth cell differentiation in the developing intestine of normal and transgenic mice. Proc. Natl. Acad. Sci. U. S. A. 91:10335–10339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mallow EB, Harris A, Salzman N, Russell JP, DeBerardinis RJ, Ruchelli E, Bevins CL. 1996. Human enteric defensins. Gene structure and developmental expression. J. Biol. Chem. 271:4038–4045 [DOI] [PubMed] [Google Scholar]
- 65.Johansson ME, Phillipson M, Petersson J, Velcich A, Holm L, Hansson GC. 2008. The inner of the two Muc2 mucin-dependent mucus layers in colon is devoid of bacteria. Proc. Natl. Acad. Sci. U. S. A. 105:15064–15069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Vaishnava S, Yamamoto M, Severson KM, Ruhn KA, Yu X, Koren O, Ley R, Wakeland EK, Hooper LV. 2011. The antibacterial lectin RegIIIγ promotes the spatial segregation of microbiota and host in the intestine. Science 334:255–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kim YS, Ho SB. 2010. Intestinal goblet cells and mucins in health and disease: recent insights and progress. Curr. Gastroenterol. Rep. 12:319–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lin J, Holzman IR, Jiang P, Babyatsky MW. 1999. Expression of intestinal trefoil factor in developing rat intestine. Biol. Neonate 76:92–97 [DOI] [PubMed] [Google Scholar]
- 69.Yu H, He Y, Zhang X, Peng Z, Yang Y, Zhu R, Bai J, Tian Y, Li X, Chen W, Fang D, Wang R. 2011. The rat IgGFcγBP and Muc2 C-terminal domains and TFF3 in two intestinal mucus layers bind together by covalent interaction. PLoS One 6:e20334. 10.1371/journal.pone.0020334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Smythies LE, Sellers M, Clements RH, Mosteller-Barnum M, Meng G, Benjamin WH, Orenstein JM, Smith PD. 2005. Human intestinal macrophages display profound inflammatory anergy despite avid phagocytic and bacteriocidal activity. J. Clin. Invest. 115:66–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lotz M, Gütle D, Walther S, Ménard S, Bogdan C, Hornef MW. 2006. Postnatal acquisition of endotoxin tolerance in intestinal epithelial cells. J. Exp. Med. 203:973–984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Maheshwari A, Kelly DR, Nicola T, Ambalavanan N, Jain SK, Murphy-Ullrich J, Athar M, Shimamura M, Bhandari V, Aprahamian C, Dimmitt RA, Serra R, Ohls RK. 2011. TGF-β2 suppresses macrophage cytokine production and mucosal inflammatory responses in the developing intestine. Gastroenterology 140:242–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Shi J, Aono S, Lu W, Ouellette AJ, Hu X, Ji Y, Wang L, Lenz S, van Ginkel FW, Liles M, Dykstra C, Morrison EE, Elson CO. 2007. A novel role for defensins in intestinal homeostasis: regulation of IL-1beta secretion. J. Immunol. 179:1245–1253 [DOI] [PubMed] [Google Scholar]
- 74.Lindén SK, Florin TH, McGuckin MA. 2008. Mucin dynamics in intestinal bacterial infection. PLoS One 3:e3952. 10.1371/journal.pone.0003952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bergstrom KS, Kissoon-Singh V, Gibson DL, Ma C, Montero M, Sham HP, Ryz N, Huang T, Velcich A, Finlay BB, Chadee K, Vallance BA. 2010. Muc2 protects against lethal infectious colitis by disassociating pathogenic and commensal bacteria from the colonic mucosa. PLoS Pathog. 6:e1000902. 10.1371/journal.ppat.1000902 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.