

Abstract
Stressor exposure has been shown to enhance host susceptibility and the severity of a plethora of illnesses, including gastrointestinal disease. In mice, susceptibility to Citrobacter rodentium has been shown to be dependent on host genetics as well as the composition of the intestinal microbiota, but the effects of stressor exposure on this gastrointestinal pathogen have not been elucidated fully. Previously, our lab showed that exposure to the prolonged-restraint stressor prior to a challenge with C. rodentium alters the intestinal microbiota community structure, including a reduction of beneficial genera such as Lactobacillus, which may contribute to stressor-enhanced C. rodentium-induced infectious colitis. To test the effects of stressor exposure on C. rodentium infection, we exposed resistant mice to a prolonged-restraint stressor concurrent with pathogen challenge. Exposure to prolonged restraint significantly enhanced C. rodentium-induced infectious colitis in resistant mice, as measured by increases in colonic histopathology, colonic inflammatory mediator gene production, and pathogen translocation from the colon to the spleen. It was further tested if the beneficial bacterium Lactobacillus reuteri could reduce the stressor-enhanced susceptibility to C. rodentium-enhanced infectious colitis. While L. reuteri treatment did not reduce all aspects of stressor-enhanced infectious colitis, it did significantly reduce pathogen translocation from the colon to the spleen. Taken together, these data demonstrate the deleterious effects that prolonged stressor exposure can have at the onset of a gastrointestinal infection by its ability to render a resistant mouse highly susceptible to C. rodentium. Probiotic treatment ameliorated the systemic manifestations of stress on colonic infection.
INTRODUCTION
Citrobacter rodentium, along with enteropathogenic and enterohemorrhagic Escherichia coli (EPEC and EHEC, respectively), belongs to the group of noninvasive pathogens collectively known as attaching and effacing (A/E) bacteria. In order to colonize its host, C. rodentium, along with its human homologs, must intimately attach to the intestinal epithelium. This interaction allows the pathogen to inject effector proteins into the host epithelial cell via a type III secretion system. These proteins, which include intimin, Tir, EspF, and Map, are located on the locus of enterocyte effacement pathogenicity island and are involved in the adherence to and effacement of the colonic epithelium, as well as the formation of actin-rich pedestals beneath the pathogen (1). Upon colonization of the colonic epithelium, A/E pathogens induce increases in leukocyte infiltration, inflammatory cytokine production, and structural changes to colonic tissue that are similar to those with EPEC and EHEC infection as well as both murine and human inflammatory bowel disease (1–3). Because mice are not naturally susceptible to intestinal EPEC or EHEC, murine infection with C. rodentium has become a widely used model of intestinal E. coli infection. C. rodentium does not normally cause disease in humans but has been shown to colonize all strains of mice. The disease can be either subclinical or fatal, depending on the host mouse strain (4). Strains that develop only subclinical symptoms, such as CD-1 and C57BL/6, are considered resistant to C. rodentium-induced infectious colitis, while mice that succumb to infection by C. rodentium, such as FVB/N and C3H/HeJ mice, are deemed susceptible (1, 4, 5). While it is recognized that host genetics contributes to enhanced susceptibility (4, 5), recent data indicate that the colonic microbiota also modulates host susceptibility to C. rodentium, in part by altering mucosal immunity (6–8). Antibiotic treatment can disrupt the natural balance of the microbiota, which can leave mice more susceptible to C. rodentium infection (9). Conversely, supplementation with beneficial bacteria can reduce susceptibility to C. rodentium infection (10–13).
Animal stressors are useful for our understanding of the impact of the host stress response on infectious diseases. Our laboratory has shown that exposure to laboratory stressors, including the most commonly used experimental stressor in biomedical and biobehavioral research, namely, physical restraint (14), significantly changes the structure of the colonic microbiota and reduces the levels of beneficial members of the genus Lactobacillus (15–18). Because lactobacilli can help to regulate mucosal inflammation and mediate resistance/susceptibility to C. rodentium (10–13, 19, 20), we hypothesized that exposing resistant CD-1 mice to a prolonged-restraint stressor during challenge with C. rodentium would significantly increase disease pathology.
It has long been known that exposure to psychological stressors can exacerbate both infectious and noninfectious diseases, and gastrointestinal (GI) diseases such as Crohn's disease, ulcerative colitis, and enteric infectious disease are often exacerbated during stressful periods (21, 22). While the mechanisms by which stressor exposure exacerbates GI diseases are still under investigation, disruptions of gut epithelial permeability are likely involved. The GI tract is lined by a single layer of epithelial cells that keeps the interior of the body separated from the contents of the gut. During stressful life events, the gut can become inflamed (23), and the tight junctions that help to control what is transported across the epithelial layer are loosened, leading to a “leaky gut.” The loosening of tight junctions, chiefly by the proinflammatory cytokine tumor necrosis factor alpha (TNF-α), allows the contents of the gut, including commensal microbes, to pass through the epithelial layer and enter into the lamina propria.
The penetration of microbes into the lamina propria results in a stronger inflammatory response, in part through activation of mucosal immune cells (24). Thus, strategies to reinforce the epithelial barrier and suppress mucosal immune activity could be beneficial in preventing stressor-induced exacerbations of GI diseases. One potentially useful strategy involves the administration of probiotic microbes, such as Lactobacillus reuteri. The Gram-positive bacterium L. reuteri can be found in both humans and laboratory animals, including mice. Many strains of L. reuteri are currently being used as probiotics, because research has shown that L. reuteri can suppress inflammation and immune cell activity (25–28). Moreover, L. reuteri has been shown to reduce experimental colitis and prevent experiment-induced intestinal permeability in rats (29, 30). Because stressor exposure reduces the levels of commensal L. reuteri found in the murine colon (J. Galley and M. T. Bailey, unpublished observations), this study tested the hypothesis that stressor exposure would increase the severity of C. rodentium-induced colonic pathology in a resistant mouse strain. This study also tested the corollary hypothesis that treating the mice with L. reuteri would prevent any stressor-induced increases in disease severity.
MATERIALS AND METHODS
Mice.
Outbred, male CD-1 mice aged 6 to 8 weeks were purchased from Charles River Laboratories (Wilmington, MA) and housed at 3 mice per cage, unless otherwise indicated, in ventilated polycarbonate cages (32 cm × 18 cm × 15 cm). For all experiments, mice were allowed to acclimate to their surroundings for 1 week prior to experimentation. Lights were maintained on a 12-h on, 12-h off cycle (lights on at 0600). Mice were housed in an Association for the Assessment and Accreditation of Laboratory Animal Care (AAALAC)-accredited facility and had free access to food and water except during experimental procedures. All procedures were approved by the Institutional Laboratory Animal Care and Use Committee (ILACUC) at the Ohio State University (protocol 2009A0235).
Prolonged restraint.
Prolonged restraint was performed as previously described (18, 31). Briefly, mice were divided into the following three groups: home-cage control (HCC), food- and water-deprived (FWD), and prolonged-restraint (RST) groups. Restrained mice were placed in well-ventilated 50-ml conical tubes at the beginning of their active cycle (i.e., at 1800) and were removed from the tubes the following morning at 0900. Because the stressor-exposed mice did not have access to food or water at the time of restraint, FWD mice had their food and water removed from their cages at the time of restraint but were allowed to move freely about their cages. Both RST and FWD mice were allowed free access to their food and water from 0900 to 1800. Mice were restrained or deprived of food and water each night for seven consecutive nights. Control mice were left undisturbed in their cages for the duration of the experiment.
Infection and bacterial enumeration.
Citrobacter rodentium strain DBS120(pCRP1::Tn5) (32) was grown overnight in Difco Lennox broth (LB) at 37°C. Cultures were centrifuged, and the resulting pellet was resuspended in sterile phosphate-buffered saline (PBS) to give a final concentration of 3 × 109 CFU/ml. Mice were then challenged at 0900 on the day following the first night of restraint via oral gavage with 100 μl containing 3 × 108 CFU and were food and water deprived for 2 h postchallenge. Mice in the RST group were restrained during the first 6 days postchallenge. The day of infection is referred to as day 0, and all data collected are referenced by the number of days postchallenge. Fecal shedding of C. rodentium was monitored on days 1, 3, 6, 12, and 24 postchallenge. Fresh stool was collected from individual mice by placing them in a sterile cage and collecting 1 or 2 stool pellets per mouse. The stool was then weighed, homogenized manually in 3 ml sterile PBS, and serially diluted at a ratio of 1:20. In order to enumerate C. rodentium only, each dilution was grown via the pour plate method in MacConkey agar supplemented with the antibiotic kanamycin (40 μg/ml) overnight at 37°C. At the time of sacrifice, spleens were flash frozen in liquid nitrogen. Spleens were then thawed, weighed, and suspended in 5 ml sterile PBS. Spleens were homogenized using a stomacher (medium speed for 1 min) and then serially diluted and plated in the same fashion as stool samples.
Experiment 1 design.
The first set of experiments was designed to test the hypothesis that prolonged stressor exposure would exacerbate C. rodentium-induced infectious colitis. To test the effects of prolonged stressor exposure on the severity of C. rodentium infection, two replicate time course experiments were performed. In the first time course experiment, all mice were housed at 3 mice/cage/group (i.e., HCC, FWD, or RST). The mice in one cage for each group were sacrificed on days 1, 3, 6, 12, and 24 postchallenge. In the replicate time course experiment, all mice were housed at 3 mice/cage/group, with the exception of mice to be sacrificed on day 12 postchallenge, which were housed at 5 mice/cage/group. The mice in one cage for each group were sacrificed on days 0, 1, 3, 6, 12, and 24 postchallenge, for a grand total of 105 mice between the two time course experiments. There were no differences in any of the measurements taken from mice that were housed at either 3 or 5 per cage on day 12 postchallenge (analysis not shown). For each experiment, all mice were infected with C. rodentium immediately following the first night of stressor exposure, with the exception of mice sacrificed on day 0, as these mice were used as noninfected baseline controls for colonic gene expression analyses. Stressor exposure continued for 6 more days, after which mice were left alone in their cages throughout the remainder of the experiment. Each time point reflects the day postchallenge. Mice were sacrificed at the designated time points by CO2 asphyxiation, and tissue samples were collected. Because there were no differences between the HCC and FWD control groups for any of the measures assessed, these groups were combined into one control group (CON).
Probiotic administration.
Lactobacillus reuteri strain 23272 (American Type Culture Collection [ATCC]) was grown overnight in Difco Lactobacilli MRS broth at 37°C with 5% CO2. Cultures were centrifuged, and the resulting pellets were resuspended in sterile PBS (vehicle [VEH]). The bacteria were then added to the drinking water to give a final concentration of 5 × 107 CFU/ml of drinking water. Mice received either probiotic or VEH in their water from 1 day prior to the first day of stressor exposure until the mice were sacrificed on day 12 postchallenge. Water supplemented with L. reuteri was monitored throughout the duration of the experiment to ensure viability. One milliliter of water was removed via a Lixit water bottle and serially diluted 1:20. Each dilution was grown on Difco Lactobacilli MRS agar at 37°C with 5% CO2. Mice typically consumed 3 ml of the fluid per day, corresponding to approximately 1.5 × 108 CFU/ml.
Experiment 2 design.
The second set of experiments was designed to test the hypothesis that probiotic intervention could prevent stressor-enhanced disease severity associated with C. rodentium challenge. Because our initial set of experiments demonstrated that the peak of C. rodentium-induced infectious colitis occurred on day 12 postchallenge, our second set of experiments was focused on day 12 postchallenge. Our hypothesis was tested over 3 replicate experiments involving 6 experimental groups (i.e., HCC/VEH, HCC/L. reuteri, FWD/VEH, FWD/L. reuteri, RST/VEH, and RST/L. reuteri). Mice were housed at 3 mice/cage/group, for a total of 54 infected mice. Noninfected control mice were used as baseline control samples for real-time PCR analysis (4 cages of noninfected control mice housed at 3 mice/cage, for a total of 12 noninfected control mice). Stressor exposure and C. rodentium challenge were the same as in the first set of experiments, but mice were given probiotic L. reuteri in their water beginning 1 day prior to stressor exposure and continuing through day 12 postchallenge. Mice were sacrificed on day 12 postchallenge by CO2 asphyxiation, and tissue samples were collected. Because there were no differences between the HCC and FWD mice within treatment groups for any of the measures assessed in the second set of experiments, these groups were combined based on treatment condition (CON/VEH and CON/L. reuteri).
Histopathology.
On days 0, 1, 3, 6, 12, and 24 postchallenge, colons were excised and cut in half longitudinally. One section was used for further analysis of colonic cytokines by real-time PCR, and the other was fixed in 10% formalin-buffered phosphate for 24 to 48 h. Samples were embedded in paraffin, and 5-μm-thick sections were stained with hematoxylin and eosin (H&E) for histopathological evaluation by a board-certified veterinary pathologist (N.M.A.P.) who was blinded to treatment groups. The severity of lesions in the colon was scored according to previously defined criteria (33). Briefly, each colon was scored on 5 different categories: inflammation, dysplasia, hyperplasia, edema, and epithelial defects. Each category received a score of 0 to 4 in increments of 0.5, based on the degree of lesion severity, as follows: 0, absent; 1, mild; 2, moderate; 3, marked; and 4, severe. All 5 categories were added together to garner a total histologic colitis index score with a maximum value of 20.
Semiquantitative real-time PCR.
Colons were harvested, and total RNA was isolated using a standard single-step isolation protocol (TRI-zol; Invitrogen). After isolation, total RNA was quantified spectrophotometrically. A reverse transcription reaction was performed to synthesize cDNA by use of an avian myeloblastosis virus (AMV) reverse transcriptase enzyme kit (Promega Corporation, Madison, WI). These samples then underwent real-time PCR in order to amplify the nucleotide sequences of interest. In all cases, the 18S rRNA gene was used as the housekeeping gene. The sequences of primers and probes used can be found in Table 1. SYBR green was used in place of a labeled probe sequence for β-defensin 3, claudin 1, and claudin 5. The master mix consisted of 2× Universal TaqMan master mix (or SYBR green), 0.9 μM (each) forward and reverse primers, and 0.250 μM sample cDNA. The PCR took place in a Prism 7000 sequence detection system and began with 2 min at 50°C, 10 min at 95°C, and then 40 amplification cycles of 15 s at 95°C and 1 min at 60°C. The relative amount of mRNA was determined using the comparative cycle threshold (CT) method as previously described (18, 31). The baseline control samples for experiment 1 were control samples from day 0, and those for experiment 2 were the day 12 noninfected control samples. Each control sample was set at a value of 1 and was used as a reference for all other experimental samples.
Table 1.
Sequences of primers and probes used for real-time PCRa
| Primer or probe | Primer direction | Sequence |
|---|---|---|
| CCL2 | Forward | TTGGCTCAGCCAGATGCA |
| Reverse | CCTACTCATTGGGATCATCTTGC | |
| CCL2 probe | AACGCCCCACTCACCTGCTGCTACT | |
| TNF-α | Forward | CTGTCTACTGAACTTCGGGGTGAT |
| Reverse | GCTCTGGGCCATAGAACTGATG | |
| TNF-α probe | ATGAGAAGTTCCCAAATGGCCTCCCTC | |
| iNOS | Forward | CAGCTGGGCTGTACAAACCTT |
| Reverse | TGAATGTGATGTTTGCTTCGG | |
| iNOS probe | CGGGCAGCCTGTGAGACCTTTGA | |
| 18S rRNA | Forward | CGGCTACCACATCCAAGGAA |
| Reverse | GCTGGAATTACCGCGGCT | |
| 18S rRNA probe | TGCTGGCACCAGACTTGCCCTC | |
| β-Defensin 3 | Forward | GCATTTGAGGAAAGGAACTCCACA |
| Reverse | GTCTCCACCTGCAGCTTTTAGCAA | |
| Claudin 1 | Forward | TTCGCAAAGCACCGGGCAGATACA |
| Reverse | GCCACTAATGTCGCCAGACCTGAA | |
| Claudin 5 | Forward | TTAAGGCACGGGTAGCACTCACG |
| Reverse | TTAGACATAGTTCTTCTTGTCGTA |
Primers were purchased from Invitrogen (Carlsbad, CA), and probes were purchased from Applied Biosystems (Foster City, CA).
Sickness assessment.
On day 12 postchallenge, mice were subjected to behavioral testing on an open field. The open field consisted of a 30-cm by 30-cm by 25-cm Plexiglas box. A 6 × 6 grid was drawn on the floor in order to test locomotion. Each mouse was placed in the open field separately and video recorded for 5 min. This test is designed to take advantage of the natural exploratory behaviors of mice. Mice are naturally curious about new environments and will explore unless the desire to explore is overridden by illness (34, 35). Sickness behavior was assessed by counting the total number of lines that were crossed by all 4 paws of each mouse within the 5-min period. Open spaces are avoided by mice due to the perceived risk of predation; however, their curious nature will cause them to eventually explore the open space of the center of the open field. The time spent in the center of the open field was recorded. Mice with more anxiety-like behaviors spend less time in the center of the open field (36).
Cytokine analysis.
Blood was collected by cardiac puncture immediately following sacrifice. Sera were collected by centrifuging samples at 1,500 × g for 10 min at 4°C and were stored at −80°C. Serum was diluted 4-fold with serum dilution buffer (Bio-Rad). A commercially available multiplex bead-based cytokine immunoassay (BioPlex; Bio-Rad Laboratories Inc., Hercules, CA) was used to measure the contents of interleukin-1β (IL-1β), IL-2, IL-6, IL-10, IL-12(p70), gamma interferon (IFN-γ), and TNF-α according to the manufacturer's protocol. Results are expressed as picograms/ml recovered from serum. The lower limits of detection were 14.1 pg/ml for IL-1β, 0.4 pg/ml for IL-2, 0.5 pg/ml for IL-6, 1.4 pg/ml for IL-10, 0.5 pg/ml for IL-12(p70), 0.1 pg/ml for IFN-γ, and 6.0 pg/ml for TNF-α.
Statistical analysis.
Stressor-induced changes in C. rodentium colonization, gene expression, and serum cytokine levels were analyzed by a two-factor analysis of variance (ANOVA), with group (CON versus RST) and day as between factors for the first set of experiments and stressor exposure (CON versus RST) and probiotic treatment (VEH versus L. reuteri) as between factors for the second set of experiments. Analyses were conducted using IBM SPSS for Windows (SPSS, Chicago, IL). Post hoc analysis comprised two-tailed Student's t test with the Bonferroni correction. Chi-square tests were performed on pathogen prevalence in the spleen. P values of <0.05 were considered significant.
RESULTS
Prolonged restraint enhances pathogen colonization and colonic histopathology.
Exposure to prolonged restraint changed C. rodentium levels throughout the 24-day experiment compared to those in control mice [F(4, 84) = 3.57; P = 0.01] (Fig. 1A). This effect was due mainly to significant differences in colonization in the RST group on days 1, 6, and 12 postchallenge (P < 0.005). While the peak of colonization was on day 6, we did not observe any significant colonic pathological changes until day 12 postchallenge. Increased colonic mass due to epithelial cell hyperplasia, a hallmark of C. rodentium infection, was significantly increased on day 12 postchallenge in the stressor-exposed group [F(1, 13) = 38.1; P < 0.001] (Fig. 1B). Total colonic histopathology, which is the combination of scores given to inflammation, edema, hyperplasia, dysplasia, and epithelial defects, was also significantly increased by prolonged restraint [F(2, 45) = 8.2; P = 0.001] (Fig. 1C). Control colons had a relatively normal appearance, with small numbers of lymphocytes infiltrating the mucosa (Fig. 1D). However, stressor-increased histopathology was evidenced by mucosal thickening, regional ulcerations in the surface epithelium, and a significant inflammatory cell infiltrate that extended from the mucosa to the edematous submucosa (Fig. 1E).
Fig 1.

Stressor exposure enhances C. rodentium-induced infectious colitis. Mice were restrained for 1 day prior to oral challenge with 3 × 108 CFU of C. rodentium. Restraint continued for 6 days postchallenge. (A) To determine the extent of colonization by C. rodentium, stool samples were collected on days 0, 1, 3, 6, 12, and 24 postchallenge. The pathogen was enumerated via the pour plate method. Exposure to restraint stress significantly increased C. rodentium colonization compared to that in CON mice. *, P < 0.005 for RST versus CON groups on indicated days postchallenge, as assessed with Student's t test with the Bonferroni correction factor as a post hoc test; †, P = 0.07 for RST versus CON groups on day 3 postchallenge, as assessed with Student's t test. Sample sizes were as follows: day 1, n = 11 (CON) and n = 5 (RST); days 3, 6, and 24, n = 12 (CON) and n = 5 (RST); and day 12, n = 16 (CON) and n = 8 (RST). (B) Colons were removed on day 12 and weighed without contents. Exposure to prolonged restraint significantly increased the weight of the colonic tissue compared to that of CON mice. *, P < 0.0001 for CON versus RST groups as assessed using ANOVA. Sample sizes were as follows: n = 10 (CON) and n = 5 (RST). (C) On days 6, 12, and 24 postchallenge, colons were removed, fixed in formalin, and subsequently embedded in paraffin. Colons were sectioned and stained with hematoxylin and eosin in order to visualize and score the pathology present in each sample. Stressor exposure significantly increased the total pathology scores. *, P < 0.00001 between RST and CON groups on day 12 postchallenge, as assessed using Student's t test with the Bonferroni correction as a post hoc test. Sample sizes were as follows: day 6, n = 6 (CON) and n = 3 (RST); day 12, n = 16 (CON) and n = 8 (RST); and day 24, n = 12 (CON) and n = 6 (RST). (D) Representative image of H&E-stained colon from a CON mouse on day 12 postchallenge. (E) Representative image of H&E-stained colon from an RST mouse on day 12 postchallenge. The asterisk marks a region with significant inflammatory cell infiltration, whereas the “X” marks regional ulcerations in the surface epithelium.
Prolonged restraint enhances inflammatory mediator gene expression in the colons of infected mice.
Colonic inflammatory mediator mRNA expression (i.e., CCL2, TNF-α, and inducible NO synthase [iNOS] mRNAs) was assessed using real-time PCR. Colonic CCL2 mRNA expression was significantly increased by stressor exposure throughout the 24-day experiment compared to the control level [F(5, 90) = 4.5; P = 0.001] (Fig. 2A). Mice exposed to prolonged restraint also had enhanced colonic TNF-α mRNA expression compared to nonstressed controls [F(5, 93) = 9.2; P < 0.001] (Fig. 2B). Similar to that of CCL2 and TNF-α, colonic iNOS gene expression was significantly increased throughout the 24-day experiment compared to colons of control mice [F(5, 93) = 45.3; P < 0.0001] (Fig. 2C). The most evident increases in colonic CCL2, TNF-α, and iNOS mRNA expression occurred on day 12 postchallenge.
Fig 2.
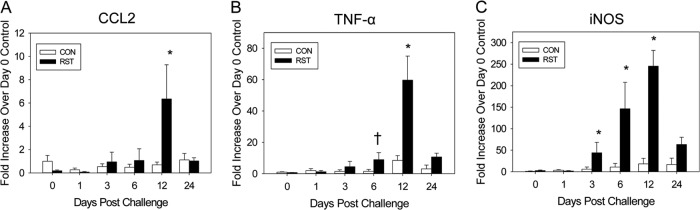
Stressor exposure during infection with C. rodentium enhances colonic inflammatory mediator gene expression. Mice were restrained for 1 day prior and 6 days after oral challenge with C. rodentium. Mice were sacrificed on days 0, 1, 3, 6, 12, and 24 postchallenge. Colons were processed in order to quantify gene expression by real-time PCR. (A) Stressor exposure significantly increased gene expression of CCL2 on day 12 postchallenge. *, P < 0.0005 between RST and CON groups on day 12 postchallenge, as assessed using Student's t test with the Bonferroni correction factor as a post hoc test. Sample sizes were as follows: day 0, n = 6 (CON) and n = 3 (RST); day 1, n = 11 (CON) and n = 5 (RST); days 3 and 6, n = 12 (CON) and n = 6 (RST); day 12, n = 16 (CON) and n = 8 (RST); and day 24, n = 11 (CON) and n = 6 (RST). (B) Prolonged restraint significantly increased TNF-α gene expression on days 6 and 12 postchallenge. †, P < 0.06 between RST and CON groups on day 6 postchallenge; *, P < 0.0005 between RST and CON groups on day 12 postchallenge. Student's t test with the Bonferroni correction factor was used as a post hoc test. Sample sizes were as follows: day 0, n = 6 (CON) and n = 3 (RST); days 1, 3, 6, and 24, n = 12 (CON) and n = 6 (RST); and day 12, n = 16 (CON) and n = 8 (RST). (C) Exposure to prolonged restraint also significantly increased iNOS mRNA expression on days 3, 6, and 12 postchallenge. *, P < 0.005 between RST and CON groups on each day postchallenge. Student's t test with the Bonferroni correction factor was used as a post hoc test. Sample sizes were as follows: day 0, n = 6 (CON) and n = 3 (RST); days 1, 3, 6, and 24, n = 12 (CON) and n = 6 (RST); and day 12, n = 16 (CON) and n = 8 (RST).
Prolonged restraint causes significant changes in spleens of infected animals.
One of the main characteristics of prolonged restraint is a reduction in spleen mass (37), which was observed on days 1, 3, and 6 postchallenge (i.e., after 2, 5, and 7 days of restraint) in stressor-exposed mice (Fig. 3A). This reduction was not evident throughout the experiment, but there were significant changes in spleen mass across the 24-day experiment [F(4, 76) = 20.1; P < 0.001]. To determine whether the unexpected increase in spleen mass on day 12 postchallenge in stressor-exposed mice was associated with an increased escape of C. rodentium from the colon and translocation to the spleen, we set out to determine if C. rodentium could be cultured from the spleen. On days 0, 1, 3, 6, and 24 postchallenge, there were no countable colonies in the CON and RST groups (data not shown). However, on day 12 postchallenge, there was an increase in the likelihood (88% versus 25%) of observing C. rodentium in the spleens of stressor-exposed mice [χ2(1) = 8.21; P < 0.005] (Fig. 3B). Not only was there an increase in the likelihood of detecting C. rodentium in the spleens of stressor-exposed mice, but there was also an enhanced pathogen burden in those mice found to have C. rodentium in the spleen [F(1, 22) = 10.0; P < 0.01] (Fig. 3C).
Fig 3.
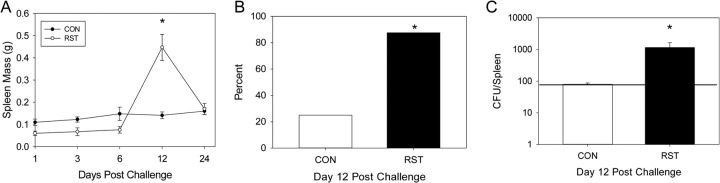
Stressor exposure enhances the translocation of C. rodentium from the colon to the spleen. Mice were restrained for 1 day prior to oral challenge with C. rodentium and then for 6 days postchallenge. Spleens were removed, weighed, and processed in order to enumerate C. rodentium by the pour plate method. (A) Stressor exposure significantly increased the size of the spleen on day 12 postchallenge compared to that for CON mice. *, P < 0.00001 between CON and RST groups on day 12 postchallenge. Student's t test with the Bonferroni correction factor was used as a post hoc test. Sample sizes were as follows: day 1, n = 6 (CON) and n = 3 (RST); days 3 and 6, n = 12 (CON) and n = 6 (RST); day 12, n = 16 (CON) and n = 8 (RST); and day 24, n = 11 (CON) and n = 6 (RST). (B) Stressor exposure increased the likelihood of pathogen translocation from the colon to spleen on day 12 postchallenge. *, P < 0.005 between CON and RST groups as assessed by chi-square (χ2) analysis. Sample sizes were as follows: n = 16 (CON) and n = 8 (RST). (C) Stressor exposure also significantly increased the pathogen burden in the spleen. *, P < 0.01 between CON and RST groups as assessed by ANOVA. Sample sizes were as follows: n = 16 (CON) and n = 8 (RST). The horizontal line in panel C represents the detection limit for C. rodentium.
Probiotic intervention does not reduce RST-enhanced pathogen colonization or colonic histopathology.
As with the previous experiment, stressor exposure significantly increased fecal C. rodentium levels on day 12 postchallenge [F(1, 50) = 17.2; P < 0.0001] (Fig. 4A). There were no changes in colonization between VEH-treated and L. reuteri-treated groups. Treatment with L. reuteri also had no effect on the stressor-induced increase in colonic histopathology. Colonic mass on day 12 postchallenge was significantly increased in both the VEH- and L. reuteri-treated stressor-exposed mice compared to control mice [F(1, 49) = 24; P < 0.001] (Fig. 4B). Total colonic histopathology was also increased in the RST mice compared to the CON mice [F(1, 28) = 9.4; P = 0.005] (Fig. 4C). In no case did treatment with L. reuteri affect colonic histopathology; histopathology was similar in VEH-treated and L. reuteri-treated mice. No lesions were observed in mice from either control group (Fig. 4D and E). Stressor-enhanced histopathology was evidenced by lesions containing inflammation, epithelial defects, hyperplasia, and dysplasia. Neutrophilic inflammation extended from the mucosa to the submucosa and was frequently associated with epithelial erosion and ulceration (Fig. 4F and G).
Fig 4.

Probiotic intervention with L. reuteri does not reduce stressor-enhanced pathogen colonization or colonic histopathology. Mice were restrained for 1 day and then orally challenged with C. rodentium. Restraint continued for 6 days postchallenge, during which the drinking water was supplemented with either PBS vehicle (VEH) or L. reuteri beginning 1 day prior to restraint. (A) To determine the extent of colonization by C. rodentium, stool samples were collected on day 12 postchallenge. Exposure to restraint stress significantly increased C. rodentium colonization compared to that in CON mice, regardless of treatment. *, ANOVA main effect for group (CON versus RST; P < 0.0001). Sample sizes were as follows: n = 18 (CON/VEH), n = 18 (CON/L. reuteri), n = 9 (RST/VEH), and n = 9 (RST/L. reuteri). (B) Colonic tissue was removed on day 12 postchallenge and weighed without contents. Exposure to prolonged restraint significantly increased the weight of the colonic tissue compared to that of CON mice, regardless of probiotic intervention. *, ANOVA main effect for group (CON versus RST; P < 0.0001). Sample sizes were as follows: n = 18 (CON/VEH), n = 17 (CON/L. reuteri), n = 9 (RST/VEH), and n = 9 (RST/L. reuteri). On day 12 postchallenge, colons were removed, fixed in formalin, and embedded in paraffin. Colons were sectioned and stained with hematoxylin and eosin in order to visualize and score the pathology present in each sample. (C) Stressor exposure enhanced colonic histopathology in both VEH- and L. reuteri-treated mice. *, ANOVA main effect for group (CON versus RST; P = 0.005). Sample sizes were as follows: n = 11 (CON/VEH), n = 12 (CON/L. reuteri), n = 5 (RST/VEH), and n = 4 (RST/L. reuteri). (D) Representative image of H&E-stained colon from a CON/VEH mouse. (E) Representative image of H&E-stained colon from a CON/L. reuteri mouse. (F) Representative image of H&E-stained colon from an RST/VEH mouse. (G) Representative image of H&E-stained colon from an RST/L. reuteri mouse. For panels F and G, an asterisk marks an area with significant inflammatory cell infiltrate, “X” marks an area with surface epithelial erosions, and “M” marks dysplasia due to disorganized mucosal glands.
Probiotic intervention reduces stressor-enhanced colonic gene expression.
Colonic inflammatory mediator gene expression was previously shown to be increased on day 12 post-C. rodentium challenge in the RST group compared to the control counterparts. Stressor exposure caused a significant increase in colonic CCL2 mRNA gene expression on day 12 postchallenge [F(1, 49) = 22.4; P < 0.001] (Fig. 5A). While L. reuteri treatment reduced the mean level of CCL2 mRNA, the L. reuteri-induced reduction was not large enough to be considered statistically significant. Similarly, colonic TNF-α gene expression also experienced a significant increase in stressor-exposed mice compared to nonstressed, infected controls [F(1, 50) = 25.3; P < 0.001] (Fig. 5B), with a nonsignificant reduction in L. reuteri-treated, stressor-exposed mice. Stressor exposure also caused a significant increase in colonic iNOS gene expression [F(1, 50) = 35.2; P < 0.001] (Fig. 5C). This stressor-enhanced increase of colonic iNOS mRNA was unchanged by L. reuteri treatment. The antimicrobial peptide β-defensin 3 was significantly reduced in the colons of C. rodentium-challenged, stressor-exposed mice compared to infected, nonstressed controls [F(1, 30) = 6.5; P < 0.05] (Fig. 5D). This stressor-enhanced 5-fold decrease in β-defensin 3 gene expression in colons of VEH-treated mice was reversed by L. reuteri treatment. There was a significant reduction in colonic claudin 1 mRNA expression in stressor-exposed mice compared to nonstressed controls [F(1, 30) = 15.0; P = 0.001] (Fig. 5E), an effect which was unchanged by L. reuteri treatment. Claudin 5 was also significantly reduced in stressor-exposed mice compared to nonstressed controls [F(1, 29) = 6.0; P = 0.01] (Fig. 5F). The observed stressor-enhanced decrease in VEH-treated mice was reversed by L. reuteri treatment.
Fig 5.

Probiotic intervention reduces stressor-enhanced colonic gene expression during the peak of infection. Mice were restrained for 1 day and then orally challenged with C. rodentium. Restraint continued for 6 days postchallenge, during which the drinking water was supplemented with either VEH or L. reuteri, beginning 1 day prior to restraint. Colons were processed in order to quantify gene expression by real-time PCR. (A) Stressor exposure significantly increased gene expression of CCL2, regardless of treatment. *, ANOVA main effect for group (CON versus RST; P < 0.001). Sample sizes were as follows: n = 17 (CON/VEH), n = 18 (CON/L. reuteri), n = 9 (RST/VEH), and n = 9 (RST/L. reuteri). (B) Stressor exposure also significantly enhanced TNF-α gene expression in both VEH- and L. reuteri-treated mice. *, ANOVA main effect for group (CON versus RST; P < 0.001). Sample sizes were as follows: n = 18 (CON/VEH), n = 18 (CON/L. reuteri), n = 9 (RST/VEH), and n = 9 (RST/L. reuteri). (C) Stressor exposure significantly increased colonic iNOS gene expression in both VEH- and L. reuteri-treated mice. *, ANOVA main effect for group (CON versus RST; P < 0.001). Sample sizes were as follows: n = 18 (CON/VEH), n = 18 (CON/L. reuteri), n = 9 (RST/VEH), and n = 9 (RST/L. reuteri). (D) Stressor exposure significantly reduced β-defensin 3 gene expression only in VEH-treated mice. *, P = 0.05 between CON/VEH and RST/VEH groups as assessed by Student's t test with the Bonferroni correction factor as a post hoc test. Sample sizes were as follows: n = 12 (CON/VEH), n = 12 (CON/L. reuteri), n = 6 (RST/VEH), and n = 4 (RST/L. reuteri). (E) Claudin 1 was also significantly reduced in RST mice, regardless of the treatment group. *, ANOVA main effect for group (CON versus RST; P = 0.001). Sample sizes were as follows: n = 12 (CON/VEH), n = 12 (CON/L. reuteri), n = 6 (RST/VEH), and n = 4 (RST/L. reuteri). (F) Claudin 5 was significantly reduced by RST only in VEH-treated mice. *, P < 0.05 between CON/VEH and RST/VEH groups. Student's t test with the Bonferroni correction factor was used as a post hoc test. Sample sizes were as follows: n = 12 (CON/VEH), n = 12 (CON/L. reuteri), n = 5 (RST/VEH), and n = 4 (RST/L. reuteri).
Probiotic intervention reduces stressor-enhanced pathogen dissemination to the spleen.
On day 12 postchallenge, spleen mass was significantly changed by L. reuteri supplementation [F(1, 50) = 7.6; P < 0.01] (Fig. 6A). There was a significant increase in day 12 spleen mass in stressor-exposed, VEH-treated mice, and this was prevented by L. reuteri treatment. On day 12 postchallenge, there was a significant increase in the likelihood of discovering C. rodentium in stressor-exposed mice compared to nonstressed mice [χ2(1) = 4.75; P < 0.05] (Fig. 6B). While there were no significant changes in pathogen dissemination in VEH-treated and L. reuteri-treated RST groups, there was a 24% drop in pathogen prevalence in spleens from L. reuteri-treated, stressor-exposed mice (78% versus 54%). Pathogen burden was also significantly increased in stressor-exposed mice compared to nonstressed control mice [F(1, 47) = 6.1; P < 0.05] (Fig. 6C). This effect was diminished in L. reuteri-fed, stressor-exposed mice.
Fig 6.
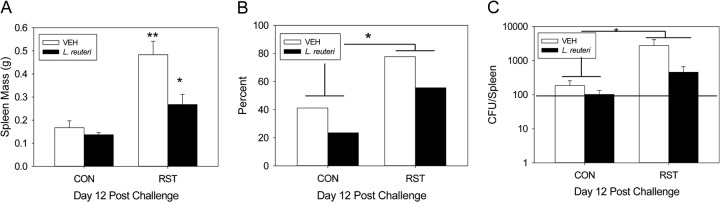
Probiotic intervention reduces stressor-enhanced pathogen translocation to the spleen. Mice were restrained for 1 day and then orally challenged with C. rodentium. Restraint continued for 6 days postchallenge, during which the drinking water was supplemented with either VEH or L. reuteri, beginning 1 day prior to restraint. Spleens were removed, weighed, and processed in order to enumerate C. rodentium by the pour plate method. (A) Stressor exposure significantly increased the size of the spleen on day 12 postchallenge in VEH-treated mice compared to CON/VEH mice. This effect was blocked in stressor-exposed mice that received L. reuteri. **, P < 0.00001 between CON/VEH and RST/VEH groups; *, P < 0.01 between RST/VEH and RST/L. reuteri groups. Student's t test with the Bonferroni correction factor was used as a post hoc test. Sample sizes were as follows: n = 18 (CON/VEH), n = 18 (CON/L. reuteri), n = 9 (RST/VEH), and n = 9 (RST/L. reuteri). (B) Stressor exposure significantly enhanced the likelihood of detecting the pathogen in the spleen in both VEH- and L. reuteri-treated mice. *, P < 0.05 between CON and RST groups as assessed by chi-square (χ2) analysis. Sample sizes were as follows: n = 17 (CON/VEH), n = 17 (CON/L. reuteri), n = 8 (RST/VEH), and n = 9 (RST/L. reuteri). (C) Stressor exposure significantly increased pathogen loads in the spleens of mice, regardless of treatment. *, ANOVA main effect for group (CON versus RST; P < 0.05). Sample sizes were as follows: n = 17 (CON/VEH), n = 17 (CON/L. reuteri), n = 8 (RST/VEH), and n = 9 (RST/L. reuteri). The horizontal line in panel C represents the detection limit for C. rodentium.
Probiotic intervention reduces anxiety-like behaviors and systemic IL-6.
Exposure to prolonged restraint during C. rodentium challenge did not affect locomotion in a novel environment (Fig. 7A). However, there was a trend for an interaction between stressor exposure and probiotic treatment (i.e., L. reuteri) in that mice that were fed L. reuteri had a reduction in the amount of time spent in the center of the open field compared to stressor-exposed, VEH-treated mice [F(1, 49) = 3.30; P = 0.075] (Fig. 7B). Even though this interaction was not quite statistically significant using a two-factor ANOVA, we tested our a priori hypothesis that VEH-treated mice exposed to the stressor would spend less time in the center of the open field than VEH-treated controls. As predicted, stressor exposure significantly reduced the time in the center of the open field in VEH-treated, stressor-exposed mice compared to VEH-treated control mice [t(25) = 2.31; P < 0.05] (Fig. 7B), an effect which was not observed in stressor-exposed mice treated with L. reuteri.
Fig 7.

Probiotic intervention reduces stressor-enhanced anxiety-like behavior and significantly reduces circulating IL-6 at the peak of infection. Mice were restrained for 1 day and then orally challenged with C. rodentium. Restraint continued for 6 days postchallenge, during which the drinking water was supplemented with either VEH or L. reuteri, beginning 1 day prior to restraint. Prior to sacrifice, mice were tested by the open field test and recorded for 5 min. Locomotion was quantified by the total number of lines crossed and the amount of time spent in the center of the open field. (A) There were no changes in the total number of lines crossed in either stressor-exposed group. Sample sizes were as follows: n = 12 (CON/VEH), n = 12 (CON/L. reuteri), n = 6 (RST/VEH), and n = 4 (RST/L. reuteri). (B) There was an increase in anxiety-like behavior, as evidenced by a reduction in the time spent in the center of the open field, in VEH-treated, stressor-exposed mice, which was restored with L. reuteri treatment. *, P < 0.05 between CON/VEH and RST/VEH groups as assessed by Student's t test with the Bonferroni correction factor as a post hoc test. Sample sizes were as follows: n = 18 (CON/VEH), n = 18 (CON/L. reuteri), n = 9 (RST/VEH), and n = 9 (RST/L. reuteri). Cytokine levels were determined by a Bio-Plex assay. (C) Circulating IL-1β was reduced by stressor exposure on day 12 postchallenge, and L. reuteri failed to modulate this reduction. *, ANOVA main effect for group (CON versus RST; P = 0.01). Sample sizes were as follows: n = 13 (CON/VEH), n = 11 (CON/L. reuteri), n = 7 (RST/VEH), and n = 7 (RST/L. reuteri). (D) Circulating IL-6, however, was significantly increased by stressor exposure in VEH-treated mice on day 12 postchallenge. Probiotic intervention with L. reuteri reduced stressor-enhanced circulating IL-6 levels. *, P < 0.001 between CON/VEH and RST/VEH groups. Student's t test with the Bonferroni correction factor was used as a post hoc test. Sample sizes were as follows: n = 14 (CON/VEH), n = 13 (CON/L. reuteri), n = 7 (RST/VEH), and n = 6 (RST/L. reuteri).
Serum IL-1β levels were significantly reduced in stressor-exposed mice compared to control groups [F(1, 34) = 10.2; P < 0.01] (Fig. 7C). Lactobacillus reuteri treatment did not change serum IL-1β levels in either control or stressor-exposed groups. However, there were changes in serum levels of IL-6 [F(1, 36) = 7.3; P = 0.01] (Fig. 7D). On day 12 postchallenge, there was a significant increase in circulating IL-6 in VEH-treated, stressor-exposed mice compared to VEH-treated control mice. This stressor-enhanced increase in circulating IL-6 was blocked in stressor-exposed mice that were treated with L. reuteri. There were no significant changes in serum levels of IL-12p70, IFN-γ, TNF-α, IL-10, or IL-2 between CON or RST mice in either treatment group (Table 2).
Table 2.
Serum cytokine levelsa
| Cytokine | Mean concn (pg/ml) ± SE |
|||
|---|---|---|---|---|
| CON-VEH | CON-L. reuteri | RST-VEH | RST-L. reuteri | |
| IL-12p70 | 25.0 ± 2.9 | 21.5 ± 4.1 | 21.7 ± 6.8 | 14.0 ± 4.0 |
| IFN-γ | 9.6 ± 1.4 | 9.7 ± 2.4 | 4.7 ± 1.6 | 6.6 ± 2.4 |
| TNF-α | 65.6 ± 12.5 | 71.1 ± 12.5 | 46.2 ± 14.3 | 44.8 ± 10.9 |
| IL-10 | 140.5 ± 18.1 | 126.2 ± 23.4 | 140.6 ± 42.6 | 84.6 ± 20.7 |
| IL-2 | 4.6 ± 0.7 | 6.8 ± 1.7 | 3.3 ± 0.9 | 3.8 ± 1.1 |
Cytokine levels were determined by Bio-Plex assay.
DISCUSSION
The results of this study confirm previous reports that outbred CD-1 mice do not develop severe colitis when challenged with C. rodentium (1, 38, 39). Low levels of the pathogen were evident in the stool of nonstressed control mice orally challenged with 300 million CFU of C. rodentium, and these control mice developed very low levels of colitis. However, simply exposing these mice to a prolonged-restraint stressor during pathogen challenge was sufficient to significantly increase disease severity. Stressor exposure significantly increased C. rodentium levels in the colon and also the expression of pathogen-induced chemokines and cytokines. These inflammatory mediators aid in the recruitment of immune cells to help fight infection and also contribute to the eradication of pathogens. However, overexpression of these genes can contribute to colonic tissue damage and immunopathology. Even though all mice were orally challenged with C. rodentium, chemokine and cytokine mRNA levels in the colons of mice exposed to the stressor during challenge with C. rodentium were 5 to 250 times higher than the levels found in nonstressed control mice. One of these chemokines, namely, CCL2, is known to recruit inflammatory monocytes to the colon (40, 41), and inflammatory monocytes are prolific producers of TNF-α and iNOS (41). Thus, it is possible that overexpression of CCL2 recruits a larger number of inflammatory monocytes to the colon, ultimately leading to higher TNF-α and iNOS levels. While this hypothesis has not yet been tested completely, the colonic histopathology is consistent with this rationale. The total colitis index, which incorporated a scoring of the infiltration of leukocytes, ranged from 0.5 to 7 for the control mice to 10 to 15 for the stressor-exposed mice. This colitis, which in stressor-exposed mice included epithelial erosion and ulceration, further demonstrates the impact that stressor exposure can have on even “nonsusceptible” hosts.
Susceptibility and resistance to C. rodentium are largely dependent upon host genetics, but studies have shown that intestinal microbes, including probiotics, can also affect disease severity. In a previous study, we demonstrated that exposure to a prolonged-restraint stressor was sufficient to significantly alter microbial communities in the cecum (18). More recent data also indicate that the colonic tissue-associated microbiota can be affected in mice exposed to prolonged restraint, with a reduction in L. reuteri often being evident in the stressor-exposed mice (data not shown). Lactobacillus reuteri has been shown to reduce colitis in germfree mice challenged with the closely related A/E pathogen EHEC (42). Thus, it was predicted that treating mice with L. reuteri would attenuate the stressor-induced increase in colonic pathology in C. rodentium-challenged mice.
Treating mice with L. reuteri had mild effects on C. rodentium-induced colitis. Treatment with L. reuteri did reduce the mean colonic levels of CCL2, TNF-α, and iNOS mRNAs in mice exposed to the stressor during challenge with C. rodentium. In addition, the mean colonic histopathology score was reduced in L. reuteri-treated mice exposed to the stressor. But, in general, these differences were not quite large enough to be statistically significant, which may have been due to the cage-level influences of supplementation of L. reuteri in the drinking water. Along with the inflammatory mediators TNF-α and iNOS, antimicrobial peptides such as β-defensins also contribute to the eradication of pathogens (43). Stressor exposure during challenge with C. rodentium caused a significant reduction in β-defensin 3 in VEH-treated mice which was reversed with L. reuteri treatment.
Throughout the duration of the experiment, it was consistently noted that stressor-exposed mice that were treated with L. reuteri exhibited fewer signs of illness (e.g., cleaner fur and more active behavior) than VEH-treated, stressor-exposed mice. Because overt illness behaviors can signify that the pathogen has escaped the colon and translocated to systemic sites, such as the spleen, we further investigated whether C. rodentium could be detected at systemic sites and whether systemic signs of illness were evident. Exposure to the stressor during challenge with C. rodentium resulted in a significant increase in the likelihood of detecting C. rodentium in the spleen. This is important, because unlike invasive gastrointestinal pathogens, such as Salmonella species, C. rodentium does not have mechanisms to invade its host (1, 44). Citrobacter rodentium and its attaching and effacing E. coli relatives stay within the digestive tract, attached to the apical surface of the colonic epithelium, during infections in an immunocompetent host (1, 44). However, during infection, C. rodentium can disrupt the tight junctions found between intestinal epithelial cells via the injection of effector proteins through a type III secretion system into the host epithelial cell. Among the inserted proteins are E. coli secreted protein F (EspF) and mitochondrion-associated protein (Map), which have been implicated in the disruption of tight junctions via changes in the expression and location of two integral proteins involved in the formation of the tight junction, namely, claudin 5 and zonula occludins 1 (45, 46). The noninvasive nature of C. rodentium, coupled with the fact that the tight junctions between intestinal epithelial cells help to regulate what can and cannot pass through the intestinal epithelial layer, led us to hypothesize that our stressor-enhanced translocation of pathogen from the colon to the spleen may have been due to the loosening of the epithelial tight junctions. In our model, we observed significant reductions in gene expression of both claudin 1 and claudin 5 in the colons of stressor-exposed mice. Importantly, treatment with L. reuteri did not change C. rodentium levels in the colon; however, it was able to ameliorate the stressor-enhanced reduction in claudin 5, which we believe contributed to the reduction of pathogen translocation from the colon to the spleen.
The ability of L. reuteri to reduce the translocation of C. rodentium from the colon to the spleen, potentially through stabilization of tight junction proteins, suggested that L. reuteri treatment would prevent the systemic manifestations of stressor exposure during C. rodentium challenge. Indeed, mice exposed to the stressor during C. rodentium challenge had significantly higher levels of circulating IL-6 but lower levels of IL-1β. Pathogen challenge often leads to behavioral changes in mice, in large part through the actions of circulating cytokines on brain biology. Previous studies have shown that elevations in circulating IL-6 can lead to the development of illness behavior and anxiety-like behavior in mice (47). In our study, treating mice with L. reuteri ameliorated the stressor-enhanced reduction in the amount of time mice spent in the center of the open field, suggesting that L. reuteri reduces anxiety-like behavior in mice exposed to the prolonged restraint stressor during C. rodentium challenge. Taken together, our data indicate that treatment with L. reuteri can attenuate the stressor-induced increases in pathogen translocation from the colon to the spleen, elevations in circulating IL-6, and anxiety-like behavior. Future studies will determine whether these factors are causally related and will determine whether stabilization of the epithelial tight junctions is necessary for the beneficial effects of L. reuteri.
Our study demonstrates the adverse effects of prolonged stressor exposure on gastrointestinal infection and disease progression and suggests that probiotics may be beneficial for some, but not all, of the stressor effects. This may be particularly important for diseases such as postinfectious irritable bowel syndrome (PI-IBS), where infection severity and stressor exposure are both known to be predisposing factors (48). Understanding the mechanisms by which probiotics can affect gastrointestinal diseases and the conditions under which they have beneficial effects will facilitate the rational design and use of probiotic microbes.
ACKNOWLEDGMENTS
These studies were generously supported by NIH grant RO1AT006552-01A1 and NIH/NIDCR grant T32 DE014320.
We also thank F. Michael Beck from The Ohio State University for help with statistical analysis, as well as Jeffrey Galley and Robert Easterling for their assistance.
Footnotes
Published ahead of print 24 June 2013
REFERENCES
- 1.Mundy R, MacDonald TT, Dougan G, Frankel G, Wiles S. 2005. Citrobacter rodentium of mice and man. Cell. Microbiol. 7:1697–1706 [DOI] [PubMed] [Google Scholar]
- 2.Higgins LM, Frankel G, Douce G, Dougan G, MacDonald TT. 1999. Citrobacter rodentium infection in mice elicits a mucosal Th1 cytokine response and lesions similar to those in murine inflammatory bowel disease. Infect. Immun. 67:3031–3039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lapointe TK, O'Connor PM, Buret AG. 2009. The role of epithelial malfunction in the pathogenesis of enteropathogenic E. coli-induced diarrhea. Lab. Invest. 89:964–970 [DOI] [PubMed] [Google Scholar]
- 4.Borenshtein D, McBee ME, Schauer DB. 2008. Utility of the Citrobacter rodentium infection model in laboratory mice. Curr. Opin. Gastroenterol. 24:32–37 [DOI] [PubMed] [Google Scholar]
- 5.Vallance BA, Deng W, Jacobson K, Finlay BB. 2003. Host susceptibility to the attaching and effacing bacterial pathogen Citrobacter rodentium. Infect. Immun. 71:3443–3453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ghosh S, Dai C, Brown K, Rajendiran E, Makarenko S, Baker J, Ma C, Halder S, Montero M, Ionescu VA, Klegeris A, Vallance BA, Gibson DL. 2011. Colonic microbiota alters host susceptibility to infectious colitis by modulating inflammation, redox status, and ion transporter gene expression. Am. J. Physiol. Gastrointest. Liver Physiol. 301:G39–G49 [DOI] [PubMed] [Google Scholar]
- 7.Willing BP, Vacharaksa A, Croxen M, Thanachayanont T, Finlay BB. 2011. Altering host resistance to infections through microbial transplantation. PLoS One 6:e26988. 10.1371/journal.pone.0026988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kamada N, Kim YG, Sham HP, Vallance BA, Puente JL, Martens EC, Nunez G. 2012. Regulated virulence controls the ability of a pathogen to compete with the gut microbiota. Science 336:1325–1329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wlodarska M, Willing B, Keeney KM, Menendez A, Bergstrom KS, Gill N, Russell SL, Vallance BA, Finlay BB. 2011. Antibiotic treatment alters the colonic mucus layer and predisposes the host to exacerbated Citrobacter rodentium-induced colitis. Infect. Immun. 79:1536–1545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen CC, Louie S, Shi HN, Walker WA. 2005. Preinoculation with the probiotic Lactobacillus acidophilus early in life effectively inhibits murine Citrobacter rodentium colitis. Pediatr. Res. 58:1185–1191 [DOI] [PubMed] [Google Scholar]
- 11.Johnson-Henry KC, Nadjafi M, Avitzur Y, Mitchell DJ, Ngan BY, Galindo-Mata E, Jones NL, Sherman PM. 2005. Amelioration of the effects of Citrobacter rodentium infection in mice by pretreatment with probiotics. J. Infect. Dis. 191:2106–2117 [DOI] [PubMed] [Google Scholar]
- 12.Gareau MG, Wine E, Reardon C, Sherman PM. 2010. Probiotics prevent death caused by Citrobacter rodentium infection in neonatal mice. J. Infect. Dis. 201:81–91 [DOI] [PubMed] [Google Scholar]
- 13.Rodrigues DM, Sousa AJ, Johnson-Henry KC, Sherman PM, Gareau MG. 2012. Probiotics are effective for the prevention and treatment of Citrobacter rodentium-induced colitis in mice. J. Infect. Dis. 206:99–109 [DOI] [PubMed] [Google Scholar]
- 14.Buynitsky T, Mostofsky DI. 2009. Restraint stress in biobehavioral research: recent developments. Neurosci. Biobehav. Rev. 33:1089–1098 [DOI] [PubMed] [Google Scholar]
- 15.Bailey MT, Coe CL. 1999. Maternal separation disrupts the integrity of the intestinal microflora in infant rhesus monkeys. Dev. Psychobiol. 35:146–155 [PubMed] [Google Scholar]
- 16.Bailey MT, Avitsur R, Engler H, Padgett DA, Sheridan JF. 2004. Physical defeat reduces the sensitivity of murine splenocytes to the suppressive effects of corticosterone. Brain Behav. Immun. 18:416–424 [DOI] [PubMed] [Google Scholar]
- 17.Bailey MT, Lubach GR, Coe CL. 2004. Prenatal stress alters bacterial colonization of the gut in infant monkeys. J. Pediatr. Gastroenterol. Nutr. 38:414–421 [DOI] [PubMed] [Google Scholar]
- 18.Bailey MT, Dowd SE, Parry NM, Galley JD, Schauer DB, Lyte M. 2010. Stressor exposure disrupts commensal microbial populations in the intestines and leads to increased colonization by Citrobacter rodentium. Infect. Immun. 78:1509–1519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen CC, Chiu CH, Lin TY, Shi HN, Walker WA. 2009. Effect of probiotics Lactobacillus acidophilus on Citrobacter rodentium colitis: the role of dendritic cells. Pediatr. Res. 65:169–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Foye OT, Huang IF, Chiou CC, Walker WA, Shi HN. 2012. Early administration of probiotic Lactobacillus acidophilus and/or prebiotic inulin attenuates pathogen-mediated intestinal inflammation and Smad 7 cell signaling. FEMS Immunol. Med. Microbiol. 65:467–480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brown CH. 1963. Acute emotional crises and ulcerative colitis. Report of seven cases. Am. J. Dig. Dis. 8:525–536 [DOI] [PubMed] [Google Scholar]
- 22.Paar GH, Bezzenberger U, Lorenz-Meyer H. 1988. The correlation of psychosocial stress and disease activity in patients with Crohn disease and ulcerative colitis. Z. Gastroenterol. 26:648–657 [PubMed] [Google Scholar]
- 23.Reber SO. 2012. Stress and animal models of inflammatory bowel disease—an update on the role of the hypothalamo-pituitary-adrenal axis. Psychoneuroendocrinology 37:1–19 [DOI] [PubMed] [Google Scholar]
- 24.Chichlowski M, Hale LP. 2008. Bacterial-mucosal interactions in inflammatory bowel disease: an alliance gone bad. Am. J. Physiol. Gastrointest. Liver Physiol. 295:G1139–G1149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lin YP, Thibodeaux CH, Pena JA, Ferry GD, Versalovic J. 2008. Probiotic Lactobacillus reuteri suppress proinflammatory cytokines via c-Jun. Inflamm. Bowel Dis. 14:1068–1083 [DOI] [PubMed] [Google Scholar]
- 26.Jones SE, Versalovic J. 2009. Probiotic Lactobacillus reuteri biofilms produce antimicrobial and anti-inflammatory factors. BMC Microbiol. 9:35. 10.1186/1471-2180-9-35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Walter J, Britton RA, Roos S. 2011. Host-microbial symbiosis in the vertebrate gastrointestinal tract and the Lactobacillus reuteri paradigm. Proc. Natl. Acad. Sci. U. S. A. 108(Suppl 1):4645–4652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thomas CM, Hong T, van Pijkeren JP, Hemarajata P, Trinh DV, Hu W, Britton RA, Kalkum M, Versalovic J. 2012. Histamine derived from probiotic Lactobacillus reuteri suppresses TNF via modulation of PKA and ERK signaling. PLoS One 7:e31951. 10.1371/journal.pone.0031951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schreiber O, Petersson J, Phillipson M, Perry M, Roos S, Holm L. 2009. Lactobacillus reuteri prevents colitis by reducing P-selectin-associated leukocyte- and platelet-endothelial cell interactions. Am. J. Physiol. Gastrointest. Liver Physiol. 296:G534–G542 [DOI] [PubMed] [Google Scholar]
- 30.Dicksved J, Schreiber O, Willing B, Petersson J, Rang S, Phillipson M, Holm L, Roos S. 2012. Lactobacillus reuteri maintains a functional mucosal barrier during DSS treatment despite mucus layer dysfunction. PLoS One 7:e46399. 10.1371/journal.pone.0046399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Head CC, Farrow MJ, Sheridan JF, Padgett DA. 2006. Androstenediol reduces the anti-inflammatory effects of restraint stress during wound healing. Brain Behav. Immun. 20:590–596 [DOI] [PubMed] [Google Scholar]
- 32.Schauer DB, Falkow S. 1993. The eae gene of Citrobacter freundii biotype 4280 is necessary for colonization in transmissible murine colonic hyperplasia. Infect. Immun. 61:4654–4661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Erdman SE, Poutahidis T, Tomczak M, Rogers AB, Cormier K, Plank B, Horwitz BH, Fox JG. 2003. CD4+ CD25+ regulatory T lymphocytes inhibit microbially induced colon cancer in Rag2-deficient mice. Am. J. Pathol. 162:691–702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kent S, Bluthe RM, Kelley KW, Dantzer R. 1992. Sickness behavior as a new target for drug development. Trends Pharmacol. Sci. 13:24–28 [DOI] [PubMed] [Google Scholar]
- 35.York JM, Blevins NA, Baynard T, Freund GG. 2012. Mouse testing methods in psychoneuroimmunology: an overview of how to measure sickness, depressive/anxietal, cognitive, and physical activity behaviors. Methods Mol. Biol. 934:243–276 [DOI] [PubMed] [Google Scholar]
- 36.Belzung C, Griebel G. 2001. Measuring normal and pathological anxiety-like behaviour in mice: a review. Behav. Brain Res. 125:141–149 [DOI] [PubMed] [Google Scholar]
- 37.Yin D, Zhang Y, Stuart C, Miao J, Zhang Y, Li C, Zeng X, Hanley G, Moorman J, Yao Z, Woodruff M. 2006. Chronic restraint stress modulates expression of genes in murine spleen. J. Neuroimmunol. 177:11–17 [DOI] [PubMed] [Google Scholar]
- 38.Barthold SW, Osbaldiston GW, Jonas AM. 1977. Dietary, bacterial, and host genetic interactions in the pathogenesis of transmissible murine colonic hyperplasia. Lab. Anim. Sci. 27:938–945 [PubMed] [Google Scholar]
- 39.Luperchio SA, Schauer DB. 2001. Molecular pathogenesis of Citrobacter rodentium and transmissible murine colonic hyperplasia. Microbes Infect. 3:333–340 [DOI] [PubMed] [Google Scholar]
- 40.Platt AM, Bain CC, Bordon Y, Sester DP, Mowat AM. 2010. An independent subset of TLR expressing CCR2-dependent macrophages promotes colonic inflammation. J. Immunol. 184:6843–6854 [DOI] [PubMed] [Google Scholar]
- 41.Bain CC, Scott CL, Uronen-Hansson H, Gudjonsson S, Jansson O, Grip O, Guilliams M, Malissen B, Agace WW, Mowat AM. 2013. Resident and pro-inflammatory macrophages in the colon represent alternative context-dependent fates of the same Ly6Chi monocyte precursors. Mucosal Immunol. 6:498–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Eaton KA, Honkala A, Auchtung TA, Britton RA. 2011. Probiotic Lactobacillus reuteri ameliorates disease due to enterohemorrhagic Escherichia coli in germfree mice. Infect. Immun. 79:185–191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.O'Neil DA. 2003. Regulation of expression of beta-defensins: endogenous enteric peptide antibiotics. Mol. Immunol. 40:445–450 [DOI] [PubMed] [Google Scholar]
- 44.Vallance BA, Khan MA, Deng W, Gruenheid S, Finlay BB. 2004. Modeling enteropathogenic and enterohemorrhagic E. coli infections and disease. Drug Discov. Today Dis. Models 1:73–79 [Google Scholar]
- 45.Muza-Moons MM, Schneeberger EE, Hecht GA. 2004. Enteropathogenic Escherichia coli infection leads to appearance of aberrant tight junctions strands in the lateral membrane of intestinal epithelial cells. Cell. Microbiol. 6:783–793 [DOI] [PubMed] [Google Scholar]
- 46.Gibson DL, Ma C, Rosenberger CM, Bergstrom KS, Valdez Y, Huang JT, Khan MA, Vallance BA. 2008. Toll-like receptor 2 plays a critical role in maintaining mucosal integrity during Citrobacter rodentium-induced colitis. Cell. Microbiol. 10:388–403 [DOI] [PubMed] [Google Scholar]
- 47.D'Mello C, Swain MG. 2011. Liver-brain inflammation axis. Am. J. Physiol. Gastrointest. Liver Physiol. 301:G749–G761 [DOI] [PubMed] [Google Scholar]
- 48.Collins SM, Barbara G, Vallance B. 1999. Stress, inflammation and the irritable bowel syndrome. Can. J. Gastroenterol. 13(Suppl A):47A–49A [DOI] [PubMed] [Google Scholar]