

Abstract
Francisella tularensis, the causative agent of tularemia, is most deadly in the pneumonic form; therefore, mucosal immunity is an important first line of defense against this pathogen. We have now evaluated the lethality of primary F. tularensis live vaccine strain (LVS) pulmonary infection in mice that are defective in IgA (IgA−/− mice), the predominant mucosal Ig isotype. The results showed that IgA−/− mice were more susceptible than IgA+/+ mice to intranasal F. tularensis LVS infection, despite developing higher levels of LVS-specific total, IgG, and IgM antibodies in the bronchoalveolar lavage specimens following infection. In addition, the absence of IgA resulted in a significant increase in bacterial loads and reduced survival. Interestingly, IgA−/− mice had lower pulmonary gamma interferon (IFN-γ) levels and decreased numbers of IFN-γ-secreting CD4+ and CD8+ T cells in the lung on day 9 postinfection compared to IgA+/+ mice. Furthermore, IgA−/− mice displayed reduced interleukin 12 (IL-12) levels at early time points, and supplementing IgA−/− mice with IL-12 prior to LVS challenge induced IFN-γ production by NK cells and rescued them from mortality. Thus, IgA−/− mice are highly susceptible to primary pulmonary LVS infections not only because of IgA deficiency but also because of reduced IFN-γ responses.
INTRODUCTION
Francisella tularensis is a facultative, intracellular Gram-negative bacterium that can invade the body via multiple routes, including intradermally, through ingestion, or by inhalation. The respiratory route of infection is most deadly with a mortality rate of as high as 30% (1). Due to its high virulence in humans, together with its high infectivity and ease of dissemination, this pathogen has been classified as a tier 1 biothreat agent (2). Consequently, there is a major effort in understanding protective immune responses against this biothreat agent, which will in turn, aid in the development of an effective vaccine strategy.
Mucosal immune responses represent the first barrier of defense against respiratory infections. Immunoglobulin A (IgA) is the predominant Ig isotype in mucosal tissues (3), and its importance in the defense against respiratory pathogens has been repeatedly shown (4–9). A lack of IgA in the human population is the most common primary immunodeficiency disease (10, 11). Although most IgA-deficient individuals are apparently healthy, there is an increased frequency of recurrent respiratory tract infections in this population. However, the heterogeneity of IgA deficiency in humans complicates investigations into the role of IgA in mucosal immunity (10).
We (8) and others (12) have previously shown that IgA-deficient mice have increased susceptibility to respiratory live vaccine strain (LVS) challenge after intranasal (i.n.) vaccination with inactivated LVS. This was interpreted as showing the importance of IgA in protection of vaccinated mice against pulmonary infection. However, in the present study, we report that mice that are defective in IgA (IgA−/− mice) are also more susceptible to primary LVS challenge than IgA+/+ mice. This loss of resistance in IgA−/− mice was associated with impaired gamma interferon (IFN-γ) responses in the lungs.
MATERIALS AND METHODS
Mice.
Six- to 7-week-old female BALB/c mice were routinely used in these studies. IgA+/+ mice were purchased from Taconic (Germantown, NY), and IgA−/− mice were bred at the Albany Medical College Animal Facility. Mice were anesthetized by intraperitoneal (i.p.) injection with 100 μl xylazine (20 mg/ml) and ketamine (1 mg/ml) in phosphate-buffered saline (PBS). For respiratory infection, anesthetized mice were i.n. inoculated with 50 μl PBS containing 500 CFU of Francisella tularensis LVS. All animal procedures were approved by the Institutional Animal Care and Use Committee.
Bacterial burden and cytokine analysis.
Lung, liver, and spleen tissue homogenates were prepared 1, 3, 5, 7, and 9 days after i.n. bacterial challenge. Serial dilutions of the samples were inoculated on chocolate agar plates and incubated for 3 days at 37°C to enumerate CFU. For cytokine analysis, organ homogenates were centrifuged at 10,000 × g for 10 min. Supernatants were collected, and they were stored at −80°C. IFN-γ and tumor necrosis factor alpha (TNF-α) levels were tested using a CBA mouse inflammatory cytokine kit (BD Biosciences). Interleukin 12 (IL-12) levels were quantitated using mouse IL-12 (p40) enzyme-linked immunosorbent assay (ELISA) set (BD Biosciences).
IFN-γ intracellular staining.
F. tularensis LVS-specific IFN-γ-producing cells were induced in the pulmonary tracts of 7-week-old IgA+/+ and IgA−/− mice by i.n. administration of 500 CFU LVS. The lungs were harvested 9 days postinfection, and single-cell suspensions were obtained by incubation with 2 mg/ml collagenase D (Roche Diagnostics), 0.25 mg/ml DNase I (Roche Diagnostics), and 10 mM MgCl2 for 1 h at 37°C. The cells were restimulated ex vivo in 96-well plates by culturing 5 × 105 cells/well with or without equal numbers of F. tularensis LVS for 1 h, followed by an additional 1 h of incubation with 10 μg/ml brefeldin A (Sigma). The cells were washed in PBS containing 2% fetal calf serum (FCS), and Fc receptors were blocked by incubation with mouse 2.4G2 (Fcγ III/II receptor) antibody for 20 min at 4°C. The cells were then stained with phycoerythrin (PE)-conjugated anti-DX5 (Biolegend), PE-Cy7-conjugated anti-CD8 (clone 53-6.7) (BD Pharmingen), or allophycocyanin (APC)-conjugated anti-CD4 (clone RM4-5) (BD Pharmingen) monoclonal antibodies (MAbs). Dead cells were labeled with 7-aminoactinomycin D (eBioscience). This was followed by 20-min incubation with BD Fixation/Permeabilization solution. The cells were washed twice with BD Perm/Wash buffer and stained for 30 min with fluorescein isothiocyanate (FITC)-conjugated anti-IFN-γ (clone XMG1.2) (Caltag). After the final wash, the cells were resuspended in 200 μl of PBS containing 2% BSA (PBS–2% BSA). Stained cells were quantitated using a FACSCanto flow cytometer.
IL-12 treatment.
In some cases, mice were inoculated i.n. with 1 μg of murine recombinant IL-12 in 25 μl PBS containing 1% normal mouse serum 24 h before F. tularensis LVS challenge. Control mice received PBS–1% normal mouse serum alone. Preliminary experiments indicated that 1 μg of IL-12 given i.n. was optimal for protective efficacy, and no toxicity was observed.
Antibody analysis.
For antibody analysis, mice were immunized i.n. with 500 CFU of F. tularensis LVS, and bronchoalveolar lavage fluid (BALF) samples were harvested at day 7 postinfection. Anti-LVS antibody levels in BALF samples were determined by ELISA as described previously (13). Briefly, microtiter plates (Nalge Nunc International, Rochester, NY) were coated at 4°C overnight with 100 μl of F. tularensis LVS (5 × 106 CFU/well) in carbonate buffer. The plates were washed with PBS containing 0.05% Tween 20 and blocked for 2 h with 200 μl of PBS containing 10% bovine serum albumin. Serial dilutions of BALF samples were added to the wells on the plates (100 μl/well), and the plates were incubated for 90 min at 37°C. After washing, biotin-conjugated goat anti-mouse antibodies specific for IgG, IgM, or IgA (Caltag, Burlingame, CA) were added and incubated for 1 h at 37°C. The plates were washed, and then streptavidin conjugated to horseradish peroxidase (Biosource, Camarillo, CA) was added (100 μl/well). The plates were incubated for 20 min and washed, and 100 μl of 3,3′,5,5′-tetramethylbenzidine (TMB) peroxidase substrate was added (KPL, Gaithersburg, MD). After 5 to 20 min, the reaction was stopped, and the plates were read at 450 nm using a PowerWave HT microplate reader (BioTek Instruments, Winooski, VT).
Histological analysis.
Lungs, spleens, and livers were harvested at day 7 following F. tularensis LVS infection and fixed overnight in 10% formaldehyde (Fisher Diagnostics). Ten-micron-thick sections were stained with hematoxylin and eosin and evaluated by light microscopy.
Statistics.
Student's t test (for comparison of two groups) or analysis of variance (ANOVA) (for comparison of multiple groups) was used to analyze bacterial burdens, cytokine levels, flow cytometric staining, and antibody levels. Survival data were analyzed by the Kaplan-Meier log rank test using GraphPad Prism 4 software (San Diego, CA). A P value of <0.05 was considered statistically significant.
RESULTS
IgA−/− mice exhibit greater susceptibility to primary LVS challenge than IgA+/+ mice.
To determine susceptibility to pneumonic tularemia in the absence of IgA, 500 CFU of F. tularensis LVS was administered i.n. to BALB/c IgA−/− and IgA+/+ mice. The mortality and morbidity of the infected mice were monitored daily for a 20-day period. Following F. tularensis LVS infection, rapid weight loss, reaching approximately 20% by day 10 postinfection, was observed in both IgA+/+ and IgA−/− mice (Fig. 1A). Infected IgA+/+ mice began to regain weight on day 11 postinfection and had fully regained their original weight by day 20. In contrast, IgA−/− mice continually lost weight with no sign of recovery during the 20-day observation period. In addition, the survival rate of IgA+/+ mice was 83%, which was significantly greater than the 17% survival rate of IgA−/− mice (P < 0.05) (Fig. 1B). These results demonstrate that IgA is indispensable for mucosal defense against sublethal i.n. F. tularensis LVS infection.
Fig 1.
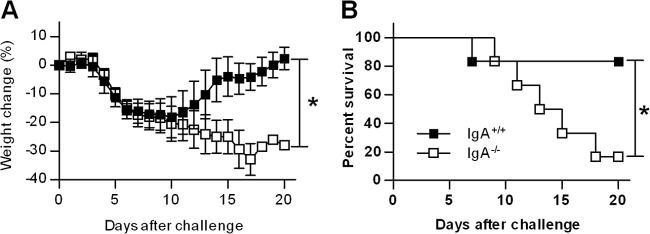
Susceptibility of IgA−/− and IgA+/+ mice to pulmonary F. tularensis LVS infection. Groups of six to eight mice were challenged i.n. with 500 CFU of F. tularensis LVS. Weight loss (A) and survival (B) of infected mice were monitored for 20 days. Values are mean percentages ± standard deviations (SD) (error bars). Values that are statistically significantly different (P < 0.05) are indicated by a bar and asterisk.
IgA−/− mice exhibit greater bacterial loads than IgA+/+ mice.
To assess whether the increased mortality in IgA−/− mice was due to uncontrolled bacterial replication, we evaluated bacterial loads in the lungs at various time points after infection. F. tularensis LVS inoculated i.n. is capable of spreading from its primary infection site, the respiratory tract, via the bloodstream, to induce systemic infection. Therefore, bacterial CFU were also enumerated in the spleens and livers. A 6-fold increase in lung LVS bacterial burden was detected in both the IgA+/+ and IgA−/− mice within 24 h postinfection. F. tularensis LVS burden reached a maximal level of approximately 107 CFU on day 3 postinfection, and the bacterial numbers in the lung were maintained until day 7 postinfection (Fig. 2A). No significant differences in lung LVS bacterial burdens were detected between IgA+/+ and IgA−/− mice. However, on day 9 postinfection, a 2-log-unit decrease in lung LVS bacterial numbers was observed only in IgA+/+ mice, while infection persisted in mice lacking IgA (P < 0.05) (Fig. 2A). Similarly, no significant differences were found in the spleen and liver CFU between the two strains on days 1, 3, 6, and 7 postinfection (Fig. 2B and C). Although a reduction in bacterial burden was observed in the spleens and livers of IgA−/− mice on day 9 postinfection, the numbers still remained significantly greater than those of IgA+/+ mice (P < 0.05). Thus, the lack of IgA does not affect bacterial burden at early time points but has a detrimental impact at later stages of LVS infection, suggesting defective clearance. These data suggest that IgA−/− mice possess intact innate immune responses but are defective in adaptive immune responses, which generally start appearing after day 7 postinfection.
Fig 2.
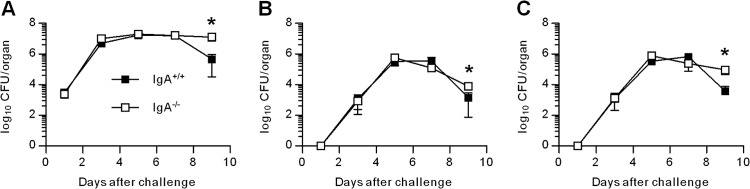
Bacterial clearance in IgA−/− and IgA+/+ mice. Groups of mice were challenged i.n. with 500 CFU of F. tularensis LVS. On days 1, 3, 5, 7, and 9 postinfection, three mice per group were sacrificed, and the bacterial CFU in the lungs (A), spleens (B), and livers (C) were enumerated. Values are means ± SD (error bars). Values that are statistically significantly different (P < 0.05) are indicated by an asterisk.
F. tularensis LVS infection causes significant tissue pathology in both IgA+/+ and IgA−/− mice.
F. tularensis LVS inoculated i.n. results in systemic infection with significant tissue injury (8). To assess the potential role of IgA in limiting organ pathology, which may account for the severe illness observed in IgA−/− mice compared to IgA+/+ mice, we analyzed lung, spleen, and liver histology on day 7 postinfection. BALB/c IgA+/+ mice infected with F. tularensis LVS had significant inflammation in all three organs examined, with severe structural damage and lesions observed in the livers and spleens and significant inflammatory cell infiltrates detected in the lungs (Fig. 3). However, the extent of organ pathology was not different between IgA+/+ and IgA−/− mice, suggesting that IgA has no protective role against LVS-induced tissue injury.
Fig 3.
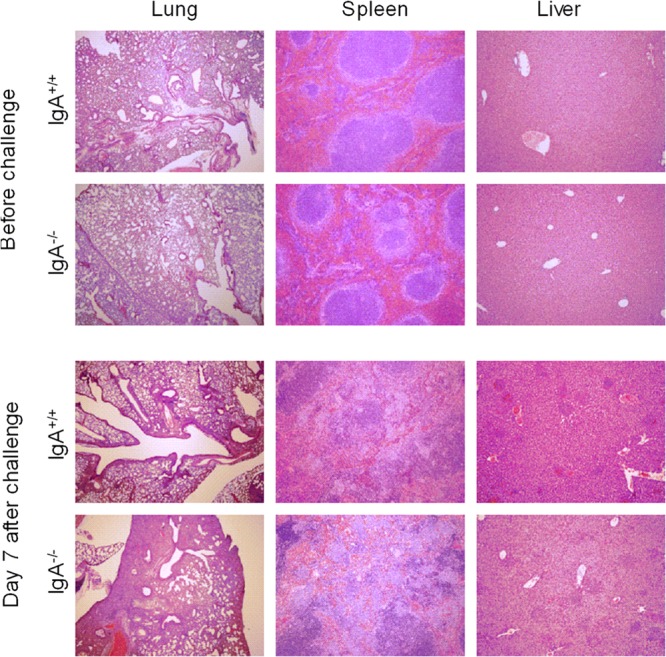
Histology of organs from naive and F. tularensis LVS-infected IgA−/− and IgA+/+ mice. Mice were infected i.n. with 500 CFU of F. tularensis LVS, and their organs were harvested at day 7 postinfection for histological analysis. Lung, spleen, and liver tissue sections were stained with hematoxylin and eosin. The sections shown are representative of the three mice in each group. Magnifications, ×40 for lungs and ×100 for spleens and livers.
IgA−/− mice have increased IgM and IgG antibody levels compared to those of IgA+/+ mice.
Cell-mediated immunity is generally considered to be essential for immunity against intracellular pathogens, and humoral immunity is thought to have more of an ancillary role. However, significant evidence in the literature indicates an important role of antibodies against F. tularensis LVS infection (13–17). Moreover, studies from our laboratory have recently reported a synergistic effect of humoral and cellular immune responses in providing sterilizing immunity against respiratory LVS infection (13). Given this, we determined the F. tularensis LVS-specific antibody titers in the respiratory tracts of infected mice. BALF samples were harvested on day 7 following infection with a low dose of F. tularensis LVS, a time point just before IgA−/− mice begin to succumb. As expected, IgA−/− mice did not generate IgA responses above the background levels (Fig. 4B). However, higher levels of total LVS-specific antibody were detected in the BALF samples from IgA−/− mice than in the samples from IgA+/+ mice (Fig. 4A). These increased levels were attributable to greater levels of both IgM and IgG antibodies (Fig. 4B and D). These results indicate that IgA−/− mice express strong IgM and IgG responses as a compensatory mechanism for the loss of IgA.
Fig 4.

Anti-LVS antibody levels in BALF samples after i.n. F. tularensis LVS infection of IgA−/− and IgA+/+ mice. F. tularensis LVS-specific total (A), IgM (B), IgA (C), and IgG (D) antibody were measured by ELISA on day 7 postinfection. Each symbol represents the mean of four or five mice per group ± SD. Values that are statistically significantly different (P < 0.05) are indicated by an asterisk. Optical densities at 450 nm (OD450) were subtracted from those of BALF samples from naive mice.
Reduced IFN-γ levels in IgA−/− mice.
Despite the increase in pulmonary antibody titers, IgA−/− mice still had greater susceptibility to primary LVS infection and compromised bacterial clearance at later time points of infection. This suggested a potential defect in cell-mediated immunity in the absence of IgA. To investigate this possibility, we measured the levels of TNF-α and IFN-γ in IgA+/+ and IgA−/− mice following F. tularensis LVS infection (Fig. 5A). These cytokines have been shown to be critical components of immunity against both systemic and pulmonary infections with F. tularensis LVS (18–22). Comparable levels of TNF-α were detected in both strains of mice at day 9 postinfection. In contrast, a striking decrease in pulmonary IFN-γ was detected in IgA−/− mice (P < 0.05). Thus, the reduced IFN-γ responses at day 9 postinfection coincide with decreased bacterial clearance in IgA−/− mice.
Fig 5.
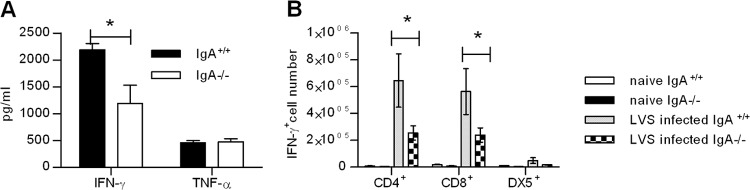
Cytokine responses in IgA−/− and IgA+/+ mice. (A) Mice were challenged i.n. with 500 CFU of F. tularensis LVS, and their lungs were harvested at day 9 postinfection. Lung homogenates were assayed to determine the levels of IFN-γ and TNF-α using the BD cytometric bead array. Values are means plus SD (error bars) for three mice in each group. (B) Lung cells from naive mice and mice infected for 9 days were isolated using collagenase digestion. The cells were restimulated in vitro with F. tularensis LVS and stained for cytoplasmic IFN-γ. Stained samples were analyzed on a FACSCanto flow cytometer. Values are means ± SD (error bars) for three mice in each group. Values that are statistically significantly different (P < 0.05) are indicated by a bar and asterisk.
To further characterize the defective IFN-γ responses in IgA−/− mice, we quantitated IFN-γ+ cells in the lungs of IgA+/+ and IgA−/−mice (Fig. 5B). The mice were challenged i.n. with 500 CFU of F. tularensis LVS, and the lungs were harvested on day 9 postinfection. Single-cell suspensions were prepared, restimulated in vitro with F. tularensis LVS, and assayed for IFN-γ-positive (IFN-γ+) CD4+ T cells, CD8+ T cells, and DX5+ NK cells by flow cytometry. The results revealed that while both strains of mice had comparable numbers of IFN-γ+ DX5+ cells, IgA−/− mice had statistically significant reductions in the numbers of IFN-γ+ CD4+ and CD8+ T cells. Collectively, these data suggest that reduced numbers of anti-LVS-specific IFN-γ-producing CD4+ and CD8+ T cells are responsible for the reduced pulmonary IFN-γ levels in IgA−/− mice.
Intranasal IL-12 treatment protects IgA−/− mice against respiratory LVS challenge.
We have previously shown that IL-12 is required for immunity against pulmonary LVS infection (18). Moreover, i.n. administration of exogenous IL-12 protects wild-type mice from lethal F. tularensis LVS infection through a mechanism that requires CD8+ T cells and IFN-γ. We hypothesized that IFN-γ induction via IL-12 treatment might compensate for the observed reduction in IFN-γ responses in IgA−/− mice and reverse the enhanced susceptibility against LVS challenge. We first evaluated pulmonary IL-12 levels in IgA−/− mice at different time points after the F. tularensis LVS infection. IL-12 levels in IgA+/+ mice peaked at day 3, remained high on day 5, and declined by days 7 to 9 postinfection (Fig. 6A). In contrast, IL-12 levels in IgA−/− mice peaked on day 5 and decreased on day 7 postinfection, indicating that IgA−/− mice have delayed IL-12 responses.
Fig 6.

Effects of IL-12 on F. tularensis infection in IgA−/− mice. (A) Mice were challenged i.n. with 500 CFU of F. tularensis LVS, and their lungs were harvested on days 1, 3, 5, 7, and 9. Lung homogenates were assayed to determine IL-12 levels using ELISA. Values are means ± SD (error bars) for three mice in each group. (B and C) Weight loss (B) and survival (C) of IgA−/− mice treated i.n. with either PBS or 1 μg of IL-12 24 h before F. tularensis LVS infection. (D to F) The surviving mice were euthanized on day 21 for CFU enumeration in the lungs (D), livers (E), and spleens (F). Each symbol represents the value for an individual mouse. The horizontal line is the mean for the group. Values that are statistically significantly different (P < 0.05) are indicated by an asterisk.
To test the protective efficacy of exogenous IL-12, we inoculated mice i.n. with 1 μg of IL-12 or PBS vehicle 24 h before challenge with F. tularensis LVS. Mice treated with IL-12 experienced less weight loss than mock-treated mice (Fig. 6B). Furthermore, IL-12-treated IgA−/− mice all survived LVS challenge, whereas mice that received PBS vehicle had a 40% survival rate (Fig. 6C). The surviving mice from this experiment were euthanized at day 21 postinfection, and their organs were harvested for bacterial burden analysis. IL-12-treated IgA−/− mice had significantly decreased lung bacterial loads than IgA−/− mice treated with vehicle only (P < 0.05) (Fig. 6D). Although statistically significant reductions in bacterial levels were not observed in the livers and spleens, there was a noticeable trend toward reduced bacterial loads in IL-12-treated mice. Thus, i.n. IL-12 treatment can effectively limit bacterial replication at the site of inoculation and thereby increase resistance to fatal F. tularensis LVS infection in IgA−/− mice.
Intranasal IL-12 treatment induces early IFN-γ production in NK cells.
To elucidate the mechanism of IL-12-induced protection against pulmonary tularemia, we determined the lung cell population(s) responsible for IFN-γ production in response to i.n. IL-12 treatment. The mice were inoculated i.n. with either PBS vehicle or IL-12 1 day before F. tularensis LVS challenge, and the lungs were harvested on day 2 postinfection. Single-cell suspensions of lung cells were prepared and analyzed by flow cytometry. The majority of IFN-γ-producing cells detected in IL-12-treated mice were DX5+ NK cells (Fig. 7A). Mice treated with IL-12 had significantly increased percentages of IFN-γ+ DX5+ cells, whereas IFN-γ+ CD4+ and CD8+ T cell percentages were comparable to mice treated with PBS (Fig. 7B). Furthermore, the overall numbers and percentages of DX5+ cells were higher in IL-12-treated mice (Fig. 7C). These data indicate that IL-12 treatment provides protection through early recruitment and activation of NK cells.
Fig 7.

Effects of IL-12 on pulmonary immunity. (A) Mice were pretreated i.n. with 1 μg of IL-12 or with PBS vehicle 24 h before infection with 500 CFU of F. tularensis LVS. Lung cells from mice infected for 2 days were isolated using collagenase digestion. The cells were restimulated in vitro with F. tularensis LVS and stained for cytoplasmic IFN-γ. Representative dot plots from each experimental group are shown. (B) Percentages of IFN-γ+ DX5+, CD4+, and CD8+ lung cells. (C) Percentages and numbers of DX5+ NK lung cells. Values are means ± SD for four mice in each group. Values that are statistically significantly different are indicated by bars and asterisks as follows: ∗, P < 0.05; ∗∗∗, P < 0.001.
DISCUSSION
We have shown that IgA−/− mice exhibit enhanced susceptibility to pulmonary F. tularensis LVS infection. This was clearly reflected in the higher mortality rate and bacterial burdens in IgA−/− mice relative to IgA+/+ controls. This finding is of clinical importance given that IgA deficiency is the most common humoral immunodeficiency, occurring at a rate of approximately 1 in 800 Caucasian individuals (11). Thus, in the event of a bioterrorist attack, in which F. tularensis may be deliberately disseminated via aerosol, individuals with IgA deficiency should be considered a high-risk group, and prioritized vaccination and/or treatment should be given. As a potential prophylactic approach for IgA-deficient individuals against pneumonic tularemia, we have utilized i.n. IL-12 treatment. Administration of exogenous IL-12 1 day before F. tularensis LVS challenge allowed IgA−/− mice to survive an otherwise fatal infection; however, significant weight loss was not prevented. This approach is based on our previous findings that i.n. administration of IL-12 significantly reduced bacterial loads and prevented the death of infected wild-type mice (18). IL-12 is a Th1 skewing cytokine that can induce IFN-γ responses, and we have previously demonstrated that the IL-12-mediated protection is partially dependent on IFN-γ expression. In the present study, we demonstrated that i.n. IL-12 treatment significantly increased DX5+ NK cell recruitment into the lungs and enhanced production of IFN-γ by these cells, but not CD4+ or CD8+ T cells.
Antibody analysis revealed that IgA−/− mice had higher levels of respiratory IgG and IgM. It is interesting to note that while IgM was almost absent in the respiratory tracts of IgA+/+ control mice, substantial levels of IgM were detected in IgA−/− mice. The enhanced production of pulmonary IgM and IgG antibodies may be a mechanism that compensates for the absence of IgA. However, despite having higher levels of IgG and IgM, IgA−/− mice still succumbed to infection, suggesting an indispensable role for mucosal IgA in resistance against pulmonary F. tularensis LVS infection. Furthermore, our data implicate a minimal role for IgG and IgM, in protection against pulmonary tularemia. These findings contrast with previous studies in which passive transfer of purified IgG antibodies, specific for F. tularensis LVS, was found to be protective against both systemic (23) and mucosal (14) challenges. Overproduction of protective IgG antibody, as observed in our model, should in theory compensate for the absence of IgA. Thus, it is possible that additional abnormalities, aside from IgA deficiency, may exist in IgA−/− mice that render them highly susceptible to pulmonary LVS infections.
Natural antibody against T cell-independent antigens is produced primarily by B1a B cells (24). There is no definitive role for B1a cells in tularemia. A study conducted by Cole et al. reported that protection afforded by purified lipopolysaccharide (LPS) against F. tularensis LVS infection is largely mediated by B1a cells (16). In contrast, B1a B cells were recently demonstrated to be detrimental against the highly virulent F. tularensis strain SchuS4 (25). In our model, we observed no consistent differences between IgA+/+ and IgA−/− mice in the recruitment of B1a cells into the lungs or spleens (unpublished observations). Thus, heightened susceptibility among IgA-deficient mice cannot be attributed to increased or decreased presence of B1a cells in the lung.
The aforementioned serum transfer studies have indicated that IgG antibodies are dependent upon host T cell immunity for optimum protection. In those studies, the beneficiary effect of F. tularensis LVS-specific IgG antibodies was lost after depletion of T cells from the recipient mice (14, 23). This observation is consistent with our previous findings that serum antibody treatment failed to protect SCID mice from F. tularensis LVS infection (13). Thus, the dependence of antibody-mediated protection on T cells prompted us to further investigate the mechanisms responsible for the increased sensitivity of IgA−/− mice to F. tularensis LVS infection. Interestingly, we observed significantly reduced numbers of IFN-γ-secreting CD4+ and CD8+ T cells in the lungs of LVS-infected IgA−/− mice compared to IgA+/+ mice. Further experiments revealed reduced pulmonary IL-12 and IFN-γ levels in IgA−/− mice. Given that mice deficient in IL-12 or IFN-γ are highly susceptible to i.n. LVS challenge (18), subnormal, antibacterial Th1 cytokine responses may be responsible, in addition to the absence of IgA, for the heightened susceptibility of IgA−/− mice.
The phenomenon of reduced IFN-γ responses in IgA−/− mice has been observed in other infection/vaccination models (5, 26). Decreased IFN-γ responses in IgA−/− mice observed by us and others are in line with previous findings that showed impaired T cell responses in IgA−/− mice. Harriman et al. showed that lymphocytes from IgA−/− mice have reduced proliferative responses to phytohemagglutinin (PHA), a T cell mitogen, and anti-CD3 treatment, compared to lymphocytes from wild-type mice (27). Further, we found that IgA−/− mice have impaired Th cell priming due to defective antigen-presenting cell functions (4). These observations in mouse models have also been reported in humans. Lymphocytes obtained from IgA-deficient individuals exhibit decreased IFN-γ (28) and proliferative responses (29) following in vitro stimulation with T cell mitogens. Collectively, the above results strongly suggest a role of IgA in regulating T cell immunity although it is currently unclear precisely how IgA might influence T cell immunity. One possibility is that IgA indirectly affects adaptive T cell immunity through modulating homeostasis of intestinal flora. We have found that IgA−/− and IgA+/+ mice have markedly different gut microbial flora (J. H. Wilson-Welder and D. W. Metzger, unpublished observations). It is well established that commensal bacteria regulate immune responses in the gut through secretion of ligands for pattern recognition receptors (30). Ichinohe et al. have recently shown that a regulatory role of commensal microbiota extends to respiratory immune responses (31). Thus, it is plausible that abnormal intestinal bacterial composition in IgA−/− mice could account for the observed impaired T cell immunity. We are currently conducting detailed investigations to test this hypothesis.
In conclusion, we have demonstrated that IgA−/− mice are highly sensitive to primary F. tularensis LVS infections. This heightened susceptibility may not simply be due to a deficiency in IgA expression but may also be attributed to impaired T cell immunity, specifically IFN-γ production by CD4 and CD8 T cells. Importantly, IgA−/− mice can be rescued from lethal F. tularensis LVS infection by i.n. IL-12 inoculation. This illustrates the potential use of IL-12 treatment as a prophylactic against lethal respiratory tularemia.
ACKNOWLEDGMENTS
This work was supported by U.S. National Institutes of Health grant PO1 AI056320.
We thank the Center for Immunology and Microbial Disease Immunology Core laboratory for technical assistance.
Footnotes
Published ahead of print 8 July 2013
REFERENCES
- 1.Oyston PC, Sjostedt A, Titball RW. 2004. Tularaemia: bioterrorism defence renews interest in Francisella tularensis. Nat. Rev. Microbiol. 2:967–978 [DOI] [PubMed] [Google Scholar]
- 2.Federal Register 2012. Possession, use, and transfer of select agents and toxins; biennial review. Final rule. Fed. Regist. 77:61083–61115 [PubMed] [Google Scholar]
- 3.Brandtzaeg P. 1989. Overview of the mucosal immune system. Curr. Top. Microbiol. Immunol. 146:13–25 [DOI] [PubMed] [Google Scholar]
- 4.Arulanandam BP, Raeder RH, Nedrud JG, Bucher DJ, Le J, Metzger DW. 2001. IgA immunodeficiency leads to inadequate Th cell priming and increased susceptibility to influenza virus infection. J. Immunol. 166:226–231 [DOI] [PubMed] [Google Scholar]
- 5.Rodriguez A, Tjarnlund A, Ivanji J, Singh M, Garcia I, Williams A, Marsh PD, Troye-Blomberg M, Fernandez C. 2005. Role of IgA in the defense against respiratory infections: IgA deficient mice exhibited increased susceptibility to intranasal infection with Mycobacterium bovis BCG. Vaccine 23:2565–2572 [DOI] [PubMed] [Google Scholar]
- 6.Tjarnlund A, Rodriguez A, Cardona PJ, Guirado E, Ivanyi J, Singh M, Troye-Blomberg M, Fernandez C. 2006. Polymeric IgR knockout mice are more susceptible to mycobacterial infections in the respiratory tract than wild-type mice. Int. Immunol. 18:807–816 [DOI] [PubMed] [Google Scholar]
- 7.Rodriguez A, Rottenberg M, Tjarnlund A, Fernandez C. 2006. Immunoglobulin A and CD8 T-cell mucosal immune defenses protect against intranasal infection with Chlamydia pneumoniae. Scand. J. Immunol. 63:177–183 [DOI] [PubMed] [Google Scholar]
- 8.Baron SD, Singh R, Metzger DW. 2007. Inactivated Francisella tularensis live vaccine strain protects against respiratory tularemia by intranasal vaccination in an immunoglobulin A-dependent fashion. Infect. Immun. 75:2152–2162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Murthy AK, Sharma J, Coalson JJ, Zhong G, Arulanandam BP. 2004. Chlamydia trachomatis pulmonary infection induces greater inflammatory pathology in immunoglobulin A deficient mice. Cell. Immunol. 230:56–64 [DOI] [PubMed] [Google Scholar]
- 10.Hanson LA, Bjorkander J, Carlsson B, Roberton D, Soderstrom T. 1988. The heterogeneity of IgA deficiency. J. Clin. Immunol. 8:159–162 [DOI] [PubMed] [Google Scholar]
- 11.Buckley RH. 1975. Clinical and immunologic features of selective IgA deficiency. Birth Defects Orig. Artic. Ser. 11:134–142 [PubMed] [Google Scholar]
- 12.Rawool DB, Bitsaktsis C, Li Y, Gosselin DR, Lin Y, Kurkure NV, Metzger DW, Gosselin EJ. 2008. Utilization of Fc receptors as a mucosal vaccine strategy against an intracellular bacterium, Francisella tularensis. J. Immunol. 180:5548–5557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kirimanjeswara GS, Golden JM, Bakshi CS, Metzger DW. 2007. Prophylactic and therapeutic use of antibodies for protection against respiratory infection with Francisella tularensis. J. Immunol. 179:532–539 [DOI] [PubMed] [Google Scholar]
- 14.Mara-Koosham G, Hutt JA, Lyons CR, Wu TH. 2011. Antibodies contribute to effective vaccination against respiratory infection by type A Francisella tularensis strains. Infect. Immun. 79:1770–1778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ray HJ, Cong Y, Murthy AK, Selby DM, Klose KE, Barker JR, Guentzel MN, Arulanandam BP. 2009. Oral live vaccine strain-induced protective immunity against pulmonary Francisella tularensis challenge is mediated by CD4+ T cells and antibodies, including immunoglobulin A. Clin. Vaccine Immunol. 16:444–452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cole LE, Yang Y, Elkins KL, Fernandez ET, Qureshi N, Shlomchik MJ, Herzenberg LA, Vogel SN. 2009. Antigen-specific B-1a antibodies induced by Francisella tularensis LPS provide long-term protection against F. tularensis LVS challenge. Proc. Natl. Acad. Sci. U. S. A. 106:4343–4348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kirimanjeswara GS, Olmos S, Bakshi CS, Metzger DW. 2008. Humoral and cell-mediated immunity to the intracellular pathogen Francisella tularensis. Immunol. Rev. 225:244–255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Duckett NS, Olmos S, Durrant DM, Metzger DW. 2005. Intranasal interleukin-12 treatment for protection against respiratory infection with the Francisella tularensis live vaccine strain. Infect. Immun. 73:2306–2311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen W, KuoLee R, Shen H, Conlan JW. 2004. Susceptibility of immunodeficient mice to aerosol and systemic infection with virulent strains of Francisella tularensis. Microb. Pathog. 36:311–318 [DOI] [PubMed] [Google Scholar]
- 20.Sjostedt A, North RJ, Conlan JW. 1996. The requirement of tumour necrosis factor-alpha and interferon-gamma for the expression of protective immunity to secondary murine tularaemia depends on the size of the challenge inoculum. Microbiology 142:1369–1374 [DOI] [PubMed] [Google Scholar]
- 21.Leiby DA, Fortier AH, Crawford RM, Schreiber RD, Nacy CA. 1992. In vivo modulation of the murine immune response to Francisella tularensis LVS by administration of anticytokine antibodies. Infect. Immun. 60:84–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Anthony LS, Ghadirian E, Nestel FP, Kongshavn PA. 1989. The requirement for gamma interferon in resistance of mice to experimental tularemia. Microb. Pathog. 7:421–428 [DOI] [PubMed] [Google Scholar]
- 23.Rhinehart-Jones TR, Fortier AH, Elkins KL. 1994. Transfer of immunity against lethal murine Francisella infection by specific antibody depends on host gamma interferon and T cells. Infect. Immun. 62:3129–3137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Alugupalli KR. 2008. A distinct role for B1b lymphocytes in T cell-independent immunity. Curr. Top. Microbiol. Immunol. 319:105–130 [DOI] [PubMed] [Google Scholar]
- 25.Crane DD, Griffin AJ, Wehrly TD, Bosio CM. 2013. B1a cells enhance susceptibility to infection with virulent Francisella tularensis via modulation of NK/NKT cell responses. J. Immunol. 190:2756–2766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang Y, Pacheco S, Acuna CL, Switzer KC, Wang Y, Gilmore X, Harriman GR, Mbawuike IN. 2002. Immunoglobulin A-deficient mice exhibit altered T helper 1-type immune responses but retain mucosal immunity to influenza virus. Immunology 105:286–294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Harriman GR, Bogue M, Rogers P, Finegold M, Pacheco S, Bradley A, Zhang Y, Mbawuike IN. 1999. Targeted deletion of the IgA constant region in mice leads to IgA deficiency with alterations in expression of other Ig isotypes. J. Immunol. 162:2521–2529 [PubMed] [Google Scholar]
- 28.Epstein LB, Ammann AJ. 1974. Evaluation of T lymphocyte effector function in immunodeficiency diseases: abnormality in mitogen-stimulated interferon in patients with selective IgA deficiency. J. Immunol. 112:617–626 [PubMed] [Google Scholar]
- 29.Cowan MJ, Fujiwara P, Ammann AJ. 1980. Cellular immune defect in selective IgA deficiency using a microculture method for PHA stimulation and limiting dilution. Clin. Immunol. Immunopathol. 17:595–605 [DOI] [PubMed] [Google Scholar]
- 30.Hooper LV, Gordon JI. 2001. Commensal host-bacterial relationships in the gut. Science 292:1115–1118 [DOI] [PubMed] [Google Scholar]
- 31.Ichinohe T, Pang IK, Kumamoto Y, Peaper DR, Ho JH, Murray TS, Iwasaki A. 2011. Microbiota regulates immune defense against respiratory tract influenza A virus infection. Proc. Natl. Acad. Sci. U. S. A. 108:5354–5359 [DOI] [PMC free article] [PubMed] [Google Scholar]