

Abstract
We previously demonstrated that intact, inactivated Streptococcus pneumoniae (unencapsulated strain R36A) inhibits IgG responses to a number of coimmunized soluble antigens (Ags). In this study, we investigated the mechanism of this inhibition and whether other extracellular bacteria exhibited similar effects. No inhibition was observed if R36A was given 24 h before or after immunization with soluble chicken ovalbumin (cOVA), indicating that R36A acts transiently during the initiation of the immune response. Using transgenic cOVA-specific CD4+ T cells, we observed that R36A had no significant effect on T-cell activation (24 h) or generation of regulatory T cells (day 7) and only a modest effect on T-cell proliferation (48 to 96 h) in response to cOVA. However, R36A mediated a significant reduction in the formation of Ag-specific splenic germinal center T follicular helper (GC Tfh) and GC B cells and antibody-secreting cells in the spleen and bone marrow in response to cOVA or cOVA conjugated to 4-hydroxy-3-nitrophenylacetyl hapten (NP-cOVA). Of note, the inhibitory effect of intact R36A on the IgG anti-cOVA response could be reproduced using R36A-derived cell walls. In contrast to R36A, neither inactivated, unencapsulated, intact Neisseria meningitidis nor Streptococcus agalactiae inhibited the OVA-specific IgG response. These results suggest a novel immunosuppressive property within the cell wall of Streptococcus pneumoniae.
INTRODUCTION
During the natural course of bacterial infections, the immune system is exposed to both cell-associated and soluble microbial components (1–3). Distinct differences exist in the immunologic properties between particulate and soluble antigens (Ags) (4–8), suggesting the possibility of cross-regulatory processes occurring upon their simultaneous encounter by the immune system. In addition, the expression of innate stimulating moieties (9, 10), scavenger receptor ligands (11), and virulence factors by intact pathogens may further influence the immune response to coimmunizing soluble antigens. A number of studies in mice have demonstrated inhibitory effects of infectious agents on antibody responses, including germinal center (GC) reactions to soluble protein antigens (12–16). Although the underlying mechanisms of these inhibitory effects were not fully clarified, pathogen-mediated inhibition of dendritic cell (DC) maturation and induction of regulatory T cells (Tregs) were implicated, depending upon the specific pathogen. An understanding of the interplay between intact microbes and immune responsiveness to soluble antigens may have implications on processes involving natural immunity, autoimmunity, and vaccination.
We previously demonstrated that intact, heat-killed Streptococcus pneumoniae inhibited the protein- and polysaccharide-specific IgG responses to a number of soluble conjugate vaccines, as well as soluble chicken ovalbumin (cOVA), upon coimmunization of mice intraperitoneally (i.p.) in the presence of alum plus CpG-containing oligodeoxynucleotides (CpG-ODN) as an adjuvant (17). In contrast, soluble conjugate vaccine had no effect on the IgG response to a pneumococcal protein expressed by the intact bacterium. Of note, coimmunization of a soluble conjugate with 1-μm latex beads failed to inhibit the subsequent IgG response, indicating that the inhibition did not depend solely on the particulate nature of the bacteria. These data suggested that some structural or biochemical feature of S. pneumoniae mediated this suppressive effect. The study, however, left unresolved the mechanism of this inhibition and whether other intact extracellular bacteria exhibited similar suppressive properties on antibody responses to a coimmunized soluble antigen. In this report, we determined potential changes in a number of key cellular parameters that could account for the suppression of the cOVA-specific IgG response following intravenous (i.v.) coimmunization of soluble cOVA with intact, heat-killed, unencapsulated S. pneumoniae. We further evaluated the potential effects of cOVA coimmunization with other extracellular bacteria (i.e., Neisseria meningitidis or Streptococcus agalactiae). Our results reveal a novel immunosuppressive property of Streptococcus pneumoniae expressed in its cell wall that acts transiently during the early stage of the immune response to cOVA. This early event has a marked inhibitory effect on the subsequent cOVA-specific T follicular helper (Tfh), germinal center, and plasma cell response, accounting for the reduction in serum titers of cOVA-specific IgG.
MATERIALS AND METHODS
Mice.
BALB/c mice were purchased from the National Cancer Institute (Frederick, MD). For studies using NP-cOVA, BALB/c mice were purchased from The Jackson Laboratory (Bar Harbor, ME) and subsequently bred and maintained within the Biological Resource Center at National Jewish Health (NJH, Denver, CO). Homozygous DO11.10 mice crossed with RAG-2−/− mice (BALB/c background) (hereafter referred to as “DO11.10 mice”), in which all CD4+ T cells express a transgenic (Tg) T-cell receptor (TCR) gene that codes for a cOVA peptide (amino acids 323 to 339), presented by major histocompatibility complex class IId (MHC-IId), were purchased from Taconic Farms (Hudson, NY). Mice were used between 7 and 12 weeks of age. These studies were conducted in accordance with the principles set forth in the Guide for the Care and Use of Laboratory Animals (18) and were approved by the Uniformed Services University of the Health Sciences and National Jewish Health Institutional Animal Care and Use Committees.
Reagents.
cOVA (Imject OVA) was purchased from Thermo Scientific (Rockford, IL). 4-Hydroxy-3-nitrophenylacetyl (NP)19-OVA was obtained from Biosearch Technologies (Novato, CA). Alum (Allhydrogel; 2%) was obtained from Brenntag Biosector (Denmark). A stimulatory 30-mer CpG-containing oligodeoxynucleotide (CpG-ODN) was synthesized (19), and a truncated (amino acids 1 to 470) Epstein-Barr virus (EBV) envelope glycoprotein (gp350) was expressed in Sf9 insect cells and purified (17).
Bacterial strains.
The unencapsulated mutant of D39 (S. pneumoniae, capsular type 2) (strain R36A) was obtained from David Briles (University of Alabama at Birmingham). The unencapsulated mutant of FAM18 C+ (Neisseria meningitidis, type C) (strain FAM18 C−) was obtained from Mustafa Akkoyunlu (FDA, Bethesda, MD) (20). The unencapsulated mutant of COH1 (Streptococcus agalactiae, type III) (strain COH1-13) was obtained from Craig Rubens (Children's Orthopedic Hospital, Seattle, WA) (21). Bacteria from frozen stocks were subcultured on BBL blood agar plates (VWR International, Bridgeport, NJ). Isolated colonies on blood agar were grown in Todd-Hewitt broth (BD Biosciences, San Jose, CA) (for R36A and group B Streptococcus [GBS]) or brain heart infusion medium (BD Biosciences) (for unencapsulated N. meningitidis, serogroup C [MenC]) to mid-log phase, collected, and heat killed by incubation at 65°C for 2 h. Sterility was confirmed by subculture on blood agar plates. Bacteria were then aliquoted at 1010 CFU/ml in phosphate-buffered saline (PBS) and frozen at −20°C.
Sonication of R36A.
A cell wall preparation of R36A was prepared by sonication of heat-killed R36A (109 CFU/ml) at an amplitude of 60 μm for 5 min using a MISONIX Ultrasonic Liquid Processor S-4000 (Farmingdale, NY). Sonicated R36A was then centrifuged at 15,000 rpm for 10 min to spin down cell wall fragments followed by two washings with PBS before immunization. Complete disruption of bacteria was confirmed by microscopy.
Immunizations.
Mice were immunized i.v. with 50 μg of cOVA or NP19-OVA adsorbed on 13 μg of alum, with or without CpG-ODN, in the presence or absence of 2 × 108 CFU heat-killed bacteria in PBS. All secondary immunizations were performed in a similar manner, but in the absence of bacteria. Serum samples for measurement of Ag-specific IgG titers, at different time points, were prepared from blood obtained through the tail vein. For adoptive transfer studies, 2.5 × 106 spleen cells (containing ∼5 × 105 Tg T cells) from DO11.10 mice were injected i.v. into wild-type (WT) BALB/c mice 24 h prior to immunization.
Measurement of antigen-specific serum titers by ELISA.
Immulon 4 enzyme-linked immunosorbent assay (ELISA) plates (Dynex Technologies, Chantilly, VA) were coated overnight with 5 μg/ml of cOVA or gp350 in PBS (50 μl/well) at 4°C. The plates were then blocked with PBS plus 1% bovine serum albumin (BSA) at 100 μl/well for 2 h at 37°C. Three-fold serial dilutions of serum samples, starting at a 1/50 serum dilution in PBS plus 1% BSA (50 μl/well), were then added and incubated overnight at 4°C followed by washing (3 times) with PBS plus 0.1% Tween 20. Alkaline phosphatase (AP)-conjugated polyclonal goat anti-mouse IgG antibody (Ab) (200 ng/ml, 50 μl/well) in PBS plus 1% BSA was then added, and the plates were incubated at 37°C for 1 h. Plates were then washed with PBS plus 0.1% Tween 20, and substrate (p-nitrophenyl phosphate, disodium; Sigma-Aldrich) was added at 1 mg/ml in TM buffer (1 M Tris plus 0.3 mM MgCl2 [pH 9.8]) for color development. Color was read at an absorbance of 405 nm on a Multiskan Ascent ELISA reader (Labsystems, Finland).
Enumeration of NP-specific ASC by ELISPOT.
For the enzyme-linked immunosorbent spot (ELISPOT) assay, NP-specific antibody-secreting cells (ASC) were measured in 96-well flat-bottom enzyme immunoassay/radioimmunoassay (EIA/RIA) high-binding plates (Costar, Corning, Sigma-Aldrich) coated overnight at 4°C with 2 μg/ml 4-hydroxy-3-iodo-5-nitrophenylacetyl (NIP)15-BSA diluted in 0.05 M K2HPO4 (pH 8.0). Plates were washed 3 times with PBS prior to blocking with warm PBS–1% gelatin (Sigma-Aldrich) at 37°C for a minimum of 1 h. Plates were washed again 3 times with PBS prior to incubation with cells. Single-cell suspensions of splenocytes or bone marrow (harvested 2 weeks postimmunization) were seeded in duplicate at 4 × 106 to 6 × 106 total viable cells per 100 μl in the first well, and 2-fold serial dilutions were carried out down the plate. Plates were incubated at 37°C in 5% CO2 for 5 to 6 h in RPMI medium (Cellgro, Manassas, VA) supplemented with 10% heat-inactivated fetal bovine serum (FBS) (BioSource, Grand Island, NY), 2 mM GlutaMAX-I (Invitrogen), 100 U/ml penicillin (Invitrogen, Grand Island, NY), 100 μg/ml streptomycin (Invitrogen), and 0.05 mM 2-mercaptoethanol (Sigma-Aldrich). Following culture, cells were lysed with H2O-0.05% Tween 20 for 10 min at room temperature and subsequently washed 3 times with PBS–0.1% Tween 20. Secreted antibody was detected by incubating plates with alkaline phosphatase (AP)-conjugated goat anti-mouse IgG (SouthernBiotech, Birmingham, AL) diluted in 1% gelatin in PBS for 1 h at 37°C. After 3 washes with PBS-Tween 20, plates were developed overnight at 4°C with 1 mg/ml 5-bromo-4-chloro-3-indolyl phosphate p-toluidine (BCIP; Sigma-Aldrich) salt substrate diluted in an alkaline buffer composed of 0.1 M 2-amino-2-methyl-1-propanol, 0.01% NaN3, 0.5 mM MgCl2, and 0.007% Triton X-405 (pH 10.25). Plates were washed 3 times with deionized H2O, allowed to dry in the dark at room temperature, and scanned (Epson Perfection 2450 photo scanner). Developed spots were counted visually from the scanned images, and the frequency of NP-specific ASCs per total number of cells plated was calculated.
Flow cytometric analysis.
Individual samples of red blood cell (RBC)-lysed spleen cells from 3 to 5 mice/group were stained using the following mouse-specific monoclonal antibodies (MAbs): Alexa Fluor 405–anti-CD4 (clone RM4-5) and allophycocyanin–anti-DO11.10 T-cell receptor (TCR) (clone KJ1-26) from Invitrogen; phycoerythrin (PE)–anti-CD69 (clone H1.2F3), PE–anti-CD25 (clone PC61), PE–Texas Red–anti-B220 (clone RA3-6B2), and fluorescein isothiocyanate (FITC)–anti-T- and B-cell activation antigen (clone GL7) from BD Biosciences, San Jose, CA; PE–Cy7–anti-PD1 (clone 29F.1A12) from Biolegend, San Diego, CA; and PE–Cy7–anti-CD25 (clone PC61.5) and PE–anti-Foxp3 (clone FJK-16S) from eBiosciences, San Diego, CA. Foxp3 staining was performed using the Foxp3 staining kit from eBiosciences as per the manufacturer's protocol. Cells were analyzed using a LSR-II flow cytometer (BD Biosciences), and results were generated using the software FlowJo (Tree Star, Ashland, OR) and FACSDiva (BD Biosciences). For detection of PC on bacteria, 1 × 105 CFU heat-killed bacteria were incubated overnight at 4°C with 0.25 μg of mouse IgG2aκ anti-PC MAb (clone PCG2a2.A1), which was obtained from J. Kenny (Beth Israel Deaconess Medical Center, Boston, MA), in PBS plus 1% BSA. This was followed by washing with PBS (2 times) and incubation with 1.25 μg biotin–goat anti-mouse IgG (Southern Biotech) on ice for 30 min followed by another washing with PBS (2 times) and incubation with 0.5 μg PE-streptavidin (BD Biosciences) on ice for 30 min. After 1 more wash with PBS, bacteria were analyzed by flow cytometry using a BD LSR-II flow cytometer. Bacteria incubated with only biotin–goat anti-mouse IgG and PE-streptavidin were used as a negative control. For studies to detect NP+ cells, the following MAbs were used: PE–Cy7–anti-B220 (clone RA3-6B2) and allophycocyanin– Cy7–anti-IgD (clone 11.26c.2a) (BioLegend); biotin-Igκ (clone 187.1, hybridoma), and PE-Cy5.5-CD11c (clone N418) [eBiosciences]. FITC-peanut agglutinin (PNA) (Vector Laboratories, Burlingame, CA) was used for detection of germinal center B cells, and 647-OVA (Invitrogen) was used to exclude non-NP binding B cells. For detection of NP-specific B cells, cells were stained with PE-NP40 (Biosearch Technologies). The secondary reagent for detecting biotin-conjugated antibodies was Pacific Blue-streptavidin (Invitrogen). Flow cytometric analyses were performed by acquiring data on a Cyan analyzer (Dako, Denmark) and with FlowJo software (Tree Star).
Measurement of T-cell proliferation by CFSE dilution.
RBC-lysed spleen cells from DO11.10 mice (2.5 × 107 cells) were incubated in 1 ml of 5 μM carboxyfluorescein succinimidyl ester (CFSE) (Vybrant CFDA-SE; Molecular Probes, Grand Island, NY) in PBS for 10 min at 37°C. Cells were then washed once, resuspended in prewarmed PBS, and incubated at 37°C for 30 min. Cells were then washed twice with PBS and transferred i.v. into WT BALB/c mice (2.5 × 106 spleen cells/mouse containing ∼5 × 105 DO11.10 Tg T cells). One day later, mice were immunized with cOVA plus alum with or without R36A. At 2.5 and 4 days postimmunization, spleen cells were obtained, and gated DO11.10 Tg T cells (CD4+ DO11.10 TCR+) were analyzed for CFSE dilution using an LSR-II flow cytometer (BD Biosciences) and ModFitLT software (Verity Software House, Topsham, ME).
Statistical analysis.
Serum antigen-specific IgG titers were expressed as geometric means ± standard errors of the means (SEM) of the individual serum titers. Significance was determined by two-tailed Student's t test. P values of ≤0.05 were considered statistically significant. Each experiment was performed at least twice to ascertain reproducibility.
RESULTS
The degree of R36A-mediated inhibition of the IgG response to cOVA is dependent on the nature of the adjuvant.
We previously demonstrated that an unencapsulated S. pneumoniae strain (R36A) inhibited IgG responses to a number of soluble proteins coinjected into mice i.p. in the presence of alum plus CpG-ODN adjuvant (17). CpG-ODN, a TLR9 agonist (22), significantly enhances antibody responses to soluble proteins in alum (23), thus making alum plus CpG-ODN a more effective adjuvant than alum alone. To extend these findings, we set out to determine whether R36A-mediated inhibition was influenced by the nature of the adjuvant used for the coinjected soluble protein. We immunized BALB/c mice i.v. with cOVA plus alum in the presence (Fig. 1A) or absence of CpG-ODN (Fig. 1B), with or without R36A at a dose of 2 × 108 CFU/mouse. Mice were similarly boosted on day 14 in the absence of R36A. R36A inhibited the IgG response to cOVA by 3- to 4-fold when cOVA was injected in alum plus CpG-ODN (Fig. 1A). In contrast R36A-mediated inhibition was ∼10-fold when cOVA was coinjected in alum alone (Fig. 1B). These data suggest an inverse relationship between the effectiveness of the adjuvant used for enhancing an antibody response to protein immunization and the degree of R36A-mediated inhibition of the IgG response. R36A also significantly inhibited the IgG anti-cOVA response to cOVA plus alum, at the lower dose of 2 × 107, but not 7 × 106, CFU/mouse (data not shown). R36A failed to inhibit the cOVA-specific IgG response when mice were first primed with cOVA plus alum alone, followed by secondary immunization with cOVA plus alum in the presence of R36A (data not shown), indicating that the inhibitory effect of R36A occurred only during the time of primary immunization. Of note, an R36A cell wall preparation produced by sonication followed by repeated washing and centrifugation also inhibited the IgG anti-cOVA response to cOVA plus alum (Fig. 1C), indicating that the inhibitory moiety was contained within the cell wall and that inhibition was not dependent on the particulate nature of the bacterium.
Fig 1.
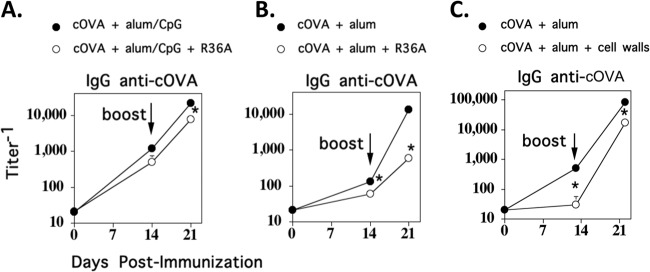
The degree of R36A-mediated inhibition of the IgG response to cOVA is dependent on the nature of the adjuvant. BALB/c mice (7 per group) were immunized i.v. with 50 μg cOVA with or without 2 × 108 CFU R36A in alum plus CpG-ODN (A), in alum alone (B), or with cell walls of R36A (C) (2 × 108 CFU equivalents/mouse). Mice were similarly boosted i.v. on day 14 in the absence of R36A or cell walls. Serum titers of cOVA-specific IgG were measured by ELISA. *, P ≤ 0.05.
Coimmunization via the subcutaneous (s.c.) route with R36A and either cOVA or truncated gp350 (an Epstein-Barr virus glycoprotein) in alum plus CpG-ODN also resulted in inhibition of the cOVA-specific and gp350-specific IgG responses, respectively (see Fig. S1 in the supplemental material). Inhibition required the injection of R36A and soluble protein at the same site, strongly suggesting that this event occurred within the draining lymph node. Thus, the inhibitory effect of R36A is mediated both within the spleen (i.v. route) and the draining lymph node (s.c. route).
R36A acts transiently within the first 24 h to cause inhibition of the cOVA-specific IgG response.
Previously, we reported that R36A was effective in mediating inhibition of an IgG antipolysaccharide response to a soluble pneumococcal conjugate vaccine in alum plus CpG-ODN when injected i.p. at the same time but not when R36A injection was delayed by 24 h (17). This indicated that the primary inhibitory event occurred during the period of initiation of the immune response. In this regard, we further wished to determine whether R36A could mediate this inhibitory effect when injected prior to cOVA immunization. We thus immunized mice with R36A 0 to 3 days prior to cOVA immunization (Fig. 2A) or 0 to 2 days following immunization (Fig. 2B). All mice were boosted with cOVA or alum alone, 14 days following primary cOVA immunization. If R36A was injected either 1 to 3 days prior to or 1 or 2 days subsequent to immunization with cOVA/alum, no inhibition of cOVA-specific IgG response was observed, in contrast to mice coimmunized with cOVA and R36A. These data demonstrated that R36A acts transiently and early during the immune response to cOVA.
Fig 2.
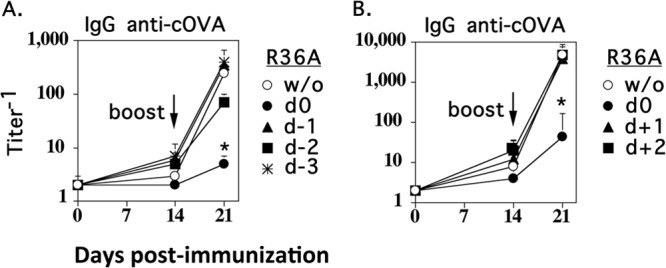
R36A acts transiently within the first 24 h to cause inhibition of the cOVA-specific IgG response. BALB/c mice (7 per group) were immunized i.v. with 50 μg cOVA in alum with or without 2 × 108 CFU R36A given 1, 2, or 3 days earlier (d−1, d−2, and d−3, respectively) than cOVA (A) or 1 or 2 days after (d+1 and d+2, respectively) cOVA immunization (B). All mice were boosted i.v. with 50 μg cOVA in alum alone 14 days after primary cOVA immunization. Serum titers of cOVA-specific IgG measured by ELISA are shown. *, P ≤ 0.05 in comparison to mice immunized with cOVA plus alum in the absence of R36A (w/o).
R36A has no apparent effect on early cOVA-specific T-cell activation and has only a modest effect on T-cell proliferation.
The cOVA-specific IgG response is dependent on CD4+ T-cell help. The observation that R36A exerted its inhibitory effect within 24 h following cOVA immunization suggested that it might be mediating an inhibitory effect on initial CD4+ T-cell priming. To determine this, we adoptively transferred cOVA-specific CD4+ Tg T cells from DO11.10 mice i.v. into BALB/c mice, followed by i.v. immunization with cOVA/alum alone or cOVA/alum plus R36A 1 day later. Splenic Tg T cells (CD4+ DO11.10 TCR+) were evaluated 24 h later by flow cytometry for expression of the T-cell activation markers CD69 and CD25 (Fig. 3A). As illustrated, R36A had no effect on the cOVA-mediated upregulation of either CD69 or CD25 on Tg T cells.
Fig 3.

R36A has no apparent effect on early cOVA-specific T-cell activation and has only a modest effect on T-cell proliferation. (A) cOVA-specific Tg T cells from DO11.10 mice were adoptively transferred into BALB/c mice and 1 day later immunized i.v. with 50 μg cOVA in alum with or without 2 × 108 CFU R36A or with PBS alone. Gated CD4+ DO11.10 TCR+ Tg T cells from spleen cell suspensions were analyzed by flow cytometry 1 day following immunization for T-cell activation markers CD69 and CD25 (3 mice/group). (B) A total of 5 × 105 cOVA-specific Tg T cells from DO11.10 mice were labeled with CFSE and adoptively transferred into BALB/c mice. One day later, mice were immunized i.v. with 50 μg cOVA in alum with or without 2 × 108 CFU R36A or with PBS alone (3 mice/group). On day 4, gated CD4+ DO11.10 TCR+ Tg T cells from spleen cell suspensions were analyzed for T-cell proliferation as reflected by CFSE dilution by flow cytometry. *, P ≤ 0.05.
To determine the proliferative response of Tg T cells, CFSE-labeled cOVA-specific Tg T cells from DO11.10 mice were adoptively transferred into BALB/c mice 1 day prior to immunization with cOVA/alum alone or cOVA/alum plus R36A. Splenic Tg T cells were analyzed by flow cytometry for CFSE dilution 2.5 and 4 days postimmunization. At day 2.5, no significant effect on the cOVA-induced Tg T-cell proliferative response was observed by coimmunized R36A (data not shown). However, by day 4 we found that R36A mediated a modest, though consistent, reduction in the percentage of Tg T cells that had undergone 9, 10, or 11 proliferative cycles (Fig. 3B). This modest R36A-mediated reduction in proliferation, however, seemed unlikely to account for the more striking inhibition of the subsequent cOVA-specific IgG response.
R36A mediates a significant reduction in the generation of GC T follicular helper cells, but has no effect on the numbers of Foxp3+ Tregs.
Regulatory T cells (Tregs) have been reported to have inhibitory effects on in vivo humoral autoimmune responses (24, 25), although we previously observed no effects of endogenous Tregs on Ig responses to intact S. pneumoniae (26). To determine if R36A affected the generation of cOVA-specific Tregs following cOVA immunization, DO11.10 Tg T cells were transferred into BALB/c mice, which were then immunized with cOVA/alum or cOVA/alum plus R36A. We then determined the numbers of Foxp3+ CD25+ Tg T cells 7 days later. As illustrated in Fig. 4A, cOVA immunization resulted in an increase in Tregs at day 7 relative to that observed in unimmunized mice. However, R36A had no significant effect on the numbers of cOVA-induced Tregs present at this time point (Fig. 4A), which is 7 days prior to the time in which mice are typically boosted with cOVA. In addition, coimmunization of cOVA-immunized mice with R36A did not consistently alter the total number of Tg T cells present on either day 7 or 8 (data not shown).
Fig 4.
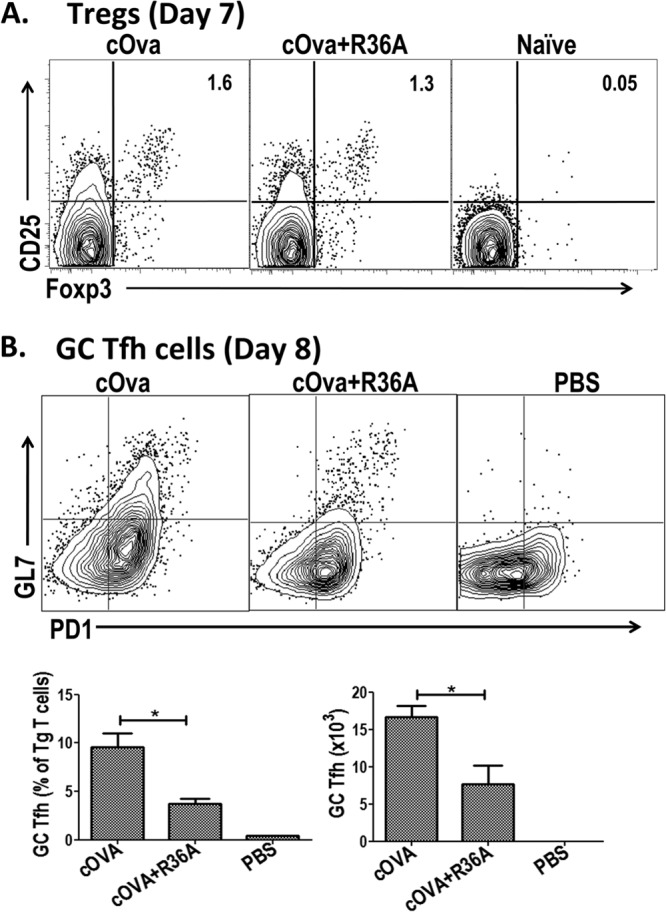
R36A mediates a significant reduction in the generation of GC T follicular helper cells but has no effect on the numbers of Foxp3+ regulatory T cells (Tregs). (A) cOVA-specific Tg T cells from DO11.10 mice were adoptively transferred into BALB/c mice and 1 day later immunized i.v. with 50 μg cOVA in alum with or without 2 × 108 CFU R36A (3 mice/group). On day 7 following immunization, gated CD4+ DO11.10 TCR+ Tg T cells from the spleen cell suspension were analyzed for Tregs (CD25+ Foxp3+). The numbers represent the mean percentage of total Tg T cells in each population. Flow cytometric dot plots are representative of one sample from each group. (B) cOVA-specific Tg T cells from DO11.10 mice were adoptively transferred into BALB/c mice and 1 day later immunized i.v. with 50 μg cOVA in alum with or without 2 × 108 CFU R36A or with PBS alone (3 to 5 mice/group). On day 8 following immunization, gated CD4+ DO11.10 TCR+ Tg T cells from spleen cell suspensions were analyzed for GC Tfh cells (GL7hi PD1hi) (top panel). Quantitation of GC Tfh data is shown in the bottom panel. *, P ≤ 0.05.
CD4+ germinal center (GC) T follicular helper (Tfh) cells play a key role in promoting T-cell-dependent (TD) antibody responses (27). These cells can be reliably identified by flow cytometry as CD4+ GL7+ PD-1hi cells, a population that is also CXCR5+ (28, 29). In this regard, we set out to determine whether R36A affected the generation of cOVA-induced GC Tfh in the adoptive transfer model using DO11.10 Tg T cells. We analyzed spleen cells 8 days postimmunization, a time point shown previously to exhibit peak numbers of GC Tfh cells (29). As illustrated in Fig. 4B and C, ∼10% of Tg T cells from mice immunized with cOVA/alum alone expressed the GC Tfh phenotype at day 8 relative to <0.5% of GC Tfh cells in mice injected with PBS. CXCR5 expression was substantially higher on CD4+ GL7+ PD-1hi Tg T cells relative to the endogenous CD4+ T cells from cOVA-immunized mice (data not shown). Of note, we observed a consistent, significant 2- to 3-fold reduction in the percentage and absolute numbers of splenic Tg T cells expressing the GC Tfh phenotype in mice immunized with cOVA/alum plus R36A relative to the levels in mice given cOVA/alum alone (Fig. 4B).
Unencapsulated variants of intact, heat-killed Streptococcus agalactiae (group B Streptococcus type III [GBS-III]) and Neisseria meningitidis (MenC) do not mediate inhibition of the cOVA-specific IgG response.
We next set out to determine whether or not the R36A-mediated inhibition of the cOVA-specific IgG response represented a more general suppressive effect of various intact bacteria on T-cell-dependent Ig responses to soluble antigens. To evaluate this, we coimmunized BALB/c mice i.v. with cOVA/alum alone or cOVA/alum plus GBS-III or MenC, followed by a boost with cOVA/alum alone on day 14. In contrast to R36A, GBS-III failed to inhibit and MenC modestly enhanced the cOVA-specific IgG response (Fig. 5A). Of note, whereas R36A induced a significant 2- to 3-fold reduction in cOVA-induced GC Tfh, 8 days postimmunization, similar to that observed in Fig. 5B, MenC had no significant effect on the generation of cOVA-specific GC Tfh (Fig. 5B).
Fig 5.
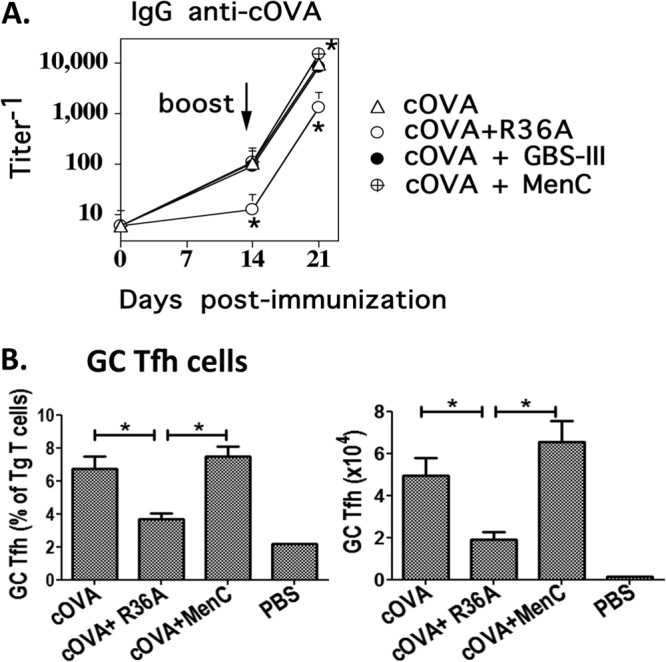
Unencapsulated variants of intact, heat-killed Streptococcus agalactiae (GBS-III) and Neisseria meningitidis (MenC) do not mediate inhibition of the cOVA-specific IgG response. (A) BALB/c mice (7 per group) were immunized i.v. with 50 μg cOVA in alum with or without 2 × 108 CFU intact heat-killed R36A, GBS-III, or MenC (all unencapsulated variants). All mice were boosted i.v. with 50 μg cOVA in alum alone on day 14. Serum titers of cOVA-specific IgG were measured by ELISA. *, P ≤ 0.05 in comparison to mice immunized with cOVA plus alum without bacteria (cOVA). (B) cOVA-specific GC Tfh cells (GL7hi PD1hi) were measured as shown in response to cOVA alone or with R36A or MenC on day 8 postimmunization (5 mice/group). *, P ≤ 0.05.
R36A, but not MenC, inhibits the generation of NP-specific GC B cells and ASC in the spleen and bone marrow when coimmunized with NP-cOVA.
The R36A-mediated reduction in the number of cOVA-induced GC Tfh cells suggested that the generation of cOVA-specific GC B cells and ASC would be correspondingly reduced. To determine this, we immunized BALB/c mice with NP-cOVA/alum alone or NP-cOVA/alum plus R36A or MenC and 14 days later measured the number of NP-specific GC B cells by flow cytometry, defined here as B220+ CD11c− NP+ IgD− PNA+. As illustrated in Fig. 6A and B, R36A, but not MenC, significantly reduced, by 2- to 3-fold, the percentage and absolute numbers of NP-specific GC B cells, as well as the absolute number of total NP+ B cells generated following NP-cOVA immunization. Consistent with these findings, the generation of NP-specific ASC in the spleen and bone marrow was reduced by R36A, but not MenC, by ∼3- to 5-fold (Fig. 6C). These data further paralleled the observed generation of cOVA-specific GC Tfh cells in the presence or absence of R36A and MenC (Fig. 5B). Collectively, these data reveal a novel immunosuppressive property of intact S. pneumoniae expressed in its cell wall that acts transiently during an early stage of the immune response to coimmunizing cOVA. This early event has a marked inhibitory effect on the subsequent cOVA-specific T follicular helper, germinal center, and plasma cell response.
Fig 6.

R36A, but not MenC, inhibits the generation of NP-specific GC B cells and ASC in the spleen and bone marrow when coimmunized with NP-cOVA. (A) BALB/c mice (3 or 4/group) were immunized i.v. with 50 μg NP-cOVA with or without 2 × 108 CFU R36A or MenC. On day 14 postimmunization, NP-specific B cells (B220+ CD11C− NP+) were analyzed for isotype-switched GC B cells (IgD−/lo PNA+) represented by the encircled populations. (B) Quantitation of NP-specific GC B cell data. (C) Quantitation of NP-specific ASC from spleen and BM in response to NP-cOVA alone or with R36A or MenC on day 14 postimmunization (3 or 4 mice/group). *, P ≤ 0.05.
DISCUSSION
During the natural course of infections with extracellular bacteria, soluble bacterial proteins are likely to be released (1–3). Little is known, however, regarding the immunologic consequences of simultaneous exposure to the immune system of the intact pathogen and these soluble antigens. We previously demonstrated that coimmunization with intact R36A resulted in a significantly reduced IgG response to a number of soluble TD antigens (17). In the present study, we investigated the mechanism of this inhibitory effect using R36A and soluble cOVA as the basis for the experimental system. We demonstrate that, although coimmunization of R36A with cOVA had no obvious effect on early cOVA-specific CD4+ T-cell activation or subsequent generation of Foxp3+ Tregs, and only a modest effect on T-cell proliferation, it led to a significant downstream reduction in the generation of cOVA-specific GC Tfh (day 8). This likely contributed to the observed decrease in GC B cells and ASC in the spleen and bone marrow (days 14). This, in turn can account, at least in large part, for the associated reduction in serum titers of cOVA-specific IgG. Of note, we show that this effect is not a general property of all intact extracellular bacteria, in that neither intact unencapsulated GBS-III nor MenC was inhibitory. Although the inhibitory moiety expressed by S. pneumoniae remains to be determined, our data indicate that it is present within the cell wall, as a cell wall preparation obtained by sonication of R36A followed by centrifugation and washing was also inhibitory. These data further indicate that inhibition does not require particulation, since sonication resulted in complete bacterial disruption.
Several other pathogens have also been reported to inhibit antibody responses to soluble TD antigens in mice, although by apparently different mechanisms. Salmonella enterica serovar Typhimurium has been reported to delay GC formation induced by haptenated proteins (15). TNP-specific IgG responses to soluble TNP-conjugated proteins are suppressed during infection with Trypanosoma cruzi, mediated by nonspecific suppressor T cells (16). Intracellular bacterial infection by Ehrlichia muris inhibited splenic, but not lymph node, NP-specific IgG responses to coadministered NP-CGG secondary to impaired generation of GC responses (12). Finally, infections with Plasmodium chabaudi (13) and foot-and-mouth disease virus (14) have each been shown to suppress OVA-specific IgG responses to soluble OVA. These suppressive effects were associated with an inhibition of DC maturation and a resultant decrease in T-cell stimulatory capacity.
A specialized class of CD4+ helper T cells, T follicular helper (Tfh) cells, has been described that initiate and maintain the GC reaction and regulate the maintenance, proliferation, and differentiation of GC B cells (27, 30, 31). GC B cells eventually give rise to long-term antibody-secreting plasma cells in the bone marrow and circulating memory B cells. Tfh cell development initiates rapidly during priming by DC and is subsequently maintained by cognate B cells by day 3 or 4 (32). The GL7 epitope defines a more differentiated subset of Tfh cells (referred to as GC Tfh cells) that localize to the GC and express higher levels of IL-4 and IL-21, key cytokines that mediate B-cell help (29). ICOS, induced on DC-primed CD4+ T cells, was shown to be a critical early signal for Tfh differentiation via upregulation of Bcl6, which then stimulated the expression of the key Tfh marker CXCR5. Of note, a bifurcation of Tfh versus effector T cells was observed as early as day 2 following a viral infection, with interleukin-2 receptor α intermediate (IL-2Rαint) cells expressing Bcl6 and CXCR5 (Tfh) and IL-2Rαhi cells expressing Blimp1, which repressed Bcl6 (effector T helper). Complete polarization was observed by 72 h even in the absence of B cells. These data indicated that the key signals initiating Tfh differentiation arise within the first 24 h during the CD4+ T-cell–DC interaction (32). Thus, the R36A-mediated reduction in cOVA-induced GC Tfh cells observed on day 8 may result from a direct or indirect effect of R36A on these early DC-mediated events affecting early Tfh differentiation. This is consistent with our data demonstrating that R36A acts to mediate this inhibition only during the first 24 h following cOVA immunization.
In addition to induction of Tfh differentiation, the generation of Foxp3+ Tregs is another event that can be controlled by DCs, early during an immune response (33), and thus could be a potential mechanism of R36A-mediated inhibition of IgG secretion in response to soluble antigens. Although Tregs are well-established negative regulators of cell-mediated immunity, their general role in humoral immunity is less clear, with few reports indicating an inhibitory effect on autoantibody production (24, 25). Certain microbial constituents that activate TLR2 on DC can induce Tregs (33). In this regard, TLR2 plays a key role in immune responses elicited by S. pneumoniae (11). Indeed, S. pneumoniae infection has been reported to suppress allergic airway disease by inducing Treg cells (34, 35), although we observed no apparent role of endogenous Tregs in regulating antibody production in response to immunization with this bacterium (26). Furthermore, in the present study we observed that although cOVA immunization induced cOVA-specific Tg Tregs, the presence of R36A had no additional effect. These data further support the notion that R36A-mediated suppression of the cOVA-specific IgG response was secondary to reduced Tfh differentiation and not enhanced generation of Treg numbers. In summary, our data strongly suggest that S. pneumoniae expresses an immunosuppressive cell wall structure that mediates inhibition of T follicular helper and associated GC and plasma cell responses to coimmunizing soluble antigens. The identity of this structure, the context in which it mediates suppression, and its cellular targets remain to be determined.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by N.I.H. R01-AI49192 (C.M.S.), R01-AI052157 (R.M.T.), and the USUHS Dean's Research and Education Endowment Fund (C.M.S.).
We also acknowledge Kateryna Lund and Karen M. Wolcott (Biomedical Instrumentation Center, USUHS) for technical assistance with flow cytometry.
Footnotes
Published ahead of print 1 July 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.00688-13.
REFERENCES
- 1.Rigden DJ, Galperin MY, Jedrzejas MJ. 2003. Analysis of structure and function of putative surface-exposed proteins encoded in the Streptococcus pneumoniae genome: a bioinformatics-based approach to vaccine and drug design. Crit. Rev. Biochem. Mol. Biol. 38:143–168 [DOI] [PubMed] [Google Scholar]
- 2.Jedrzejas MJ. 2004. Extracellular virulence factors of Streptococcus pneumoniae. Front. Biosci. 9:891–914 [DOI] [PubMed] [Google Scholar]
- 3.Lopez R, Garcia JL, Garcia E, Ronda C, Garcia P. 1992. Structural analysis and biological significance of the cell wall lytic enzymes of Streptococcus pneumoniae and its bacteriophage. FEMS Microbiol. Lett. 79:439–447 [DOI] [PubMed] [Google Scholar]
- 4.Kovacsovics-Bankowski M, Clark K, Benacerraf B, Rock KL. 1993. Efficient major histocompatibility complex class I presentation of exogenous antigen upon phagocytosis by macrophages. Proc. Natl. Acad. Sci. U. S. A. 90:4942–4946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guermonprez P, Valladeau J, Zitvogel L, Thery C, Amigorena S. 2002. Antigen presentation and T cell stimulation by dendritic cells. Annu. Rev. Immunol. 20:621–667 [DOI] [PubMed] [Google Scholar]
- 6.Ziegler HK, Orlin CA, Cluff CW. 1987. Differential requirements for the processing and presentation of soluble and particulate bacterial antigens by macrophages. Eur. J. Immunol. 17:1287–1296 [DOI] [PubMed] [Google Scholar]
- 7.Vidard L, Kovacsovics-Bankowski M, Kraeft SK, Chen LB, Benacerraf B, Rock KL. 1996. Analysis of MHC class II presentation of particulate antigens of B lymphocytes. J. Immunol. 156:2809–2818 [PubMed] [Google Scholar]
- 8.Nayak JV, Hokey DA, Larregina A, He Y, Salter RD, Watkins SC, Falo LD., Jr 2006. Phagocytosis induces lysosome remodeling and regulated presentation of particulate antigens by activated dendritic cells. J. Immunol. 177:8493–8503 [DOI] [PubMed] [Google Scholar]
- 9.Kawai T, Akira S. 2011. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity 34:637–650 [DOI] [PubMed] [Google Scholar]
- 10.Franchi L, Munoz-Planillo R, Reimer T, Eigenbrod T, Nunez G. 2010. Inflammasomes as microbial sensors. Eur. J. Immunol. 40:611–615 [DOI] [PubMed] [Google Scholar]
- 11.Paterson GK, Orihuela CJ. 2010. Pneumococci: immunology of the innate host response. Respirology 15:1057–1063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Racine R, Jones DD, Chatterjee M, McLaughlin M, Macnamara KC, Winslow GM. 2010. Impaired germinal center responses and suppression of local IgG production during intracellular bacterial infection. J. Immunol. 184:5085–5093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Millington OR, Di Lorenzo C, Phillips RS, Garside P, Brewer JM. 2006. Suppression of adaptive immunity to heterologous antigens during Plasmodium infection through hemozoin-induced failure of dendritic cell function. J. Biol. 5:5. 10.1186/jbiol34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ostrowski M, Vermeulen M, Zabal O, Geffner JR, Sadir AM, Lopez OJ. 2005. Impairment of thymus-dependent responses by murine dendritic cells infected with foot-and-mouth disease virus. J. Immunol. 175:3971–3979 [DOI] [PubMed] [Google Scholar]
- 15.Cunningham AF, Gaspal F, Serre K, Mohr E, Henderson IR, Scott-Tucker A, Kenny SM, Khan M, Toellner KM, Lane PJ, Maclennan IC. 2007. Salmonella induces a switched antibody response without germinal centers that impedes the extracellular spread of infection. J. Immunol. 178:6200–6207 [DOI] [PubMed] [Google Scholar]
- 16.Reed SG, Roters SB, Goidl EA. 1983. Spleen cell-mediated suppression of IgG production to a non-parasite antigen during chronic Trypanosoma cruzi infection in mice. J. Immunol. 131:1978–1982 [PubMed] [Google Scholar]
- 17.Chattopadhyay G, Chen Q, Colino J, Lees A, Snapper CM. 2009. Intact bacteria inhibit the induction of humoral immune responses to bacterial-derived and heterologous soluble T cell-dependent antigens. J. Immunol. 182:2011–2019 [DOI] [PubMed] [Google Scholar]
- 18.National Research Council 1996. Guide for the care and use of laboratory animals. National Academies Press, Washington, DC [Google Scholar]
- 19.Sen G, Flora M, Chattopadhyay G, Klinman DM, Lees A, Mond JJ, Snapper CM. 2004. The critical DNA flanking sequences of a CpG oligodeoxynucleotide, but not the 6 base CpG motif, can be replaced with RNA without quantitative or qualitative changes in Toll-like receptor 9-mediated activity. Cell. Immunol. 232:64–74 [DOI] [PubMed] [Google Scholar]
- 20.Kocabas C, Katsenelson N, Kanswal S, Kennedy MN, Cui X, Blake MS, Segal DM, Akkoyunlu M. 2007. Neisseria meningitidis type C capsular polysaccharide inhibits lipooligosaccharide-induced cell activation by binding to CD14. Cell. Microbiol. 9:1297–1310 [DOI] [PubMed] [Google Scholar]
- 21.Wessels MR, Haft RF, Heggen LM, Rubens CE. 1992. Identification of a genetic locus essential for capsule sialylation in type III group B streptococci. Infect. Immun. 60:392–400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hemmi H, Takeuchi O, Kawai T, Kaisho T, Sato S, Sanjo H, Matsumoto M, Hoshino K, Wagner H, Takeda K, Akira S. 2000. A Toll-like receptor recognizes bacterial DNA. Nature 408:740–745 [DOI] [PubMed] [Google Scholar]
- 23.Cooper CL, Davis HL, Morris ML, Efler SM, Adhami MA, Krieg AM, Cameron DW, Heathcote J. 2004. CPG 7909, an immunostimulatory TLR9 agonist oligodeoxynucleotide, as adjuvant to Engerix-B HBV vaccine in healthy adults: a double-blind phase I/II study. J. Clin. Immunol. 24:693–701 [DOI] [PubMed] [Google Scholar]
- 24.Iikuni N, Lourenco EV, Hahn BH, La Cava A. 2009. Cutting edge: regulatory T cells directly suppress B cells in systemic lupus erythematosus. J. Immunol. 183:1518–1522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jang E, Cho WS, Cho ML, Park HJ, Oh HJ, Kang SM, Paik DJ, Youn J. 2011. Foxp3+ regulatory T cells control humoral autoimmunity by suppressing the development of long-lived plasma cells. J. Immunol. 186:1546–1553 [DOI] [PubMed] [Google Scholar]
- 26.Lee KS, Sen G, Snapper CM. 2005. Endogenous CD4+ CD25+ regulatory T cells play no apparent role in the acute humoral response to intact Streptococcus pneumoniae. Infect. Immun. 73:4427–4431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Crotty S. 2011. Follicular helper CD4 T cells (TFH). Annu. Rev. Immunol. 29:621–663 [DOI] [PubMed] [Google Scholar]
- 28.Kerfoot SM, Yaari G, Patel JR, Johnson KL, Gonzalez DG, Kleinstein SH, Haberman AM. 2011. Germinal center B cell and T follicular helper cell development initiates in the interfollicular zone. Immunity 34:947–960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yusuf I, Kageyama R, Monticelli L, Johnston RJ, Ditoro D, Hansen K, Barnett B, Crotty S. 2010. Germinal center T follicular helper cell IL-4 production is dependent on signaling lymphocytic activation molecule receptor (CD150). J. Immunol. 185:190–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McHeyzer-Williams LJ, Pelletier N, Mark L, Fazilleau N, McHeyzer-Williams MG. 2009. Follicular helper T cells as cognate regulators of B cell immunity. Curr. Opin. Immunol. 21:266–273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Deenick EK, Ma CS, Brink R, Tangye SG. 2011. Regulation of T follicular helper cell formation and function by antigen presenting cells. Curr. Opin. Immunol. 23:111–118 [DOI] [PubMed] [Google Scholar]
- 32.Choi YS, Kageyama R, Eto D, Escobar TC, Johnston RJ, Monticelli L, Lao C, Crotty S. 2011. ICOS receptor instructs T follicular helper cell versus effector cell differentiation via induction of the transcriptional repressor Bcl6. Immunity 34:932–946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Manicassamy S, Pulendran B. 2011. Dendritic cell control of tolerogenic responses. Immunol. Rev. 241:206–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Preston J, Thorburn A, Starkey M, Beckett E, Horvat J, Wade M, O'Sullivan B, Thomas R, Beagley K, Gibson P. 2011. Streptococcus pneumoniae infection suppresses allergic airways disease by inducing regulatory T-cells. Eur. Respir. J. 37:53–64 [DOI] [PubMed] [Google Scholar]
- 35.Thorburn AN, Foster PS, Gibson PG, Hansbro PM. 2012. Components of Streptococcus pneumoniae suppress allergic airways disease and NKT cells by inducing regulatory T cells. J. Immunol. 188:4611–4620 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.