

Abstract
Functional exhaustion of CD8+ T cells due to increased expression of inhibitory molecule PD-1 (Programmed Death-1) causes reactivation of latent disease during later phases of chronic toxoplasmosis. Onset of disease recrudescence results in decreased parasite cyst burden concomitant with parasites undergoing stage conversion from a primarily encysted, quiescent bradyzoite to a fast-replicating, highly motile tachyzoite. Thus, reduced cyst burden is one of the early hallmarks of disease recrudescence. This was further validated by depleting gamma interferon (IFN-γ), a cytokine known to control latent toxoplasmosis, in chronically infected prerecrudescent mice. Since CD8+ T cells (an important source of IFN-γ) lose their functionality during the later phases of chronic toxoplasmosis, we next examined if adoptive transfer of functional CD8+ T cells from acutely infected donors to the chronically infected prerecrudescent hosts could impede parasite de-encystation and rescue exhausted CD8+ T cells. While the transfer of immune CD8+ T cells temporarily restricted the breakdown of cysts, the exhausted endogenous CD8+ T cell population was not rescued. Over time, the donor population got deleted, resulting in parasite de-encystation and host mortality. Considering that donor CD8+ T cells fail to become long-lived, one of the cardinal features of memory CD8+ T cells, it bears the implication that memory CD8 differentiation is impaired during chronic toxoplasmosis. Moreover, our data strongly suggest that while adoptive immunotherapy can prevent parasite de-encystation transiently, reduced antigen burden in the chronic phase by itself is insufficient for rescue of exhausted CD8+ T cells. The conclusions of this study have profound ramifications in designing immunotherapeutics against chronic toxoplasmosis.
INTRODUCTION
Toxoplasma gondii is an obligate intracellular parasite of the phylum apicomplexan which infects approximately 30% to 80% of humans worldwide (1–3). According to a recent CDC report, toxoplasmosis is considered to be a leading cause of food-borne mortality in the United States and ranks as one of the five neglected parasitic infections that have been targeted by the CDC for public health action (http://www.cdc.gov/parasites/toxoplasmosis/). Acute infection of immunocompetent adults remains largely asymptomatic, and immune control results in parasite encystation at immune-privileged sites, including the brain, where it apparently persists quiescently for the life of the host (4, 5). Loss of immune competence results in parasite reactivation in infected hosts, leading to encephalitis, which was a major problem globally for HIV-infected populations in the pre-highly active antiretroviral therapy (HAART) era (4, 5). Although the incidence of Toxoplasma encephalitis (TE) has declined substantially in the United States and other developed countries due to anti-Toxoplasma prophylactic treatment and antiretroviral HAART therapy, it remains a major problem in AIDS patients in developing countries due to the lack of appropriate therapy and health care infrastructure (6–8). Alarmingly, in sub-Saharan Africa, 25 million people are HIV positive (http://www.unaids.org/bangkok2004/GAR2004_html/ExecSummary_en/Execsumm_en.pdf), and coinfection with T. gondii is highly underdiagnosed (9). Based on the high seroprevalence in sub-Saharan Africa combined with the high rate of HIV infection, it has been estimated that 2.5 to 10 million people in African countries are at risk of dying from toxoplasmosis (6). Beyond the coprevalence with AIDS, Toxoplasma meningoencephalitis has been noted in malnourished HIV-negative immunocompetent adults in India (10). Apart from these regions, atypical Toxoplasma strains have been associated with significant human morbidity in countries in South and Central America (11–13). Thus, this understudied pathogen remains a severe problem in developing countries.
Although innate immune responses play an important role during early T. gondii infection, long-term protection against this parasite is mediated by the adaptive immune response (4). Among the T cell populations involved, gamma interferon (IFN-γ)-producing CD8+ T cells are critical for keeping chronic infections under control (4). Depletion of IFN-γ or CD8+ T cells in chronically infected mice leads to reactivation of latent infection and the ultimate death of the host (4, 5, 14, 15). Recent studies from our laboratory have reported that chronic infection with T. gondii in the genetically susceptible C57BL/6 mouse results in a graded increase in the level of the inhibitory receptor, PD-1 (Programmed Death-1), on CD8+ T cells (16–20). This leads to elevated CD8 apoptosis and progressive attrition of their functionality in terms of IFN-γ production. This phenomenon of CD8 “exhaustion” is concomitant with parasite reactivation and host mortality. Administration of blocking anti-PD-L1 antibody to chronically infected animals reinvigorated CD8+ T cell functionality and prevented their death (16). Based on these studies, it can be concluded that functional CD8+ T cells play a key role in keeping T. gondii in a quiescent stage and preventing the reactivation of latent infection. As most of the mortality due to Toxoplasma infection is due to reactivation of latent infection, immunotherapeutic agents that arrest the process would prove highly beneficial to the infected host. Previous reports, including those from our laboratory, have demonstrated that adoptive transfer of immune CD8+ T cells can protect naive animals against the lethality of acute infection (4). In the current study, we determined if exogenous immune CD8+ T cell treatment could rescue the endogenous population and thus ensure host survival. Interestingly, while administration of exogenous CD8+ T cells from acutely infected animals to chronically infected mice initially prevented parasite reactivation in the recipients, it did not rescue the endogenous CD8 population or result in differential long-term survival results. Additionally, donor CD8+ T cells could not be detected during the later stages of infection, suggesting that development of memory, a hallmark of robust CD8 immunity, is impaired during chronic toxoplasmosis. Overall, the conclusions of this study have important implications for designing immunotherapeutic approaches that target chronic toxoplasmosis.
MATERIALS AND METHODS
Mice.
Female C57BL/6 mice (6 to 8 weeks of age) were purchased from the National Cancer Institute. CD90.1 mice were purchased from Jackson Laboratories. Animal studies were carried out in agreement with the Institutional Animal Care and Use Committee (IACUC)-approved guidelines at George Washington University Medical Center. Mice were scored daily on a scale of 1 (asymptomatic) to 5 (most severe) based on several aspects. Moribund mice were characterized by severe loss of mobility, hunched back, piloerection or ruffled fur, and weight loss. All experiments were approved by the George Washington University Medical Center Animal Resource Facility IACUC under protocol approval number A052 in accordance with American Association for Accreditation of Lab Animal Care (AAALAC)-certified guidelines.
Parasites and infection.
T. gondii cysts of ME49 strain were prepared from the brains of infected mice. Unless mentioned otherwise, animals were infected with an average of 10 cysts/mouse via intragastric route and sacrificed by CO2 inhalation at different time points postinfection (pi).
TLA preparation.
Toxoplasma lysate antigen (TLA) was extracted from the RH strain of the parasite, and preparation was carried out as previously described (21).
Cyst enumeration.
Brains were homogenized in 1.5 ml phosphate-buffered saline (PBS) using a Dounce homogenizer. All cyst counts were performed immediately after preparation of brain homogenates. Cysts were then counted by examining 10 μl of the brain homogenate under a coverslip using light microscopy under ×40 magnification. Total cyst numbers were determined per brain based on counting 6 to 9 slides prepared from each brain homogenate.
Real-time RT-PCR for parasite stage-specific markers.
Total RNA was harvested from flash-frozen infected mouse brains by homogenization in TRIzol reagent (Invitrogen) followed by extraction using phenol chloroform, digestion with DNase-1 (Roche), and then further purification with RNeasy spin columns (Qiagen). cDNAs were generated using Moloney murine leukemia virus (MMLV) reverse transcriptase (RT) (Invitrogen), and semiquantitative real-time PCR was performed using iQ SYBR green Supermix (Bio-Rad) with primers for SAG-1 (IDT), BAG-1 (IDT), and Toxoplasma-specific actin (TgACT-1; IDT) on an iCycler iQ thermal cycler (Bio-Rad). Samples used for RT-PCR were first normalized for constitutively expressed Tg-ACT1 (22). Primer sequences are as follows: for 5′TgACT-1, TCCCGTCTATCGTCGGAAAG; for 3′TgACT-1, CCATTCCGACCATGATACCC; for 5′SAG-1, ATCGCCTGAGAAGCATCACTG; for 3′SAG-1, CGAAAATGGAAACGTGACTGG; for 5′BAG-1, GACGTGGAGTTCGACAGCAAA; for 3′BAG-1, ATGGCTCCGTTGTCGACTTCT; and for 3′ENO-2, ACTCGTTCTTAGTTCCATCG.
PCRs were carried out using 1 cycle at 95°C for 10 min followed by a two-step cycle, one at 95°C for 15 s and then another at 60°C for 1 min repeated 40 times. PCR products were then analyzed for quality by melt curve analysis. Quantitation of transcripts for SAG-1 and BAG-1 relative to day 10 postinfection (day 10 postinfection level = 1.0) were calculated according to the Pfaffl method of quantitation (23).
Lymphocyte isolation and staining.
A single-cell suspension was generated from spleen and brain using a standard protocol (16, 24). Splenic single-cell suspensions were made by mechanical disruption followed by red blood cell lysis. Brain lymphocyte enrichment was carried out as follows: brains were individually washed in Hanks balanced salt solution (HBSS)-heparin (2 IU/ml) and then mashed through a 70-μm-pore-size cell strainer followed by the use of a gradient centrifuge (2,000 rpm for 20 min) and a 30% Percoll solution containing 100 IU/ml of heparin. The pellet was then resuspended in cold PBS–2% fetal calf serum (FCS). The following antibodies were used in cell surface staining and intracellular staining of lymphocytes: αCD8β (clone H35-17.2; eBioscience), αPD-1 (clone J43; eBioscience), αCD90.1 (clone OX-7; BioLegend), αIFN-γ (clone XMG1.2; BD Biosciences), αKi-67 (clone B56; BD Biosciences), and αPD-L1 (clone MIH5; eBioscience). Intracellular staining was performed after surface staining, using a Cytofix/Cytoperm kit (BD Biosciences) per the manufacturer's protocol. Cell fluorescence was measured with a BD FACSCalibur or a Cytek-upgraded 8-color BD FACSCalibur cytometer which accounts for differences in fluorescence scale. Data were analyzed using FlowJo (TreeStar) software. Cells were leukocyte gated based on forward scatter and side scatter. Gating for positive and negative populations was set up based on FMO (fluorescence minus one) controls. MFI denotes mean fluorescence intensity.
Intracellular cytokine detection.
For cytokine detection, restimulation was carried out for 16 h with 30 μg/ml of TLA in supplemented Iscove's Complete Dulbecco's modified Eagle's medium (DMEM) at 37°C in 5% CO2. Monensin (BD Biosciences) (0.65 μl/ml) and brefeldin A (BD Biosciences) were added during the final 9 h of stimulation.
Flow cytometric detection of T. gondii-infected cells.
Intracellular staining was performed on single-cell suspensions as described above with some modifications. Postpermeabilization, staining with fluorescein isothiocyanate (FITC)-labeled polyclonal anti-Toxoplasma antibody (Abcam) was performed. This was followed by incubation with biotinylated anti-FITC antibody (FIT-22; BioLegend) and subsequent labeling with streptavidin FITC (eBioscience). Cells were analyzed based on leukocyte gating and forward and side scatter.
In vivo antibody treatment.
For in vivo IFN-γ depletion, rat anti-IFN-γ (clone XMG1.2; BioXCell) (4 mg) or rat IgG1 isotype-matched control antibody (BioXCell) (4 mg) was injected intraperitoneally (i.p.) daily for 4 days starting at day 31 pi.
Adoptive transfer of CD8+ T cells.
ME49-infected CD90.1 mice (or wild-type mice for some experiments) or naive animals were sacrificed at day 14 pi, splenic single-cell suspensions were prepared, and T cells from the tissues were isolated by magnetic purification using αCD8β antibody (Stem Cell Technology) according to the manufacturer's instructions. Purified CD8+ T cells (>90% pure; 4 × 106 cells/mouse) were adoptively transferred to chronically infected (week 7 pi) recipient mice (wild type; CD90.2) via the intravenous (i.v.) route. For some experiments, control “recipients” were left untreated or injected with PBS. For adoptive transfer of memory CD8+ T cells, cells from brains of infected CD45.1 mice were first purified by magnetic purification at day 24 pi. Purified CD8+ T cells were subsequently stained with αCD8β, αKLRG1 (clone 2F1; eBioscience), and αCD127 (clone A7R34; eBioscience) and were sorted using a BD FACSAria instrument. Sorted effector or memory CD8+ T cells (1.5 × 104) were then adoptively transferred to chronically infected (week 5 pi) recipient (CD45.2) mice.
Statistical analysis.
Differences in cyst burden, parasite gene expression levels, percentages, absolute numbers, and MFI for each experiment were evaluated using Student's t test with P < 0.05 taken as statistically significant. Error bars in graphs represent standard deviations of values for individual mice in the group from one experiment. Comparison of survival curves was performed using the log-rank (Mantel-Cox) test. All computations were performed using GraphPad Prism Software.
RESULTS
Toxoplasma de-encystation correlates with parasite reactivation during chronic toxoplasmosis.
In the intermediate hosts (including humans), Toxoplasma undergoes stage conversion between the rapidly proliferating and highly motile tachyzoite which is considered to be responsible for acute toxoplasmosis and the relatively quiescent, slowly replicating, encysted bradyzoite that can persist lifelong (25, 26). Host innate immunity and adaptive immunity are thought to cause differentiation of tachyzoites into bradyzoites (27). Several steps are involved in tachyzoite-to-bradyzoite differentiation, including changes in metabolism and replacement of the parasitophorous vacuole with a cyst wall (25, 28). These processes require coordinated transcription of specific genes that are highly dynamic and occur prior to full encystation and have the potential to revert, during parasite reactivation, to tachyzoite-like levels (29). Hence, in immunocompromised individuals such as HIV-infected individuals and bone marrow transplant recipients, loss of immune controls results in parasite reconversion from the bradyzoite to the tachyzoite stage, ultimately leading to TE (25). Similarly, a recent study from our group, using the same strain of Toxoplasma (as well as the same infective dose, route of infection, and time points of analysis) has demonstrated that host mortality in chronically infected C57BL/6 mice correlates with bradyzoite-to-tachyzoite conversion as assessed by expression of tachyzoite-specific ENO-2 and bradyzoite-specific ENO-1 (16). Since bradyzoites remain primarily encysted and parasite reactivation involves stage conversion from the bradyzoite to the tachyzoite stage, we hypothesized that an initial parasite reactivation would result in a decreased cyst burden in brain (16, 25). Notably, the kinetics of the cyst burden determined from the brains of Toxoplasma-infected mice indicates a dynamic pattern (Fig. 1A). Cyst numbers reach their peak at week 5 postinfection (pi), are significantly reduced at week 7 pi, increase modestly at week 10, and then sharply decrease in moribund mice. To further validate if the reduced cyst burden noted at week 7 pi correlated with parasite stage conversion, we assayed for alterations in stage-specific parasite expression by measuring transcription of tachyzoite-specific SAG-1 and bradyzoite-specific BAG-1 genes. As shown in Fig. 1B and C, reduced cyst burden at week 7 correlated with elevated SAG-1 and decreased BAG-1 levels. This is in agreement with our previous study which showed that chronically infected mice at week 7 pi exhibit increased ENO-2 (tachyzoite specific) and decreased ENO-1 (bradyzoite specific). Interestingly, we observed a modestly increased cyst burden (Fig. 1A) along with elevated expression of both ENO-2 and ENO-1 at week 10 pi (16), indicating that despite limited tachyzoite-to-bradyzoite conversion, the parasites were still in a reactivated stage. Overall, our data strongly argue that the onset of disease recrudescence during chronic toxoplasmosis involves parasite de-encystation concomitant with bradyzoite-to-tachyzoite-stage conversion.
Fig 1.
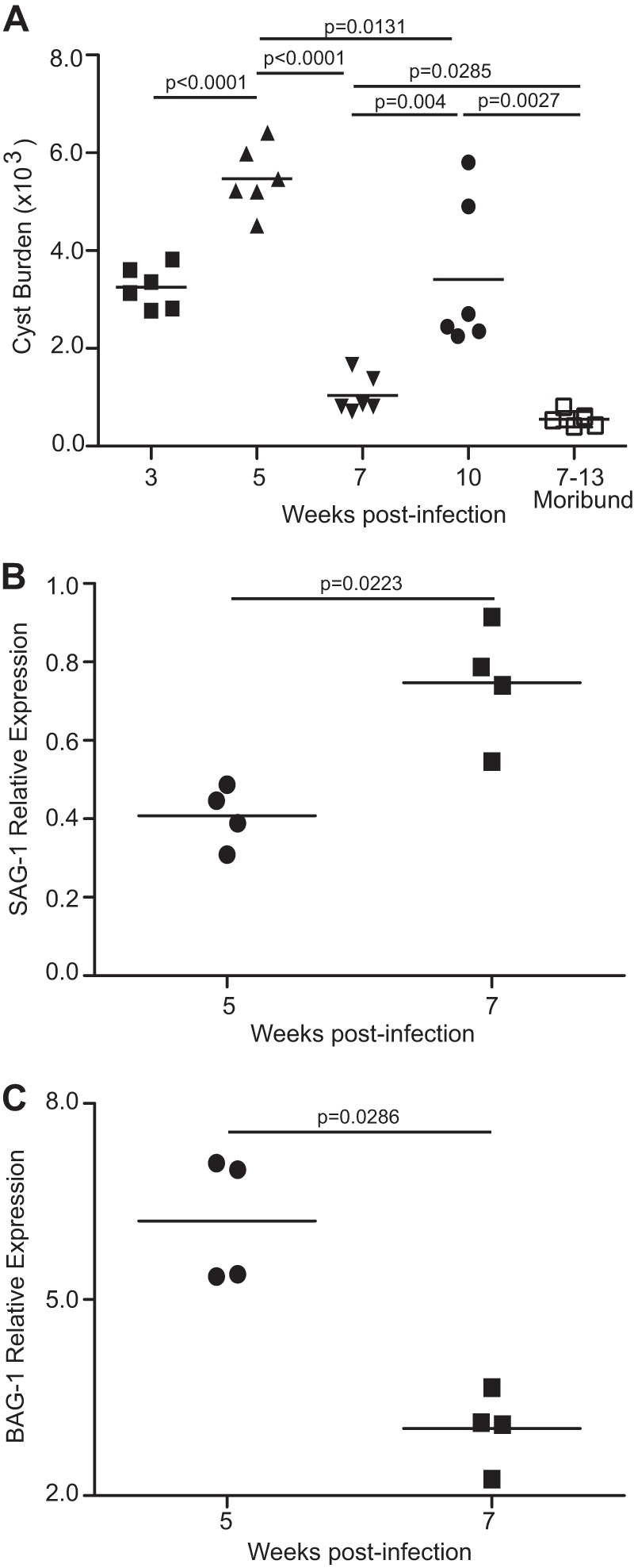
Decreased cyst burden during late-chronic T. gondii infection correlates with parasite reactivation. (A) C57BL/6 mice were infected with 10 ME49 cysts orally, and the number of brain cysts was enumerated at weeks 3, 5, 7, and 10 postinfection. A similar trend in cyst kinetics was observed in 3 independent experiments. (B and C) SAG-1 and BAG-1 relative expression levels were measured in brain at week 7 pi. The transcript level at day 10 was taken as 1. The data represent at least two experiments with at least 4 mice per group. Error bars represent standard deviations throughout.
Revalidation of parasite de-encystation as an early hallmark of Toxoplasma reactivation.
Next, we wanted to revalidate whether de-encystation is indeed an early hallmark of parasite reactivation. The role of IFN-γ in maintaining T. gondii parasites in the quiescent stage in vivo is well established (4). Since IFN-γ depletion during chronic toxoplasmosis has been reported to induce parasite reactivation and host mortality, we determined if chronically (early-phase) infected anti-IFN-γ-treated mice showed a reduced cyst burden, akin to mice at the late phase (week 7) of chronic infection (14). To address this, mice were treated with αIFN-γ antibody or isotype control starting at day 31 pi and brains were analyzed for various parameters 4 days later (Fig. 2A). In agreement with the parasite kinetic data presented above, anti-IFN-γ treatment resulted in a marked decrease in cyst burden (Fig. 2B). Next, we wanted to address whether this decreased cyst burden in αIFN-γ-treated mice was due to parasite reactivation or due to clearance of cysts by IFN-γ-independent immune mechanisms. To address this, we assayed for nonencysted parasites. Unlike free parasites, Toxoplasma has been shown to disseminate in the infected host using parasitized leukocytes (30). Interestingly, a recent study from our laboratory has also demonstrated that stage conversion during later phases of chronic toxoplasmosis results in a sharp increase in levels of parasitized leukocytes of myeloid lineage, especially in moribund mice (16). Based on this and taking into consideration the fact that encysted parasites in brain are primarily restricted to neurons in chronically infected mice (26), we hypothesized that if the reduced cyst burden in IFN-γ-depleted mice was due to parasite reactivation, it would result in an increase in the number of nonencysted parasites, thereby leading to an elevated frequency of parasitized leukocytes. In contrast, if the decreased cyst burden was due to immune response-mediated cyst clearance, no increase in the parasitized leukocyte frequency would be noted. In agreement with our primary hypothesis, the reduced cyst burden in αIFN-γ-treated mice was found to be associated with an elevated frequency of parasitized leukocytes (Fig. 2C and D). These observations suggest that a decrease in the cyst number is a hallmark of initiation of disease recrudescence in chronically infected hosts. Although a 4-day αIFN-γ treatment led to a decreased cyst burden, prolonged (9-day) antibody administration did indeed result in an increased cyst burden (along with elevated morbidity; 4,560 to 6,440 cysts/brain) akin to that seen with week 10 infected untreated mice (Fig. 1A), suggesting that that there is a continuous cycle of de-encystation and re-encystation (i.e., nonencysted parasites forming cysts) taking place (data not shown).
Fig 2.
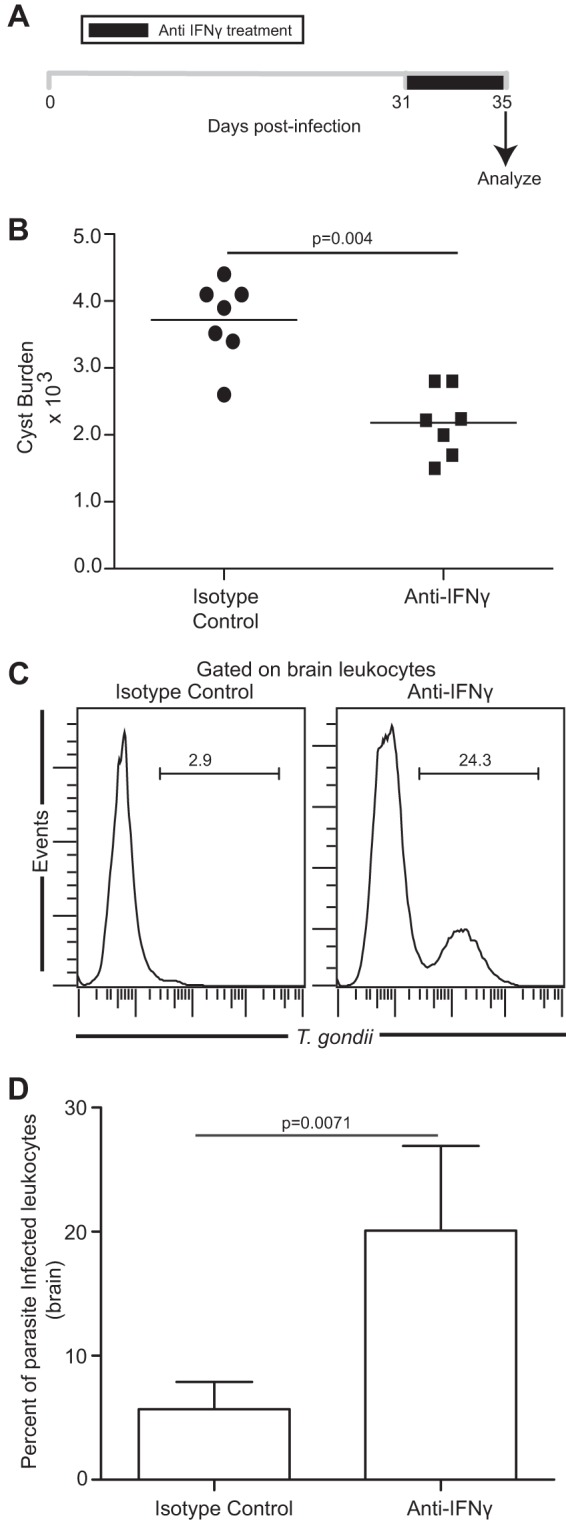
Reduced cyst burden and increased leukocyte parasitization in mice treated with αIFN-γ during early-chronic infection. (A) αIFN-γ antibody was injected daily, starting at day 31 pi (n = 7). Animals were sacrificed 4 days after initiation of treatment. (B) The number of brain cysts was enumerated in isotype- or αIFN-γ antibody-treated mice. (C and D) T. gondii-infected cells in brain leukocyte-gated samples (n = 5) were assayed by flow cytometry, and the results are depicted as histograms (C) or a bar graph (D). The data represent two experiments with at least 4 mice per group.
Adoptive transfer of immune CD8+ T cells controls the reactivation of latent Toxoplasma infection.
As mentioned earlier, IFN-γ is critical for preventing Toxoplasma reactivation (14). Incidentally, CD8+ T cells via IFN-γ production play a pivotal role in controlling chronic infection (14, 31). Since CD8+ T cells become exhausted and lose their ability to produce optimal IFN-γ during the later phases of chronic toxoplasmosis (16), we hypothesized that adoptive transfer of immune and healthy (i.e., not exhausted) CD8+ T cells from acutely infected mice to a chronically infected host, prior to disease recrudescence, would prevent parasite reactivation. To address this, CD8+ T cells were purified from acutely infected congenic animals (CD90.1) at week 2 pi and transferred to chronically infected recipients (CD90.2) which had been challenged with T. gondii cysts 5 weeks earlier (Fig. 3A). Control animals received an equal number of CD8+ T cells isolated from naive donors or were left untreated. As expected, chronically infected animals prior to receiving immune CD8+ T cells exhibited a high cyst number in the brain which was significantly reduced 2 weeks later (week 7 pi) in control mice (untreated mice or recipients of naive CD8+ T cells) (Fig. 3B). However, treatment with immune CD8+ T cells enabled the recipients to maintain the cyst number at week 5 (early-chronic) levels, suggesting that the de-encystation process was controlled at least up to 2 weeks posttransfer. Although the cyst burden in immune CD8+ T cell transfer recipients was maintained at week 5 levels, the possibility of parasites de-encysting and subsequently re-encysting due to immune pressure cannot be entirely ruled out. Such a process would mask any potential de-encystation. However, parasite de-encystation, no matter how transient, would expose antigens from nonencysted parasites to the immune system. KLRG1 expression on CD8+ T cells has been shown to be a marker of repeated antigen stimulation (32, 33), and prevention of antigen release results in reduced KLRG1 expression (34). As shown in Fig. 3C and D, concomitant with a reduced cyst burden in control recipients (week 2 posttransfer), endogenous CD8 T cells in brain exhibited elevated KLRG1 expression vis-à-vis pretransfer levels (week 5, early chronic) at both week 2 and week 4 posttransfer. In contrast, KLRG1 expression on endogenous CD8+ T cells at week 2 posttransfer was maintained at early-chronic levels in mice that received immune CD8+ T cells. Similar trends were also noted with total CD8 populations (i.e., both donor and recipient CD8+ T cells; data not shown). This strongly suggests that donor CD8 T cells maintain cyst burdens at week 5 levels by preventing parasite de-encystation. However, at week 4 posttransfer, KLRG1 expression on endogenous CD8+ T cells in immune CD8+ T cell transfer recipients was elevated to the same level as in control animals (Fig. 3D). This bears the implication that while donor CD8+ T cells prevent parasite de-encystation early on, they fail to control parasite reactivation at the later stages of infection. In agreement with this notion, the cyst burden at week 4 posttransfer was reduced in immune CD8+ T cell transfer recipients and was equivalent to that seen in the control group (Fig. 3E). The role of immune CD8+ T cells in controlling chronic Toxoplasma reactivation was further established by measuring the frequency of infected leukocytes in brain and blood of recipient animals. Severalfold decreases in the percentage of infected leukocytes in immune CD8+ T cell-administered animals were noted in both tissues at week 2 posttransfer (Fig. 3F and G). This further reinforces the paradigm that donor CD8+ T cells are indeed able to control parasite reactivation. However, this ability of immune CD8+ T cells to control de-encystation extended to a limited time frame. At week 4 posttransfer, the percentages of infected leukocytes were elevated to similar levels in both groups of mice (Fig. 3G). The limited ability of immune CD8+ T cells to control de-encystation over an extended time frame was further established by survival studies. As shown in Fig. 3H, no difference in the long-term survival of immune CD8+ T cell transfer recipients and control group mice was noted.
Fig 3.

Control of parasite reactivation by adoptive transfer of immune CD8+ T cells. (A) Splenic CD8+ T cells (4 × 106) purified from donor mice at week 2 pi were adoptively transferred via the i.v. route into chronically infected recipient mice at week 5 pi. (B) Cyst burden in brains of recipient mice (n = 5) was evaluated 2 weeks posttransfer. (C and D) Frequencies of KLRG1-expressing recipient CD8+ T cells (n = 4) in brain are shown as density plots (C) (2 weeks posttransfer) or as a bar graph (D). (E) Cyst burden in brains of recipient mice was assessed at 4 weeks posttransfer. (F and G) T. gondii-infected cells in brain and blood leukocyte-gated samples from recipient mice (n = 5) were assayed by flow cytometry. (F) A histogram depicts frequency of parasite-infected leukocytes at 2 weeks posttransfer. (G) A bar graph (left panel, brain; right panel, blood) shows percentages of infected leukocytes at weeks 2 and 4 posttransfer in recipient animals that received immune CD8+ T cell transfer or PBS. (H) Survival of immune CD8+ T cell transfer recipients (n = 8) and saline-treated controls (n = 7) was monitored on a daily basis. Data represent two experiments.
CD8+ T cells from immune donors fail to rescue the endogenous CD8+ T cell population in chronically infected hosts.
As stated earlier, CD8+ T cells exhibit elevated PD-1 expression concomitant with CD8 dysfunction and parasite reactivation during the later stages of chronic infection (16). This is accompanied by upregulation of PD-L1, a PD-1 receptor, on leukocytes (16). Considering that blockade of PD-1 interaction with its receptor PD-L1 has been shown to reinvigorate CD8+ T cell response, we next explored if control of parasite reactivation by donor CD8+ T cells resulted in downregulation of these inhibitory molecules in the endogenous population. At first, the level of PD-L1 expression in the spleen and brain leukocytes of recipient mice was measured at weeks 2 and 4 posttransfer. Similar to their ability to impede the reactivation process, immune CD8+ T cells were able to reduce PD-L1 expression on the splenic and brain leukocytes of the infected recipients at 2 weeks posttransfer (Fig. 4A and B). However, this ameliorative effect was transient and at week 4 posttransfer no difference in PD-L1 expression was noted between the two groups (Fig. 4A and B). Since blockade of PD-1–PD-L1 signaling via αPD-L1 treatment has been shown to augment CD8+ T cell proliferation during chronic toxoplasmosis (16) and the current CD8 immunotherapy regimen was able to downregulate PD-L1, albeit transiently, we hypothesized that the absolute number of endogenous CD8+ T cells would be elevated at least at early time points in immune CD8+ T cell transfer recipients. In contrast to our hypothesis, irrespective of the presence or absence of donor CD8+ T cell transfer, the absolute number of endogenous CD8+ T cells was not augmented at week 2 or week 5 posttransfer. Additionally, at the later time point, donor CD8+ T cells could not be detected, suggesting that Toxoplasma infection fails to elicit a long-term CD8 response. Next, we determined if the transfer of immune CD8+ T cells had any effect on PD-1 expression of endogenous or donor CD8 populations. Due to the very low number of donor cells, we could not reliably assess expression of PD-1 or other molecules on these cells. As shown in Fig. 4E and F, transfer of immune CD8+ T cells to the infected mice did not alter PD-1 expression of the endogenous cells, suggesting their inability to rescue exhaustion in these cells. The combined results of this study demonstrate that despite initial control of parasite reactivation and attenuation of PD-L1 expression, immune donor CD8+ T cells not only are unable to restore endogenous CD8+ T cell numbers but also fail to downregulate CD8-expressed PD-1 levels.
Fig 4.

Donor CD8+ T cells do not ameliorate endogenous CD8+ T cell exhaustion during late-chronic toxoplasmosis. (A and B) PD-L1 expression was assayed on total splenic and brain leukocytes by flow cytometry at weeks 2 and 4 posttransfer. Data are presented as representative histograms (A) or bar graphs (B). (C) The absolute number of endogenous CD8+ T cells in spleen and brain in the recipient mice was computed. (D) A bar graph represents the absolute number of donor CD8+ T cells in spleen (left panel) and brain (right panel) in immune CD8+ T cell transfer recipients. N.D. denotes “not detectable.” (E and F) PD-1 expression on endogenous CD8+ T cells in spleen and brain was evaluated by flow cytometry. Data are presented as a histogram (E) or as a bar graph (F) (left panel, spleen; right panel, brain). The data represent 2 experiments with at least 4 mice per group.
Donor CD8+ T cells fail to rescue the functionality of endogenous populations.
Although adoptive transfer of immune CD8+ T cells failed to restore the absolute number of endogenous CD8+ T cells in recipient mice, this does not necessarily rule out an elevated proliferative potential of CD8+ T cells in such animals. Augmented apoptosis or differential CD8+ T cell recruitment can potentially result in unaltered numbers of CD8+ T cells in these recipients. Hence, to definitively address if control of parasite reactivation had any effect on in situ proliferation, CD8+ T cells were assessed for Ki-67, a widely used maker for cell proliferation (16, 17). As shown in Fig. 5A and B, independently of immune CD8+ T cell administration, endogenous CD8+ T cell proliferation remained unaltered in both spleen and brain. As mentioned earlier, CD8+ T cell-produced IFN-γ plays a critical role in the control of chronic infection (4). Hence, we next examined if prevention of parasite reactivation resulted in augmented IFN-γ production by endogenous CD8+ T cells. Adoptive transfer of immune or naive CD8+ T cells did not result in differential IFN-γ production by CD8+ T cells in spleen or brain (Fig. 5C and D). Taken together, our data suggest that control of antigen or PD-L1 downregulation by immune donor CD8+ T cells is not sufficient for rescue of endogenous CD8 functionality in chronically infected mice.
Fig 5.

Donor CD8+ T cells fail to rescue endogenous CD8+ T cell functionality during late-chronic toxoplasmosis. (A and B) In vivo endogenous CD8+ T cell proliferation was assessed in spleen and brain of recipient animals at 2 weeks posttransfer by measuring Ki-67 expression via intracellular staining. Data are presented as pseudocolor plots (A) or as bar graphs (B) (left panel, spleen; right panel, brain). (C and D) Splenocytes and brain mononuclear cells were stimulated with TLA as described in Materials and Methods and then evaluated for IFN-γ production by endogenous CD8+ T cells at 2 weeks posttransfer. Data are depicted as a pseudocolor plot (C) (left panel, unstimulated; right panel, TLA stimulated) or as bar graphs (D) (left panel, spleen; right panel, brain). The data are representative of 2 experiments with 3 to 4 mice per group.
Exogenous memory CD8+ T cells fail to rescue endogenous populations.
Antigen-independent long-term maintenance and rapid recall response are the hallmarks of memory CD8+ T cells generated during acute infection (4). Given that adoptive transfer of immune CD8+ T cells from acutely infected animals had no long-term impact on the endogenous CD8+ T cell response, we reasoned that this effect may be enhanced by transferring memory cells as opposed to effector CD8 T cells. A previous study had demonstrated that CD8+ T cells home to the brain in a strictly antigen-specific manner (35). Hence, in order to ensure that recipient animals received similar numbers of antigen-specific effector or memory CD8+ T cells, donor CD8+ T cells were purified from brains of infected congenic animals (CD45.1) at week 4 postinfection. Subsequently, effector (KLRG1hi CD127lo) and memory (KLRG1lo CD127hi) CD8+ T cell subsets were sorted based on well-established subset-specific phenotypic markers and adoptively transferred to chronically infected (week 5 pi) recipients (Fig. 6A) (18). A previous study has demonstrated in the Borna disease virus model that lymphocytes harvested from brain indeed home to the recipient brain upon adoptive transfer (36). Five weeks posttransfer, brains of recipient animals were assessed to determine if treatment with memory as opposed to effector CD8+ T cells resulted in differential long-term effects on parasite control and endogenous CD8+ T cell rescue. As shown in Fig. 6B, independently of the adoptively transferred CD8 subset, no significant decrease in the percentage of infected brain leukocytes was noted in the recipients vis-à-vis untreated mice (Fig. 3G). This suggests that poor long-term control of chronic toxoplasmosis is not specific to the donor CD8 subset. Further, levels of PD-L1 expression on brain leukocytes were similar in the two groups of recipient animals (Fig. 6C). Similarly, the absolute numbers of endogenous brain resident CD8+ T cells (Fig. 6D) as well their PD-1 expression levels remained unaltered (Fig. 6E). This suggests that exhausted cells cannot be rescued even in the presence of memory CD8+ T cells from healthy donors. Lastly, to determine if transfer of donor memory CD8+ T cells improved the functionality of exhausted endogenous T cell populations, IFN-γ production was examined. Independently of the CD8+ T cell subset transferred, no change in IFN-γ production by endogenous CD8+ T cells was noted (Fig. 6F). These results demonstrate that the transient ameliorative effect provided by donor CD8+ T cells in chronically infected animals is not improved or maintained by treating animals with memory CD8+ T cells.
Fig 6.

Donor memory CD8+ T cells fail to rescue endogenous CD8+ T cells during late-chronic toxoplasmosis. (A) Donor CD45.1 mice were infected with T. gondii. Four weeks later, brain CD8+ T cells were purified from donor CD45.1 mice and subsequently sorted into effector and memory T cell subsets. Sorted cells (1.5 × 104) were transferred into chronically infected CD45.2 recipient mice (n = 4) at week 5 postinfection via the i.v. route. (B) T. gondii-infected cells in brain leukocyte-gated samples from recipient mice were assayed by flow cytometry. A bar graph shows percentages of parasite-infected leukocytes at 5 weeks posttransfer (10 weeks postinfection) in recipient animals that received effector CD8+ T cells or memory CD8+ T cells. (C) PD-L1 expression was assayed on total brain leukocytes by flow cytometry at week 5 posttransfer in recipient animals. (D) A bar graph represents the absolute number of endogenous CD8+ T cells in brain of CD8+ T cell recipients. (E) PD-1 expression on endogenous CD8+ T cells in brain was evaluated by flow cytometry. (F) Brain mononuclear cells were stimulated with TLA and then evaluated for IFN-γ production by endogenous CD8+ T cells at 5 weeks posttransfer. Data were acquired using a Cytek-upgraded 8-color BD FACSCalibur flow cytometer which accounts for differences in fluorescence scale.
DISCUSSION
CD8+ T cells play a critical role in long-term T. gondii infection, and their optimal maintenance is essential for controlling the chronic infection (4). Mice lacking CD8+ T cells are unable to withstand chronic infection (37), and anti-CD8 treatment of chronically infected wild-type animals leads to reactivation of latent disease (14). Reactivation of parasitic infection in HIV-infected populations is believed to occur at later stages of viral infection when the adaptive immune compartment gets compromised (4). Similarly, wild-type C57BL/6 mice, in spite of evoking a robust CD8+ T cell response during the acute phase (38), are unable to control chronic infection (16). These animals ultimately become moribund and succumb to reactivation of disease. The inability of these mice to contain chronic infection was attributed to CD8+ T cell dysfunction mediated by increased expression of a coinhibitory molecule, PD-1 (16). This is the first report describing host mortality due to PD-1-mediated CD8+ T cell exhaustion in a parasitic infection. Similar to observations made in chronic viral models of CD8 exhaustion (39), reinvigoration of CD8+ T cell response via αPD-L1 therapy could restore the exhausted CD8+ T cells and prevent death.
As stated above, immune CD8+ T cells have the ability to protect naive or immunodeficient animals against acute Toxoplasma infection which can otherwise prove to be lethal to these animals (4, 5). In the current study, we planned to determine if exogenous treatment with immune CD8+ T cells can control parasite reactivation and rescue exhausted CD8+ T cells. At first, we established that de-encystation as shown by reduced cyst burden in chronically infected mice (week 7) is a hallmark of disease onset. This was reconfirmed using multiple approaches such as assessment of parasite stage-specific markers, frequency of parasitized leukocytes, and CD8-mediated expression of KLRG1, a marker of repeated antigen encounter (34). Since IFN-γ is considered to be pivotal for keeping chronic infection under control and depletion of this cytokine results in rapid parasite reactivation (14), we further validated if transient IFN-γ depletion resulted in parasite de-encystation and reactivation. Akin to disease recrudescence during “natural” infection, αIFN-γ treatment resulted in decreased cyst burden and increased leukocyte parasitization.
Multiple studies have interpreted higher cyst burden as a readout of poor immune response (40, 41). However, most of these studies using knockout mice or antibody-based depletion or immune cell transfer or cytokine addition have used experimental setups which have a strong impact on the acute-phase infection (as well as chronic). Thus, the absence of an immune factor such as interleukin-12 (IL-12) in p40−/− mice would lead to poor control of tachyzoites in the acute phase, resulting in an elevated number of tachyzoites getting converted into cysts in the chronic phase (40). In the former context, the elevated cyst burden can be interpreted only as a readout for poor immune control during the acute phase and not necessarily during the chronic phase of infection. In contrast, the current study exclusively focused on the chronic infection phase and addressed factors that ensure that cysts remain quiescent. Based on our data, it seems that reduced cyst burden is a good maker for the onset of disease recrudescence during natural infection. Although after initial reactivation at week 7 the cyst burden modestly increased at week 10, presumably due to partial immune control, the parasites were still undergoing stage conversion. Nevertheless, moribund mice in all cases exhibited low cyst numbers and a high degree of leukocyte parasitization (4). Similar results in terms of reduced cyst burden were noted in another study involving a dexamethasone-induced model of reactivated toxoplasmosis (42). It is possible that in moribund mice, due to severely attenuated CD8 response as well other potential immune deficits (16), nonencysted parasite are not forced to re-encyst like mice infected at week 10. This perhaps results in a sharp decrease in cyst burden with a corresponding increase in the percentage of parasitized leukocytes.
Since CD8 T cells undergo exhaustion concomitant with parasite reactivation during the later phases of chronic toxoplasmosis (16), we next explored if adoptive transfer of healthy immune CD8+ T cells to chronically infected prerecrudescent mice was able to control parasite reactivation. Although such donor cells were able to prevent Toxoplasma reactivation in chronically infected hosts, the effect was not long-term. During the later stages, immune CD8+ T cell transfer recipients underwent parasite de-encystation akin to control mice. This was further evidenced by the observation that administration of immune CD8+ T cells did not confer a long-term survival advantage to these animals. Also, surprisingly, treatment of chronically infected animals with memory phenotype CD8+ T cells from healthy donors failed to rescue the exhausted CD8+ T cells or decrease the parasite load in the recipient animals. Considering that memory T cell imprinting can take place as early as the first cell division (43), this raises the issue of whether the donor memory CD8 T cells even at week 4 pi are already predisposed to CD8 exhaustion. Alternatively, it is possible that upon transfer to a highly exhausted environment (where the PD-1 receptor PD-L1 is expressed at high levels on hematopoietic cells), these memory CD8 T cells may become rapidly dysfunctional and deleted. These possibilities may not be mutually exclusive. This is not entirely unexpected based on our previous finding that PD-1 is not only expressed (albeit at a lower level than in chronically infected mice) very early during the course of Toxoplasma infection (16) but is also expressed preferentially on memory phenotype CD8+ T cells (18).
This transient control of reactivation was concomitant with reduced PD-L1 expression. Since the PD–PD-L1 pathway is involved in CD8 exhaustion and treatment with αPD-L1 rescues CD8+ T cell functionality (16), it is indeed surprising that attenuated PD-L1 levels failed to augment endogenous CD8+ T cell proliferation or functionality or absolute numbers. Moreover, irrespective of the source of donor CD8+ T cells, PD-1 expression in endogenous CD8+ T cell populations remained unaffected. This discrepancy may be due to failure of immune CD8+ T cells to bring down PD-L1 expression sufficiently to ablate PD-1–PD-L1 interaction.
Since donor CD8+ T cells fail to rescue the endogenous CD8 population, it is likely that effector mechanisms exhibited by donor CD8+ T cells are directly responsible for short-term control of parasite reactivation. As mentioned earlier, previous studies in chronic T. gondii model have demonstrated that cytotoxicity and, especially, IFN-γ production are critical effector mechanisms for CD8+ T cell-mediated parasite control (15, 37, 44). The differential contributions of these effector mechanisms to short-term parasite control by donor CD8+ T cells need to be assessed. CD8+ T cell-produced IFN-γ also plays a critical role in eliciting nitric oxide production by macrophages via induction of inducible nitric oxide synthase (iNOS) (45). Incidentally, nitric oxide (NO) plays a critical role in control of chronic toxoplasmosis (45). Recently, PD-L1 expression has been shown to downregulate iNOS expression and NO production by peritoneal macrophages in Trypanosoma cruzi-infected mice (46). In light of this finding and the observation that donor CD8+ T cells downregulate PD-L1 expression on endogenous leukocytes in our model, it is tempting to speculate that another mechanism of CD8-mediated parasite control may be increased NO production by macrophages via PD-L1 downregulation.
A recent study in a viral model has suggested that the antigen burden is a critical determinant of CD8 exhaustion (47). However, our current data strongly suggest that control of antigen during chronic phase of infection is not sufficient for CD8+ T cell rescue. Although donor immune CD8+ T cells failed to rescue exhausted CD8+ T cells, these cells themselves do not exhibit a cardinal feature of robust memory CD8+ T cell long-term survival. As such, bulk donor CD8+ T cells or even sorted memory phenotype donor CD8+ T cells cannot be detected at later time points. A previous study has demonstrated that memory/effector imprinting of T cell occurs early in the course of T cell development (43). A subsequent investigation demonstrated that antigen encounter history diversifies the CD8 T cell transcriptome while preserving a core signature (48). This raises the possibility that donor CD8+ T cells are not appropriately programmed for memory development or that their memory lineage commitment gets corrupted after transfer into a dysfunctional environment. Addressing these potential mechanisms will be critical for development of robust immunotherapy against this pathogen. Although αPD-L1 treatment rescued exhausted CD8+ T cells during chronic toxoplasmosis (16), a caveat of this study is that prolonged blockade of the PD–PD-L1 pathway carries the risk of autoimmunity (49). Additionally, will the discontinuation of anti-PD-L1 antibody restart the process of exhaustion? Nevertheless, as our data clearly demonstrate, immunotherapy with immune CD8+ T cells is not an appropriate solution for the rescue of dysfunctional CD8+ T cells. Recent studies from our group have demonstrated that anti-PD-L1 treatment of chronically infected mice augments the levels of CD8+ T cell-expressed Eomes (16, 17), a T-box family transcription factor critical for memory development (50). Taking this into consideration, it will be interesting to determine if adoptive transfer of retrovirally transfected Eomes overexpressing CD8+ T cells is able to elicit a more robust and long-lasting outcome. Since one of the factors downregulating Eomes is BLIMP-1, a zinc finger transcription factor (51), alternatively, adoptive transfer of immune CD8+ T cells from conditional BLIMP-1 homozygous or heterozygous animals may be better at controlling disease recrudescence. Addressing these possibilities will be important for development of better immunotherapeutics against this pathogen. Nevertheless, the current report provides strong evidence that CD8+ T cells can control parasite reactivation, albeit transiently, via prevention of de-encystation. Identifying new molecular targets that make donor CD8+ T cells long-lived will be critical for preventing TE from a clinical perspective.
ACKNOWLEDGMENT
This work was supported by US National Institutes of Health grant AI-33325 to I.A.K.
Footnotes
Published ahead of print 1 July 2013
REFERENCES
- 1.Kur J, Holec-Gasior L, Hiszczynska-Sawicka E. 2009. Current status of toxoplasmosis vaccine development. Expert Rev. Vaccines 8:791–808 [DOI] [PubMed] [Google Scholar]
- 2.Falusi O, French AL, Seaberg EC, Tien PC, Watts DH, Minkoff H, Piessens E, Kovacs A, Anastos K, Cohen MH. 2002. Prevalence and predictors of Toxoplasma seropositivity in women with and at risk for human immunodeficiency virus infection. Clin. Infect. Dis. 35:1414–1417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Grant IH, Gold JW, Rosenblum M, Niedzwiecki D, Armstrong D. 1990. Toxoplasma gondii serology in HIV-infected patients: the development of central nervous system toxoplasmosis in AIDS. AIDS 4:519–521 [PubMed] [Google Scholar]
- 4.Bhadra R, Gigley JP, Khan IA. 2011. The CD8 T-cell road to immunotherapy of toxoplasmosis. Immunotherapy 3:789–801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gigley JP, Bhadra R, Khan IA. 2011. CD8 T cells and Toxoplasma gondii: a new paradigm. J. Parasitol. Res. 2011:243796. 10.1155/2011/243796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mui EJ, Schiehser GA, Milhous WK, Hsu H, Roberts CW, Kirisits M, Muench S, Rice D, Dubey JP, Fowble JW, Rathod PK, Queener SF, Liu SR, Jacobus DP, McLeod R. 2008. Novel triazine JPC-2067-B inhibits Toxoplasma gondii in vitro and in vivo. PLoS Negl. Trop. Dis. 2:e190. 10.1371/journal.pntd.0000190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jones JL, Hanson DL, Dworkin MS, Alderton DL, Fleming PL, Kaplan JE, Ward J. 1999. Surveillance for AIDS-defining opportunistic illnesses, 1992–1997. MMWR CDC Surveill. Summ. 48:1–22 [PubMed] [Google Scholar]
- 8.Vidal JE, Hernandez AV, de Oliveira AC, Dauar RF, Barbosa SP, Jr, Focaccia R. 2005. Cerebral toxoplasmosis in HIV-positive patients in Brazil: clinical features and predictors of treatment response in the HAART era. AIDS Patient Care STDS 19:626–634 [DOI] [PubMed] [Google Scholar]
- 9.Lindström I, Kaddu-Mulindwa DH, Kironde F, Lindh J. 2006. Prevalence of latent and reactivated Toxoplasma gondii parasites in HIV-patients from Uganda. Acta Trop. 100:218–222 [DOI] [PubMed] [Google Scholar]
- 10.Pradhan S, Yadav R, Mishra VN. 2007. Toxoplasma meningoencephalitis in HIV-seronegative patients: clinical patterns, imaging features and treatment outcome. Trans. R. Soc. Trop. Med. Hyg. 101:25–33 [DOI] [PubMed] [Google Scholar]
- 11.Lehmann T, Marcet PL, Graham DH, Dahl ER, Dubey JP. 2006. Globalization and the population structure of Toxoplasma gondii. Proc. Natl. Acad. Sci. U. S. A. 103:11423–11428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Carme B, Demar M, Ajzenberg D, Darde ML. 2009. Severe acquired toxoplasmosis caused by wild cycle of Toxoplasma gondii, French Guiana. Emerg. Infect. Dis. 15:656–658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Khan A, Jordan C, Muccioli C, Vallochi AL, Rizzo LV, Belfort R, Jr, Vitor RW, Silveira C, Sibley LD. 2006. Genetic divergence of Toxoplasma gondii strains associated with ocular toxoplasmosis, Brazil. Emerg. Infect. Dis. 12:942–949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gazzinelli R, Xu Y, Hieny S, Cheever A, Sher A. 1992. Simultaneous depletion of CD4+ and CD8+ T lymphocytes is required to reactivate chronic infection with Toxoplasma gondii. J. Immunol. 149:175–180 [PubMed] [Google Scholar]
- 15.Suzuki Y, Conley FK, Remington JS. 1989. Importance of endogenous IFN-gamma for prevention of toxoplasmic encephalitis in mice. J. Immunol. 143:2045–2050 [PubMed] [Google Scholar]
- 16.Bhadra R, Gigley JP, Weiss LM, Khan IA. 2011. Control of Toxoplasma reactivation by rescue of dysfunctional CD8+ T-cell response via PD-1-PDL-1 blockade. Proc. Natl. Acad. Sci. U. S. A. 108:9196–9201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bhadra R, Gigley JP, Khan IA. 2011. Cutting edge: CD40-CD40 ligand pathway plays a critical CD8-intrinsic and -extrinsic role during rescue of exhausted CD8 T cells. J. Immunol. 187:4421–4425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bhadra R, Gigley JP, Khan IA. 2012. PD-1-mediated attrition of polyfunctional memory CD8+ T cells in chronic toxoplasma infection. J. Infect. Dis. 206:125–134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bhadra R, Khan IA. 2012. Redefining chronic toxoplasmosis—a T cell exhaustion perspective. PLoS Pathog. 8:e1002903. 10.1371/journal.ppat.1002903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bhadra R, Cobb DA, Weiss LM, Khan IA. 2013. Psychiatric disorders in toxoplasma seropositive patients—the CD8 connection. Schizophr. Bull. 39:485–489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Khan IA, Kasper LH. 1996. IL-15 augments CD8+ T cell-mediated immunity against Toxoplasma gondii infection in mice. J. Immunol. 157:2103–2108 [PubMed] [Google Scholar]
- 22.Wetzel DM, Hakansson S, Hu K, Roos D, Sibley LD. 2003. Actin filament polymerization regulates gliding motility by apicomplexan parasites. Mol. Biol. Cell 14:396–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pfaffl MW. 2001. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 29:e45. 10.1093/nar/29.9.e45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bhadra R, Khan IA. 2012. IL-7 and IL-15 do not synergize during CD8 T cell recall response against an obligate intracellular parasite. Microbes Infect. 14:1160–1168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lyons RE, McLeod R, Roberts CW. 2002. Toxoplasma gondii tachyzoite-bradyzoite interconversion. Trends Parasitol. 18:198–201 [DOI] [PubMed] [Google Scholar]
- 26.Ferguson DJ, Hutchison WM. 1987. The host-parasite relationship of Toxoplasma gondii in the brains of chronically infected mice. Virchows Arch. A Pathol. Anat. Histopathol. 411:39–43 [DOI] [PubMed] [Google Scholar]
- 27.Dubey JP, Lindsay DS, Speer CA. 1998. Structures of Toxoplasma gondii tachyzoites, bradyzoites, and sporozoites and biology and development of tissue cysts. Clin. Microbiol. Rev. 11:267–299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dzierszinski F, Mortuaire M, Dendouga N, Popescu O, Tomavo S. 2001. Differential expression of two plant-like enolases with distinct enzymatic and antigenic properties during stage conversion of the protozoan parasite Toxoplasma gondii. J. Mol. Biol. 309:1017–1027 [DOI] [PubMed] [Google Scholar]
- 29.Fux B, Nawas J, Khan A, Gill DB, Su C, Sibley LD. 2007. Toxoplasma gondii strains defective in oral transmission are also defective in developmental stage differentiation. Infect. Immun. 75:2580–2590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Courret N, Darche S, Sonigo P, Milon G, Buzoni-Gatel D, Tardieux I. 2006. CD11c- and CD11b-expressing mouse leukocytes transport single Toxoplasma gondii tachyzoites to the brain. Blood 107:309–316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang X, Claflin J, Kang H, Suzuki Y. 2005. Importance of CD8(+)Vbeta8(+) T cells in IFN-gamma-mediated prevention of toxoplasmic encephalitis in genetically resistant BALB/c mice. J. Interferon Cytokine Res. 25:338–344 [DOI] [PubMed] [Google Scholar]
- 32.Bengsch B, Spangenberg HC, Kersting N, Neumann-Haefelin C, Panther E, von Weizsacker F, Blum HE, Pircher H, Thimme R. 2007. Analysis of CD127 and KLRG1 expression on hepatitis C virus-specific CD8+ T cells reveals the existence of different memory T-cell subsets in the peripheral blood and liver. J. Virol. 81:945–953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Voehringer D, Koschella M, Pircher H. 2002. Lack of proliferative capacity of human effector and memory T cells expressing killer cell lectinlike receptor G1 (KLRG1). Blood 100:3698–3702 [DOI] [PubMed] [Google Scholar]
- 34.Bustamante JM, Bixby LM, Tarleton RL. 2008. Drug-induced cure drives conversion to a stable and protective CD8+ T central memory response in chronic Chagas disease. Nat. Med. 14:542–550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Galea I, Bernardes-Silva M, Forse PA, van Rooijen N, Liblau RS, Perry VH. 2007. An antigen-specific pathway for CD8 T cells across the blood-brain barrier. J. Exp. Med. 204:2023–2030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sobbe M, Bilzer T, Gommel S, Noske K, Planz O, Stitz L. 1997. Induction of degenerative brain lesions after adoptive transfer of brain lymphocytes from Borna disease virus-infected rats: presence of CD8+ T cells and perforin mRNA. J. Virol. 71:2400–2407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Suzuki Y, Remington JS. 1988. Dual regulation of resistance against Toxoplasma gondii infection by Lyt-2+ and Lyt-1+, L3T4+ T cells in mice. J. Immunol. 140:3943–3946 [PubMed] [Google Scholar]
- 38.Bhadra R, Guan H, Khan IA. 2010. Absence of both IL-7 and IL-15 severely impairs the development of CD8 T cell response against Toxoplasma gondii. PLoS One 5:e10842. 10.1371/journal.pone.0010842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wherry EJ. 2011. T cell exhaustion. Nat. Immunol. 12:492–499 [DOI] [PubMed] [Google Scholar]
- 40.Yap G, Pesin M, Sher A. 2000. Cutting edge: IL-12 is required for the maintenance of IFN-gamma production in T cells mediating chronic resistance to the intracellular pathogen, Toxoplasma gondii. J. Immunol. 165:628–631 [DOI] [PubMed] [Google Scholar]
- 41.Fox BA, Falla A, Rommereim LM, Tomita T, Gigley JP, Mercier C, Cesbron-Delauw MF, Weiss LM, Bzik DJ. 2011. Type II Toxoplasma gondii KU80 knockout strains enable functional analysis of genes required for cyst development and latent infection. Eukaryot. Cell 10:1193–1206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Djurković-Djaković O, Milenkovic V, Nikolic A, Bobic B, Grujic J. 2002. Efficacy of atovaquone combined with clindamycin against murine infection with a cystogenic (Me49) strain of Toxoplasma gondii. J. Antimicrob. Chemother. 50:981–987 [DOI] [PubMed] [Google Scholar]
- 43.Chang JT, Palanivel VR, Kinjyo I, Schambach F, Intlekofer AM, Banerjee A, Longworth SA, Vinup KE, Mrass P, Oliaro J, Killeen N, Orange JS, Russell SM, Weninger W, Reiner SL. 2007. Asymmetric T lymphocyte division in the initiation of adaptive immune responses. Science 315:1687–1691 [DOI] [PubMed] [Google Scholar]
- 44.Suzuki Y, Wang X, Jortner BS, Payne L, Ni Y, Michie SA, Xu B, Kudo T, Perkins S. 2010. Removal of Toxoplasma gondii cysts from the brain by perforin-mediated activity of CD8+ T cells. Am. J. Pathol. 176:1607–1613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schlüter D, Deckert-Schluter M, Lorenz E, Meyer T, Rollinghoff M, Bogdan C. 1999. Inhibition of inducible nitric oxide synthase exacerbates chronic cerebral toxoplasmosis in Toxoplasma gondii-susceptible C57BL/6 mice but does not reactivate the latent disease in T. gondii-resistant BALB/c mice. J. Immunol. 162:3512–3518 [PubMed] [Google Scholar]
- 46.Dulgerian LR, Garrido VV, Stempin CC, Cerban FM. 2011. Programmed death ligand 2 regulates arginase induction and modifies Trypanosoma cruzi survival in macrophages during murine experimental infection. Immunology 133:29–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mueller SN, Ahmed R. 2009. High antigen levels are the cause of T cell exhaustion during chronic viral infection. Proc. Natl. Acad. Sci. U. S. A. 106:8623–8628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wirth TC, Xue HH, Rai D, Sabel JT, Bair T, Harty JT, Badovinac VP. 2010. Repetitive antigen stimulation induces stepwise transcriptome diversification but preserves a core signature of memory CD8(+) T cell differentiation. Immunity 33:128–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kasagi S, Kawano S, Kumagai S. 2011. PD-1 and autoimmunity. Crit. Rev. Immunol. 31:265–295 [DOI] [PubMed] [Google Scholar]
- 50.Banerjee A, Gordon SM, Intlekofer AM, Paley MA, Mooney EC, Lindsten T, Wherry EJ, Reiner SL. 2010. Cutting edge: the transcription factor eomesodermin enables CD8+ T cells to compete for the memory cell niche. J. Immunol. 185:4988–4992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rutishauser RL, Martins GA, Kalachikov S, Chandele A, Parish IA, Meffre E, Jacob J, Calame K, Kaech SM. 2009. Transcriptional repressor Blimp-1 promotes CD8(+) T cell terminal differentiation and represses the acquisition of central memory T cell properties. Immunity 31:296–308 [DOI] [PMC free article] [PubMed] [Google Scholar]