

Abstract
Legionella pneumophila uses the Icm/Dot type 4B secretion system (T4BSS) to deliver translocated protein substrates to the host cell, promoting replication vacuole formation. The conformational state of the translocated substrates within the bacterial cell is unknown, so we sought to determine if folded substrates could be translocated via this system. Fusions of L. pneumophila Icm/Dot-translocated substrates (IDTS) to dihydrofolate reductase (DHFR) or ubiquitin (Ub), small proteins known to fold rapidly, resulted in proteins with low translocation efficiencies. The folded moieties did not cause increased aggregation of the IDTS and did not impede interaction with the adaptor protein complex IcmS/IcmW, which is thought to form a soluble complex that promotes translocation. The translocation defect was alleviated with a Ub moiety harboring mutations known to destabilize its structure, indicating that unfolded proteins are preferred substrates. Real-time analysis of translocation, following movement during the first 30 min after bacterial contact with host cells, revealed that the folded moiety caused a kinetic defect in IDTS translocation. Expression of an IDTS fused to a folded moiety interfered with the translocation of other IDTS, consistent with it causing a blockage of the translocation channel. Furthermore, the folded protein fusions also interfered with intracellular growth, consistent with inefficient or impaired translocation of proteins critical for L. pneumophila intracellular growth. These studies indicate that substrates of the Icm/Dot T4SS are translocated to the host cytosol in an unfolded conformation and that folded proteins are stalled within the translocation channel, impairing the function of the secretion system.
INTRODUCTION
Legionella pneumophila is a pathogen of amoebae that grows in an intravacuolar niche in these cells (1–4). Upon aerosolization of contaminated water supplies, inhalation of L. pneumophila and subsequent internalization by alveolar macrophages lead to disease (2, 5, 6). The bacterium replicates within a membrane-bound vacuole in macrophages that is identical to the replication compartment found in amoebae (7). Formation of the Legionella-containing vacuole (LCV) requires a functional type IV secretion system (T4SS) known as the Icm/Dot complex (8, 9), which transports a massive collection of effectors into the host cytosol. To date, over 275 of these protein substrates have been identified (10–19). Shortly after internalization of the bacteria into either macrophages or amoebae, mitochondria and markers of the endoplasmic reticulum are recruited to the LCV to establish a vacuole that allows replication before the cell lyses, releasing bacteria into the extracellular milieu for a second round of infection (20–22).
Type IV secretion systems (T4SS) are utilized by a wide variety of Gram-negative and Gram-positive bacteria to translocate effector molecules across the bacterial membrane. This diverse group of secretion complexes is defined by ancestral relatedness to bacterial conjugation machines that are able to translocate nucleic acids, proteins, and nucleoprotein complexes (23–26). T4SS can be broadly classified into two subgroups based on similarity to the archetypal Agrobacterium tumefaciens VirB/VirD4 (VirB/D4) complex (24). Type IVA systems (T4ASS) share significant similarity to the VirB/D4 system and can be found in pathogens such as Helicobacter pylori and Brucella abortus (24, 25). The type IVB systems (T4BSS) are distinct from the VirB/D4 system and are best represented by Legionella species and Coxiella burnetii (24, 25, 27, 28). Although sequence similarities may be limited between the T4ASS and T4BSS members, there is evidence for functional conservation between these two subclasses as both appear to have core protein components that form a complex spanning the inner and outer membranes of Gram-negative organisms (29–31). In addition, each has a coupling ATPase complex, which is thought to act as a receptor for translocated substrates, transferring them to other components of the secretion system as a first step in transit to target cells (32).
In L. pneumophila, the earliest steps in the translocation process involve interactions between the Icm/Dot-translocated substrate (IDTS) and the IcmS/IcmW chaperone (33, 34), which is a soluble heteromeric component of the (IcmS/W) apparatus. As IcmS/W associates with the coupling ATPase complex DotL/DotM/DotN, it is likely that this five-member multiprotein complex presents translocated substrates to core secretion system components that span the bacterial envelope. Inactivating mutations in IcmS or IcmW result in highly reduced efficiencies of translocation of most IDTSs although a subset appears to translocate into host cells independently of this chaperone. It is likely that structural determinants control the dependence on IcmS/W for translocation of a protein and that IDTSs are capable of interacting directly with the secretion system in the absence of the chaperone. The critical components of the translocated substrates that allow Icm/Dot recognition are still somewhat obscure although it is clear that each IDTS contains a carboxyl-terminal signal of less than 30 amino acids that targets proteins into the secretion complex (13, 35). Both short polar amino acids and a glutamate-rich region appear to be common sequence determinants for this signal (13, 19, 35). The proteins that recognize this signal and target the protein to the complex are not known.
In many secretion systems, there are determinants other than the recognition signal that are required for successful movement through the membrane. In Escherichia coli, chimeric proteins that fused secretion signal sequences onto normally cytoplasmic proteins resulted in “jamming” of the secretion apparatus, presumably by entry of an impassable substrate into the secretion channel (36). Similarly, translocation of proteins from the cytosol into mitochondria, chloroplasts, and lysosomes can all be blocked by introduction of tightly folded protein moieties into coding sequences, consistent with there being a secretion-competent conformation required for translocation (37–39). The necessity of a secretion-competent conformation, however, does not appear evident in many systems, such as the bacterial twin-arginine translocation (Tat) pathway and the eukaryotic peroxisomal import pathway, in which the insertion of folded domains has no effect on translocation (40–42).
Specialized bacterial secretion systems that deliver proteins to target cells provide a very special problem for maintaining a secretion-competent conformation. The translocated substrates of these systems are often synthesized and stored prior to contact of bacteria with their secretion targets, and a large bolus of protein must be injected via these systems quickly. In spite of these issues, there is evidence that a tightly folded moiety interferes with translocation of type III secretion substrates, either because it is rejected by the complex or jams the apparatus (43, 44). There is also some evidence that in the case of some proteins, the secretion apparatus itself can unfold a substrate, converting a nontranslocatable form into a secretion-competent conformation (45, 46). In contrast, little is known regarding the tertiary structure of T4SS substrates, and structural analysis of the T4ASS core complex does not necessarily demand that substrates be unfolded prior to entering into the channel (30, 47). In this study, we investigate whether the L. pneumophila Icm/Dot system can tolerate substrates containing domains known to poison secretion by other translocation apparatuses. The results obtained are consistent with the presence of conformational states that interfere with efficient translocation.
MATERIALS AND METHODS
Bacterial strains, plasmids, and cell culture.
All bacterial strains and plasmids used in this work are listed in Table 1. All PCR primers used in this work are available from the authors. Growth medium for L. pneumophila was as described previously (8, 48). For L. pneumophila, chloramphenicol was used at a concentration of 20 μg/ml. For E. coli strains, antibiotics were used at the following concentrations: kanamycin, 40 μg/ml; ampicillin, 100 μg/ml; and chloramphenicol, 30 μg/ml.
Table 1.
Strains and plasmids used in this study
| Strain or plasmid | Genotype or relevant characteristic(s)a | Oligonucleotide(s) | Reference or source |
|---|---|---|---|
| Strains | |||
| L. pneumophila | |||
| Lp01 | Philadelphia-1 rpsL hsdR | 8 | |
| Lp02 | Philadelphia-1 rpsL hsdR thyA mutant | 8 | |
| Lp03 | Lp02 dotA03 | 8 | |
| Lp02 ΔicmS | 62 | ||
| E. coli | |||
| DH5α | λ− ϕ80dlacZΔM15 Δ(lacZYA-argF)U169 recA1 endA1 hsdR17(rK− mK−) supE44 thi-1 gyrA relA1 | ||
| Plasmids | |||
| pJB908 | pMMB66EH oriRSF1010 oriT bla tdΔi | 66 | |
| pJB2581 | cyaA in pJB908-tdΔi, Cmr | 49 | |
| pCyaA-DHFR | pJB2581 dhfr+ | 35, 36 | This study |
| pCyaA-MavU | pJB2581 mavU | 63, 58 | This study |
| pCyaA-DHFR-MavU | pCyaA dhfr mavU | 63, 64, 65, 58 | This study |
| pCyaA-MavU(C50) | pJB2581 mavU(348-end) | 44, 45 | This study |
| pCyaA-DHFR-MavU(C50) | pCyaA dhfr mavU(348-end) | 44, 45 | This study |
| pCyaA-MavU(C186) | pJB2581 mavU(212-end) | This study | |
| pCyaA-DHFR-MavU(C186) | pCyaA dhfr mavU(212-end) | 63, 58, 71, 72 | This study |
| pCyaA-MavU(C118) | pJB2581 mavU(280-end) | This study | |
| pCyaA-DHFR-MavU(C118) | pCyaA dhfr mavU(280-end) | 63, 58, 73, 74 | This study |
| pCyaA-MavU(C78) | pJB2581 mavU(320-end) | This study | |
| pCyaA-DHFR-Mavu(C78) | pCyaA dhfr mavU(320-end) | 64, 58, 75, 76 | This study |
| pMavU | pJB908 mavU | 99, 58 | This study |
| pCyaA-SidG | pJB2581 sidG | 60, 54b | This study |
| pCyaA-DHFR-SidG | pCyaA dhfr sidG | 60, 54b | This study |
| pJB-CyaA-RalF | pJB2581 ralF | 66, 67 | This study |
| pCyaA-DHFR-RalF | pCyaA dhfr ralF | 66, 67 | This study |
| pCyaA-LidA | pJB2581 lidA | 25, 31 | This study |
| pCyaA-DHFR-LidA | pCyaA dhfr lidA | 25, 31, 17, 18, 20, 21 | This study |
| pCyaA-SidC | pJB2581 sidC | 7, 10 | This study |
| pCyaA-DHFR-SidC | pCyaA dhfr sidC | 7, 10 | This study |
| pCyaA-VIpD | pJB2581 vipD | 22, 24 | This study |
| pCyaA-DHFR-VipD | pCyaA dhfr vipD | 22, 24 | This study |
| pCyaA-Ub | pJB2581 ubi4p | 93, 95 | This study |
| pCyaA-Ub(G3,13) | pJB2581 ubi4p(G3,13) | 94, 95 | This study |
| pCyaA-Ub-RalF | pJB2581 ubi4p ralF | 93, 95, 66, 67 | This study |
| pCyaA-Ub(G3,13)-RalF | pJB2581 ubi4p(G3,13) ralF | 94, 95, 66, 67 | This study |
| pCyaA-Ub(V76)-RalF | pJB2581 ubi4p(V76) ralF | 108, 95 | This study |
| pFLAG-SidG | pJB908 FLAG-sidG | 104b, 43 | This study |
| pFLAG-SidG(Cterm) | pJB908 FLAG-sidG(936-end) | 43 | This study |
| pFLAG-MavU | pJB908 FLAG-mavU | 110 | This study |
| pFLAG-RalF | pJB908 FLAG-ralF | 109 | This study |
| pJB5809 | pGST-IcmS/W | J. Vogel | |
| pGST | pJB908 GST | This study | |
| pSR47s | oriTRP4 oriPR6K sacB Kanr | 14 |
For the plasmids, amino acid positions or substitutions are given in parentheses with the gene designations.
L. pneumophila strain Lp02 (thyA-hsdR rpsL) (49), a thymine (Thy)-auxotrophic derivative of the Philadelphia-1 isolate, was used as the wild type in experiments. Lp03 (Lp02 dotA03) was used as a translocation-deficient control where indicated (49). The icmS deletion mutant (Lp02 ΔicmS) was generated as previously described (50).
All CyaA fusions were constructed by cloning IDTS open reading frames (ORFs) into pJB2581 (51). Dihydrofolate reductase (DHFR) fusions were constructed by inserting the coding sequence of Mus musculus dhfr (GenBank NM_010049.3) into pJB2581, in between the cyaA and IDTS sequences unless otherwise indicated. Fusions were generated using splicing by overlap extension PCR (SOE-PCR). Ubiquitin fusions were constructed similarly to those with DHFR, using the 67-amino-acid monomer of ubi4p from Saccharomyces cerevisiae S288c (NM_001181859.1). FLAG-tagged fusions were constructed by adding FLAG sequence (ATG-GATTACAAGGACGACGATGACAAG) to the amino terminus of IDTS ORFs by PCR and cloning into pJB908. The glutathione S-transferase (GST)-IcmS/IcmW plasmid (pJB5809) was a generous gift of J. Vogel, Washington University, St. Louis, MO.
The U937 cell line (ATCC CRL-1593.2) was passaged and differentiated as described previously (8). HEK293T cells (ATCC CRL-11268) were grown in Dulbecco's modified Eagle's medium (DMEM) containing 4.5g/liter d-glucose (catalog number 11995; Invitrogen-Gibco) supplemented with 10% heat-inactivated HyClone fetal bovine serum (FBS; Thermo Scientific). Mouse bone marrow-derived macrophages (BMDM) were prepared from the femurs of female A/J mice (Jackson Laboratories) according to a previously published protocol (52) and cultivated in RPMI 1640 medium supplemented with glutamate (Invitrogen-Gibco) and 10% heat-inactivated FBS (Gibco). All manipulations with animals were approved by the Tufts University IACUC, and the research approach was approved the Tufts University Institutional Biosafety Review Board.
CyaA translocation assay.
Translocation assays using CyaA protein fusions were adapted from previous protocols (51). Unless otherwise indicated, L. pneumophila was grown overnight in AYE [N-(2-acetamido)-2-aminoethanesulfonic acid-buffered yeast extract] broth with appropriate additives to exponential phase (A600 of ∼2.2 to 2.4, with less than 10% of the bacteria motile). CyaA protein expression was induced with 200 μM isopropyl-β-d-thiogalactopyranoside (IPTG) and incubated for an additional 3 to 5 h until late exponential phase (A600 ∼3.2 to 3.6 with more than 50% motility). A total of 2.5 × 106 phorbol ester-differentiated U937 cells were incubated in 24-well plates and challenged with L. pneumophila strains at a multiplicity of infection (MOI) of 1. After a 1-h incubation with bacteria (unless otherwise indicated), U937 cells were washed three times with warm phosphate-buffered saline (PBS), and extracts were prepared by the addition of 200 μl of lysis buffer (50 mM HCl, 0.1% Triton X-100) and incubation on ice for 10 min. Lysates were collected, boiled for 5 min, and then neutralized by the addition of 12 μl of 0.5 M NaOH. Protein was precipitated by adding 400 μl of cold 95% ethanol (EtOH) and incubated at −20°C for at least 5 min. Insoluble material was removed by centrifugation for 5 min at 13,000 rpm. Supernatants were dried and resuspended in assay buffer, and the cyclic AMP (cAMP) concentration was measured in extracts diluted 1:500 using a GE Healthcare Life Sciences Biotrak cAMP enzyme-linked immunosorbent assay (ELISA) kit (RPN225). Data were normalized to the amount of the housekeeping protein isocitrate dehydrogenase (ICDH) or the Icm/Dot system protein DotF to determine bacterial load, and data were expressed as a ratio of the cyclase reaction rate relative to the steady-state levels of CyaA reporter fusions in each well, as determined by Western blotting of bacterial lysates. Data are presented as fmol/well/[fusion protein].
Antibodies used in this work.
For detection of cyclase fusions by Western blotting probing, anti-CyaA (3D1) mouse monoclonal IgG (Santa Cruz) was used at a concentration of 1:15,000 diluted in 5% milk with a goat anti-mouse IgG conjugated to horseradish peroxidase (HRP) secondary antibody used at a concentration of 1:10,000. For detection using the FLA-9000 developer, anti-CyaA was used at a concentration of 1:5,000 diluted in NAP (non-animal protein) Blocker (GBiosciences) with a Cy5-goat anti-mouse IgG at a concentration of 1:10,000 or Cy3-goat anti-mouse IgG at a concentration of 1:5,000 (Invitrogen). Serum specific for Bacillus subtilis isocitrate dehydrogenase (ICDH) was generously provided by A. L. Sonenshein, Tufts University Medical School, Boston, MA.
Real-time assay of translocation rate.
A GloSensor cAMP assay (TM076; Promega) was adapted for the real-time analysis of translocation using L. pneumophila harboring CyaA fusions. Two days prior to infection, 293T cells were resuspended at a density of 1.5 × 105 cells/ml in a solid white 96-well plate (Costar 3917) and incubated overnight. The next day, cells were transfected with pGloSensor20F cAMP plasmid (E1171; Promega) at a concentration of 12.5 ng/μl, as well as with a plasmid expressing FcγRIIA at a concentration of 18.75 ng/μl, using FuGENE HD, according to the manufacturer's instructions. After 24 h of incubation, the medium was carefully removed from the 293T cells and replaced with 200 μl of GloSensor cAMP reagent (E1290; Promega) equilibration medium (88% CO2-independent medium [Invitrogen 18045], 10% FBS, 2 to 3% [vol/vol] freshly thawed cAMP reagent) supplemented with 100 μg/ml thymidine and 200 μM IPTG, as necessary. The cells were incubated for 1 h further at 37°C prior to challenge at an MOI of 10 with bacteria that were diluted in opsonizing medium (CO2-independent medium, 10% FBS, 100 μg/ml Thy, 200 μM IPTG, and anti-Legionella antibody [1:1,000]). The wells containing the opsonized bacteria were subjected to 5 min of centrifugation at 1,000 rpm and immediately placed in a luminescence plate reader preheated to 37°C. Luminescence measurements were taken every 2 min for up to 1 h. As a positive control, forskolin, an activator of adenylyl cyclase, was added at a concentration of 10 μM. Data were recorded as relative light units, after the subtraction of background from wells that were not incubated with the cAMP reagent.
Determination of DHFR enzymatic activity.
DHFR activity of the CyaA-IDTS fusions was measured with a colorimetric assay based on the NADPH-dependent reduction of dihydrofolic acid to tetrahydrofolic acid. For measurement of DHFR activity, 100-ml overnight cultures of L. pneumophila expressing IDTS fusions to murine DHFR were pelleted, resuspended in 0.5 ml of ice-cold lysis buffer (50 mM Tris, pH 7.5, 1 mM EDTA, 50 μg/ml DNase, 200 μg/ml lysozyme, protease inhibitor cocktail [Roche]), and incubated on ice for 30 min. Samples were lysed by three freeze-thaw cycles, alternating 1- to 2-min incubations in liquid nitrogen, and a 37°C water bath. Cell debris was cleared by centrifugation at 15,000 × g at 4°C for 30 min. Total protein in the extracts was quantified using a Bradford protein assay (Bio-Rad) according to standard protocols.
For each reaction, 100 μl of protein extracts (0.5 to 1.0 mg of total protein) was incubated in assay buffer (50 mM Tris-HCl, pH 7.5, 500 mM NaCl, 5 mM β-mercaptoethanol) containing 100 μM β-2′-NADPH (Sigma) and 100 μM dihydrofolic acid (Sigma). In each reaction mixture, 100 μM trimethoprim (TMP) or 100 μM methotrexate (MTX) was added. The substrate (dihydrofolic acid) was then added, the reaction solution was mixed by inverting the cuvette several times; the A340 was measured immediately and every 15 s thereafter for 3 min. Purified mammalian DHFR (1.5 × 10−3 units) was used as a positive control. Specific activity in μmol/min/mg of protein is calculated by the following formula: units/mg protein = [(ΔOD/minsample) − (ΔOD/minblank)]/ε(mM−1 cm−1)V (mg protein/ml), where ε is 12.3, V is the volume of enzyme (ml), and mg protein/ml is the enzyme concentration of the original sample. To normalize specific activity to the DHFR present in the fusions, steady-state levels of DHFR were determined by Western blotting to obtain protein amounts. Quantification by densitometry compared to a standard curve made from serial dilutions of pure DHFR on the same immunoblot was used to determine protein concentrations.
Cyclase assay on bacterial cultures.
A total of 1 × 108 bacteria of each culture were pelleted at 8,000 rpm for 5 min, resuspended in 190 μl of lysis buffer (60 mM Tris [pH 8], 10 mM MgCl2, 50 μM CaCl2, 50 μg/ml DNase, 200 μg/ml lysozyme, protease inhibitor cocktail [Roche]) and incubated on ice for 45 min. Samples were lysed by three freeze-thaw cycles, alternating 1 to 2 min incubations in liquid nitrogen, and a 37°C water bath. In both lysed and unlysed samples, cell debris was cleared by centrifugation at 15,000 × g at 4°C for 5 min. For the reaction mixture, 100 μl of supernatant was mixed with 1 mM ATP in 50 mM Tris and 50 μM CaCl2; 10 μM calmodulin (CaM) was added to each tube, and reaction mixtures were incubated at 37°C for 5 to 30 min before reactions were stopped with 2× stop buffer (100 μM HCl–0.2% Triton X-100) and boiling for 5 min, followed by neutralization with 12 μl of 0.5 M NaOH. Protein was precipitated with 400 μl of ice-cold 95% EtOH and incubated at −20°C for at least 5 min. Extracts were dried under vacuum and mixed with assay buffer, and cAMP concentration was measured as for all cyclase assays. Data were normalized to the expression of CyaA fusions as determined by steady-state levels of lysates on a Western blot and displayed as fmol/[fusion protein].
Quantification of protein aggregation.
Overnight cultures of L. pneumophila harboring fusion proteins were grown as for cyclase assays. A total of 2.5 × 109 bacteria were pelleted at 10,000 × g for 5 min, resuspended in 1 ml of lysis buffer (50 mM Tris, pH 7.5, 10 mM EDTA [pH 8.0], 50 μg/ml DNase, 200 μg/ml lysozyme, and protease inhibitor cocktail [Roche]), and incubated on ice for 30 min. Samples were lysed as described for bacterial cyclase assays. Cellular debris was separated by a slow centrifugation at 3,000 × g for 1 min. The pellets were resuspended in 500 μl 2× sample buffer and saved. Total protein samples were obtained by mixing 80 μl of supernatant with 20 μl of 5× sample buffer. To precipitate proteins, 800 μl of supernatant was subjected to centrifugation at ∼100,000 × g for 1 h. Pellets (aggregated proteins) were resuspended in 400 μl of 2× sample buffer, and 160 μl of supernatant was mixed with 40 μl of 5× sample buffer. All samples were boiled, and the equivalent of 2.5 × 107 cells for each sample was separated by SDS-PAGE. CyaA-IDTS protein in each sample was detected using antibody against CyaA and quantified by densitometry. Percent aggregation was expressed as the amount of protein in the pellet fractions relative to the total protein.
Copurification of the IDTS and IcmS/W complex.
E. coli DH5α was transformed with the plasmid pJB5809, encoding GST-IcmS-IcmW (J. Vogel, Washington University, St. Louis, MO), and a second plasmid that encodes a FLAG-tagged IDTS. A plasmid expressing GST without IcmS/IcmW was used as a control. Overnight cultures (500 ml) were grown in LB medium supplemented with kanamycin and carbenicillin. Protein expression was induced by the addition of 0.5 M IPTG, and cells were incubated at 30°C for 3 h with slow shaking. Cultures were harvested by centrifugation at 10,000 × g for 15 min at 4°C, and pellets were resuspended in 10 ml of PBS (pH 7.4), 5 mM MgCl2, 50 μg/ml DNase, 1 mM dithiothreitol (DTT), and protease inhibitor cocktail (Roche), followed by lysing with a French press. Lysates were clarified by centrifugation at 15,000 × g for 15 min at 4°C, and cleared lysates were incubated with 0.5 ml of glutathione beads for 2 h at 4°C. Columns were washed three times with cold wash buffer (PBS [pH 7.4], 5 mM MgCl2, 1 mM DTT, and protease inhibitor cocktail [Roche]). Bound GST-IcmS/IcmW and associated proteins were eluted with four 0.75-ml aliquots of 10 mM reduced glutathione in 50 mM Tris (pH 8.0). Elutions were combined and concentrated by trichloroacetic acid (TCA) precipitation. All fractions were mixed with 2× sample buffer and probed after Western blotting, with eluate fractions enriched 20×.
Translocation assayed by bacterial fractionation.
Differentiated U937 cells were plated in a six-well tissue culture dish at a density of 1.5 × 107 cells/well. L. pneumophila strains were cultured as for other translocation assays and used to challenge U937 cells at an MOI of 5. After 1.5 h, wells were washed three times with warm medium to remove nonassociated bacteria. Cells were lifted by pipetting up and down in 2 to 3 ml of PBS, pelleted by centrifugation for 5 min at 1,000 rpm, resuspended in 200 μl of PBS containing 0.5% digitonin and protease inhibitor cocktail (Roche), and incubated for 20 min with rotation at room temperature. The detergent lysate was separated by centrifugation at 10,000 rpm for 15 min at 4°C. Supernatant, representing the detergent soluble fraction, was mixed with 5× sample buffer and boiled. Pellets were further solubilized in 2% SDS and mixed with 2× sample buffer.
Intracellular growth assay.
Bone marrow-derived macrophages (BMDM) from A/J mice were replated on glass coverslips in a 24-well plate at 2 × 105 cells per well. Postexponential phase L. pneumophila was used to challenge cells at an MOI of 1, and plates were subjected to centrifugation for 5 min at 1,000 rpm to promote bacterial association with cells. After 2 h, cells were washed with RPMI medium, and growth was allowed to proceed for an additional 12 h. Coverslips were washed in PBS, fixed with 3.7% formaldehyde for 10 min, and blocked in 4% goat serum in PBS. After samples were permeabilized with 0.1% Triton X-100, bacteria were stained by anti-L. pneumophila rat serum and secondary anti-rat Texas Red (1:500) (Invitrogen). The number of intracellular bacteria in individual phagosomes was counted by visual inspection under a microscope for 150 to 300 cells.
RESULTS
Fusion of dihydrofolate reductase (DHFR) to Icm/Dot-translocated substrates interferes with translocation.
The fusion of secreted proteins to DHFR, a rapidly folding enzyme that can be stabilized in its folded conformation with folate analogs, is commonly used to monitor whether proteins can be secreted or translocated in a folded state (33, 37–39, 43, 53). To analyze if translocation via the Legionella pneumophila Icm/Dot type IVB secretion system can occur in the presence of folded substrates, fusions of several Icm/Dot-translocated substrates (IDTSs) to the carboxyl terminus of the murine DHFR were constructed. We chose four different substrates to analyze. RalF and SidG were interrogated because there is considerable information regarding the nature of the C-terminal translocation signals of these proteins as well as the effects of the mature protein sequences on translocation efficiency (34, 54). MavU was chosen because in a previous study we found that its C-terminal signal was one of the most effective targeting sequences identified among the IDTSs, so we wanted to determine if a high-efficiency signal could overcome deficits caused by the presence of a folded domain (13). Finally, we wanted to investigate whether the introduction of a folded domain internal to an IDTS could have different effects from placing the domain upstream from the complete coding sequence. For this reason, we used LidA, because in previous studies we had identified important junctions between domains in the protein that we thought would likely tolerate added sequences (55).
To detect translocation, each construction was tagged with the calmodulin-dependent adenylyl cyclase enzymatic domain encoded by the Bordetella pertussis cyaA gene (Fig. 1A; Table 1). Production of cAMP by these constructions requires translocation into eukaryotic cells as bacteria do not express calmodulin (56). Expression of these CyaA constructs was induced by the addition of IPTG to cultures of the wild-type L. pneumophila strain Lp02, and the bacteria were used to challenge the human macrophage-like cell line U937. The translocation efficiency of each protein fusion into target U937 cells was quantified as intracellular cAMP produced by the CyaA-IDTS fusions, normalized to relative steady-state levels of the fusion within each strain (see Materials and Methods) (13).
Fig 1.

Insertion of DHFR interferes with translocation. (A) Schematic representation of CyaA-IDTS fusion proteins used in translocation assays. For fusions containing the DHFR moiety, DHFR was fused to the carboxyl terminus of CyaA immediately upstream of an IDTS or translocation signal. (B) The presence of DHFR inhibits translocation. Translocation levels of the IDTS were quantified by measuring intracellular cAMP levels resulting from CyaA. The wild-type L. pneumophila strain Lp02 (black and gray bars) or the translocation-defective dotA strain (striped bar) expressing the indicated CyaA protein fusions was used to challenge U937 cells. After 1 h, intracellular cAMP levels were determined by ELISA in cell lysates (see Materials and Methods). The values represent the mean cAMP determined in seven separate experiments (21 independent infections), normalized to the amount of the T4SS protein DotF and steady-state levels of CyaA in the vector-alone control absent the IDTS, as determined by immunoblotting of bacterial lysates. Black bars, fusions to CyaA alone, as indicated; gray bars, the same IDTS fused to DHFR as shown in panel A; striped bar, Cya-RalF expressed in a dotA mutant strain. Error bars indicate ± standard errors. *, P < 0.02; **, P < 0.004; ***, P < 0.001.
Each of the IDTSs tested exhibited translocation defects when fused to DHFR (Fig. 1B), with each DHFR fusion showing translocation levels that were between 8-fold (for DHFR-MavU) and 40-fold (for DHFR-LidA) lower than the comparable fusions lacking the DHFR moiety. In the case of DHFR-LidA, it appeared that DHFR totally blocked translocation; its translocation efficiency was no greater than that observed with a mutant strain (dotA) containing a defective type IV system unable to translocate IDTS (Fig. 1B).
Interestingly, the CyaA protein that was not fused to any IDTS still translocated at levels ∼5-fold higher in a wild-type strain than in a dotA mutant background (Fig. 1B), and this level was higher than what we observed with wild-type strains harboring the DHFR-LidA fusion. The CyaA construction had a short tail added to its carboxyl-terminal end to facilitate construction of the fusions, and this may have acted as a cryptic signal since the identical construction lacking this short carboxyl-terminal sequence showed translocation levels as low as those of the dotA control (J. Vogel, personal communication). This leakage through the Icm/Dot complex was abolished when CyaA was fused to DHFR, indicating that the DHFR moiety alone is inhibitory to translocation through the Icm/Dot complex even when a cryptic translocation signal is present in a protein (Fig. 1B).
There was considerable translocation of the protein having a DHFR fusion to MavU, presumably due to the extremely effective C-terminal signal, so we tested whether the relatively high levels of translocation were due to protein structure or the nature of the signal. To this end, serial amino-terminal deletions of the L. pneumophila IDTS MavU (Lpg1798) were constructed to selectively remove domains in the protein. MavU is a 398-amino-acid protein predicted to contain a Rho-GTPase-activating protein (RhoGAP) domain and a coiled-coil region (Fig. 2A). Therefore, truncations were made immediately downstream of the RhoGAP domain (Fig. 2, C186), flanking the predicted coiled-coil domain (C118 and C78), and just upstream of the predicted disordered carboxyl terminus (C50) (Fig. 2A; Table 1). CyaA fusions to these truncated proteins were all similarly expressed, and all exhibited translocation efficiencies that were similar to the translocation efficiency of the full-length MavU (Fig. 2B and C, black bars). Translocation of each of the truncated proteins fused to DHFR was then determined relative to the amount of full-length steady-state protein found on gels (see Materials and Methods). All of these DHFR fusion proteins were translocated at similar levels and showed similar levels of defects, consistent with the idea that a specific protein structure cannot ameliorate DHFR-mediated translocation defects (Fig. 2C). The observation that translocation of CyaA-DHFR-MavU(C50), encoding only the C-terminal 50 amino acids of MavU, was similar to that of the full-length MavU-DHFR fusion indicates that the sensitivity of an IDTS to translocation inhibition resulting from insertion of the DHFR moiety does not depend on the size of the hybrid protein but may be connected to the efficiency at which the C-terminal signal can allow recognition by the Icm/Dot translocation system.
Fig 2.
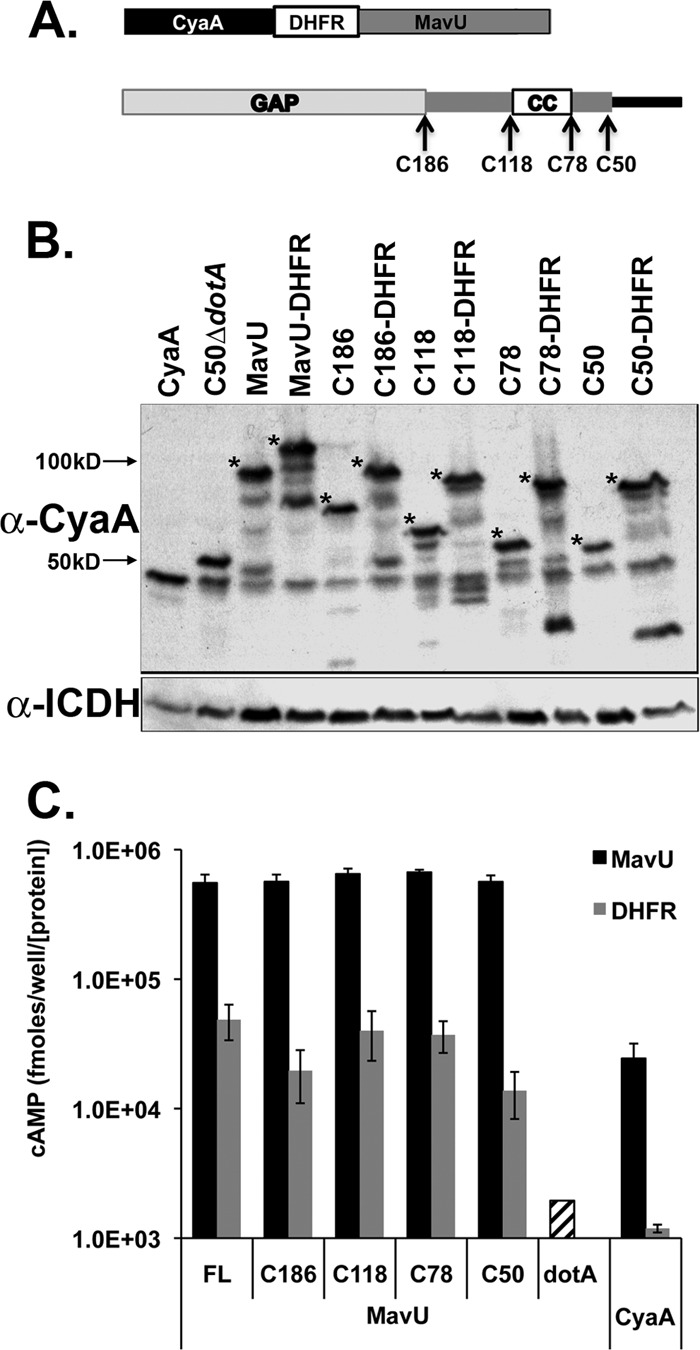
Serial truncations of MavU fused to DHFR exhibit equivalent defects in translocation competence. (A) A schematic of MavU fusions to CyaA and DHFR (top) and of the MavU protein with the RhoGAP domain (light gray), coiled-coil domain (white box), and predicted disordered carboxyl terminus (black) (bottom) is shown. Arrows indicate individual truncation sites for CyaA fusions. (B) Steady-state levels of MavU protein fusions. Fusion protein expression was detected using a monoclonal antibody against CyaA. Asterisks note full-length fusion bands. Bacterial load was quantified using antibody against the housekeeping protein ICDH. The blot is a representative of strains used in one typical CyaA translocation assay. (C) The presence of the DHFR moiety interferes with truncated translocation-competent proteins. U937 cells were challenged with the wild-type L. pneumophila strain Lp02 harboring truncations (black and gray bars) or a translocation-defective dotA3 strain harboring full-length CyaA-MavU (striped bars). After 1 h, intracellular cAMP levels in host cells were determined (see Materials and Methods). The values represent the average amount of cAMP determined in seven separate experiments (21 independent infections) normalized to the amount of the housekeeping protein ICDH in bacteria and then normalized to the amount of CyaA in the fusion protein, as determined by immunoblotting of bacterial lysates. α, anti; FL, full-length.
The DHFR moiety in IDTS fusions is enzymatically active.
In many of the previous studies examining the effects of protein folding on secretion, a DHFR inhibitor such as methotrexate (MTX), which inhibits both mammalian and bacterial proteins, was necessary to stabilize the DHFR in a folded conformation (37). As DHFR blocked IDTS translocation in the absence of MTX, we tested whether the inhibitor would cause further depression of translocation and whether the enzyme was folded in an active confirmation, consistent with its ability to block translocation. As expected, MTX had no effect on the growth in culture of the thy mutant strain used in these studies (data not shown). Furthermore, the addition of MTX did not depress further the already low levels of translocation of the CyaA-DHFR-MavU fusion (data not shown). This raised the possibility that the DHFR moiety was not assuming the conformation of the native active protein in these protein fusions. Therefore, to determine whether DHFR placed within the chimeric proteins was in its native folded form, we tested if the derivatives were enzymatically active.
DHFR activity of the CyaA-IDTS fusions was measured using a colorimetric assay that detects the NADPH-dependent reduction of dihydrofolic acid to tetrahydrofolic acid (Fig. 3A) (see Materials and Methods). Cell lysates of L. pneumophila expressing CyaA-DHFR-IDTS fusions were incubated with NADPH and dihydrofolic acid, and the accumulation of tetrahydrofolate was measured (see Materials and Methods). To determine the specific activity, the rate of hydrolysis as a function of protein concentration was determined by quantitating the amount of fusion protein expressed for each sample (see Materials and Methods) (Fig. 3B). Trimethoprim was added to eliminate the endogenous L. pneumophila DHFR activity as, unlike MTX (57), this antibiotic inhibits the prokaryotic protein specifically and has no effect on the mammalian DHFR present in the fusions (Fig. 2C). The specific activities of DHFR in the protein fusions were comparable to those of purified DHFR, consistent with proper folding of this moiety (Fig. 2C). As expected, no activity was seen in a CyaA-IDTS fusion that did not have DHFR, and the enzymatic activity in all reactions was abolished by MTX (Fig. 2C). The data are consistent with a folded protein moiety causing defective substrate translocation.
Fig 3.
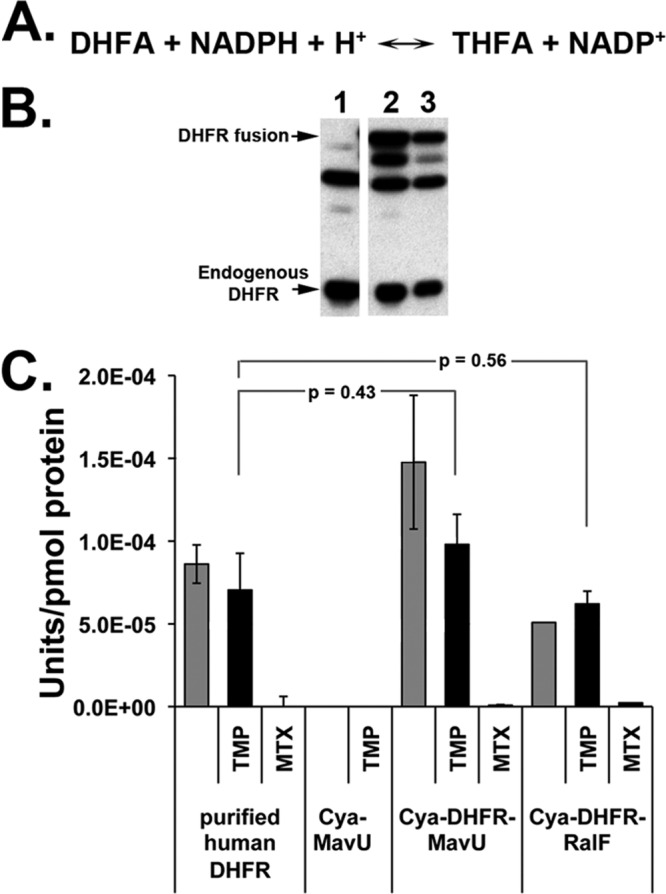
The DHFR moiety within the DHFR-IDTS fusions is catalytically active. (A) NADPH-dependent reduction of dihydrofolic acid (DHFA) to tetrahydrofolic acid (THFA) catalyzed by DHFR. (B) Immunoblot of L. pneumophila soluble extracts from strains overexpressing CyaA-MavU (lane 1) or two different cultures of CyaA-DHFR-MavU proteins (lanes 2 and 3). Antibody against DHFR was used to determine expression of the CyaA-DHFR-IDTS or endogenous DHFR. Densitometry of the CyaA-DHFR-IDTS bands allowed for quantification of protein concentration for determining specific activity. (C) DHFR fusion proteins are enzymatically active. Bacterial cultures were grown in the presence of IPTG to induce expression of the indicated protein fusions. Cells were lysed, and the soluble protein extracts were combined with 100 μM NADPH, 100 μM dihydrofolic acid, and either 50 μM TMP or MTX to inhibit endogenous bacterial DHFR activity or all DHFR activity, respectively. Purified human DHFR was used as a positive control. Activity was normalized to DHFR protein levels as determined by Western blotting. P values are determined by Student's t test, comparing DHFR activity of fusions in the presence of TMP to the purified human DHFR control.
DHFR does not interfere with cyclase activity or increase aggregation of the chimeric proteins.
One complication of using CyaA fusions is that the assay is contingent on the enzymatic activity of the CyaA moiety being unaffected by the IDTS sequence. Therefore, it was necessary to exclude the possibility that the presence of DHFR inhibits adenylate cyclase activity. A modified, in vitro version of the CyaA translocation assay was established to quantify the enzymatic activity of CyaA-DHFR-IDTS fusions relative to the respective CyaA-IDTS. L. pneumophila cultures were grown to late exponential phase, induced to express the CyaA constructs, and lysed. The supernatants of the bacterial lysates were incubated for 30 min in the presence of calmodulin and the adenylate cyclase substrate ATP, and the resulting cAMP production was measured via ELISA (see Materials and Methods). No significant difference was observed in the CyaA activities of any of the protein fusions, regardless of DHFR, confirming that DHFR is not inhibiting the activity of the marker enzyme (Fig. 4A). Similar data were seen when the incubation period was shortened to 5 min, indicating that the longer reaction time did not overload the system (data not shown). Background levels of cAMP produced by L. pneumophila not expressing any CyaA fusion and a reaction mixture lacking calmodulin confirmed that adenylate cyclase activity was due to the B. pertussis CyaA. The culture supernatant from a strain that was not lysed also did not produce significant levels of cAMP, consistent with previous research showing that Icm/Dot secretion occurs only after contact with a host cell (58). These data are consistent with the DHFR moiety having no effect on cyclase activity, with the caveat that it is possible that after translocation into host cells, the folding properties of the translocated substrate may differ significantly from that observed in bacterial extracts.
Fig 4.

Translocation deficiency is not due to loss of adenylate cyclase activity or aggregation of fusion proteins. (A) Adenylate cyclase activity is unaffected by DHFR. Bacterial lysates were incubated in the presence of ATP and calmodulin for 30 min at 37°C to allow for the production of cAMP. Samples were harvested and assayed as described in Materials and Methods. (B) Aggregation of IDTS protein fusions does not correlate with translocation defects of DHFR-IDTS fusions. L. pneumophila wild-type strains expressing the fusions were lysed, and cell debris was cleared by low-speed centrifugation. The resulting supernatant fraction was subjected to centrifugation at 90,000 × g for 1 h to precipitate any aggregated protein. Supernatant and pellet fractions after centrifugation were analyzed by Western blotting with anti-CyaA and anti-ICDH antibody probes. D, low-speed pellet; T, low-speed supernatant; S, high-speed supernatant; P, high-speed pellet. (C) The intensity of the fusion proteins in panel B was determined by densitometry, and protein expression is graphed as a percentage of the total protein after high-speed centrifugation.
We next determined if DHFR hinders translocation by causing aggregation of the fusions. To compare aggregation of DHFR fusions and IDTS alone, we lysed cells and compared the relative amounts of soluble and pelleted protein after high-speed centrifugation. The presence of substrate proteins in postcentrifugation fractions was visualized via immunoblots probed with anti-CyaA (Fig. 4B) (see Materials and Methods). The percentage of aggregation was determined as the amount of CyaA fusion protein that precipitated after high-speed centrifugation relative to the total protein retrieved, as quantified by densitometry of the immunoblots. In all strains tested, no significant difference in aggregation was seen between the IDTS fusions in the presence or absence of DHFR, demonstrating that DHFR does not cause protein aggregation (Fig. 4C). Interestingly, a majority of the protein remained soluble after centrifugation in the strains expressing RalF and MavU fusions, but in the strains expressing SidG fusions, nearly all protein recovered from the high-speed centrifugation was seen in the pellets. Although a relatively small amount of soluble SidG protein appears to be available, our results show that SidG is still readily translocated. These results are consistent with the model that the DHFR moiety does not cause aggregation and that aggregation does not correlate with decreased translocation.
Interference of IDTS translocation by ubiquitin requires folding of the interfering moiety.
Although the data presented argue that the DHFR moiety was folded and did not interfere with the reporter readout, we could not analyze the consequences of conditional folding of an interfering moiety. Therefore, we analyzed ubiquitin (Ub) fusions to IDTSs as mutant Ub derivatives exist that are poorly folded, allowing us to compare folded and unfolded internal domains. As with DHFR, ubiquitin (Ub) is a compact, rapidly folding protein that can serve as a disruptive domain for secreted substrates in multiple systems (43, 59, 60). Furthermore, simple mutations can be used to destabilize the protein and yield an unfolded control for secretion assays (43, 59). To this end, we constructed a fusion of RalF tagged with CyaA to a Ub protein in which two residues in the hydrophobic core (Ile3 and Ile13) are replaced with Gly (UbG3,13) to destabilize the protein conformation (Fig. 5A) (59).
Fig 5.
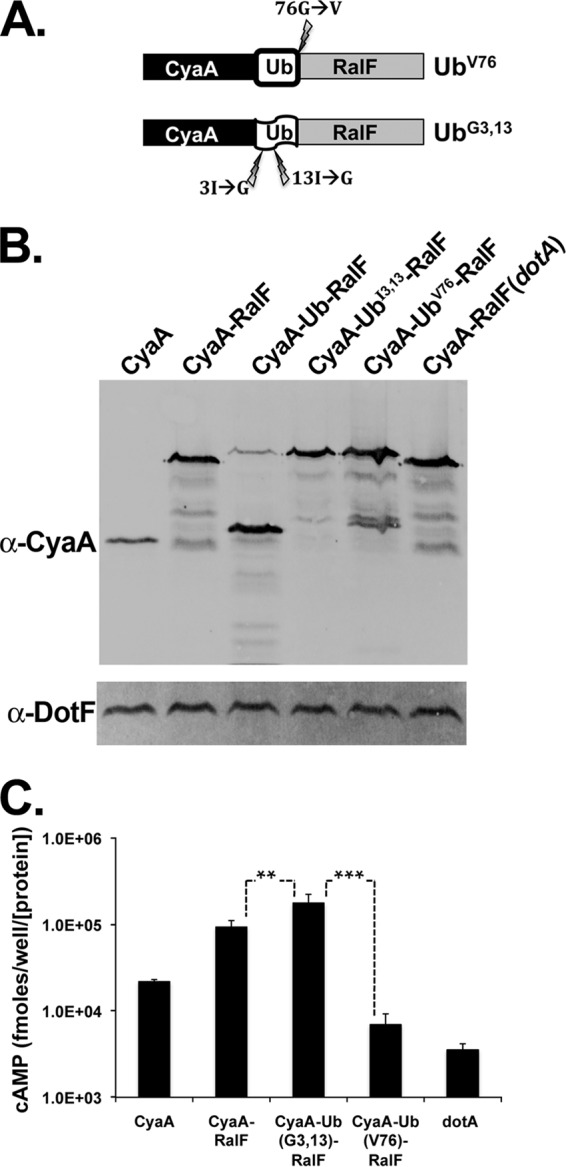
Folded ubiquitin inhibits translocation of fused Icm/Dot substrates. U937 cells were challenged with the wild-type L. pneumophila strain Lp02 or a translocation-defective dotA strain expressing CyaA-tagged RalF fused to either ubiquitin, a nonfolding variant of ubiquitin (UbG3,13), or a variant that cannot be cleaved by DUBs (UbV76). (A) Schematic of the RalF-Ub fusions. Lightning bolts indicate locations of the mutations used to create an uncleavable (UbV76) or destabilized (UbG3,13) ubiquitin. (B) Wild-type Ub is cleaved in L. pneumophila. Immunoblotting of fusion protein expression was performed using a monoclonal antibody against CyaA. Bacterial load was quantified using antibody against the Icm/Dot complex protein DotF. The blot is a representative of strains used in one typical CyaA translocation assay. (C) Folded Ub inhibits translocation. A CyaA translocation assay was performed as described in the legend of Fig. 1. The values represent the average cAMP amounts produced from four independent experiments (9 to 12 independent infections) normalized to DotF and then normalized to the expression of CyaA in the vector. Asterisks mark significance based on Student's t test (*, P ≤ 0.02; **, P ≤ 0.0002; ***, P ≤ 0.0009).
Interestingly, we found that the CyaA-RalF fusion to wild-type Ub was cleaved in the cytosol of L. pneumophila (Fig. 5B). This may be due to a deubiquitinating protease (DUB) that was predicted bioinformatically to be encoded by L. pneumophila (Lpg2907; MavW) (13, 61). In contrast, the UbG3,13 protein was not cleaved, consistent with the interpretation that these mutations prevent folding of the Ub domain. As cleaved Ub fusion proteins would be uninformative in our experiments, we constructed an additional Ub construct by replacing the terminal residue (Gly76) with a Val residue (UbV76). This mutation alters the site of proteolytic cleavage by DUBs without perturbing the folding of Ub (62).
CyaA-RalF fused to the folded, uncleavable UbV76 demonstrated very low translocation efficiency (Fig. 5C), resulting in cAMP levels similar to those of the dotA strain. In contrast, the fusion protein having the unfolded UbG3,13 derivative was translocated at a small but significantly (P = 0.02) higher efficiency than CyaA-RalF alone. Taking these results together with the DHFR data, we conclude that the extent to which an IDTS maintains a region with a folded conformation inversely correlates with its translocation efficiency.
The DHFR-MavU fusion protein exhibits a kinetic defect in translocation.
The above analysis shows that accumulation of the fusion proteins is lowered by the presence of a poison domain but does not investigate whether there is premature termination of secretion or whether the domain slows the rate of translocation. To carry out real-time, kinetic analysis of translocation to answer this question, we adapted the GloSensor cAMP assay (see Materials and Methods). The system uses a modified Photinus pyralis luciferase that contains a cAMP-binding domain. Binding of cAMP causes a conformational change that allows the luciferase to interact with its substrate to produce light. For our purposes, monolayers of 293T cells transiently transfected with a plasmid expressing the modified luciferase and the FcγRIIA receptor (to facilitate uptake of IgG-opsonized L. pneumophila) were preincubated with the luciferase substrate and then challenged with L. pneumophila strains harboring IDTSs fused to CyaA. Intracellular cAMP resulting from translocation of the adenylate cyclase was monitored by the luminescent output over the course of the infection (see Materials and Methods). The kinetic analysis agreed strongly with the endpoint cAMP assay. DHFR fusions to RalF and SidG, as well as the CyaA gene itself, resulted in a substantial kinetic delay in luminescence signal (decreased rate of transfer as represented by depressed slope and increased time-to-half-maximal signal). (Fig. 6A and B). In each case, the time to half-maximal luminescence for the wild-type proteins fused to Cya was approximately 5 min (Fig. 6A to C). In contrast, for the DHFR fusions to RalF and SidG, the time to half-maximal luminescence was at least 30 min (Fig. 6A and B). The MavU-DHFR fusion, however, exhibited a more modest kinetic delay, with a time to half-maximal luminescence of approximately 18 min although the total accumulation of luminescence was clearly lower than that of the wild type (Fig. 6C). Similar results were obtained with the quantitative endpoint CyaA translocation assay using individual time points over the course of infection (Fig. 6D). Both assays are consistent with an initial kinetic delay caused by DHFR fusion to MavU of about 15 to 20 min. This indicates that for at least some IDTSs, a folded moiety within the protein interferes with the efficiency of its passage through the Icm/Dot channel, lowering the yields of translocated product due to depressed rates of translocation and possibly delayed initiation of transit.
Fig 6.

The DHFR-IDTS fusion proteins exhibit kinetic defects in translocation. Results of real-time translocation assays of CyaA and Cya-DHFR moieties fused to IDTS RalF (A), SidG (B), and MavU (C) are shown. HEK293 cells transiently transfected with the pGloSensor cAMP biosensor plasmid and FcγRIIA were incubated with the pGloSensor luciferase substrate reagent for 1 h (see Materials and Methods). Cells were challenged with bacterial strains expressing the indicated proteins that had been opsonized with anti-Legionella IgG. The fluorescence was measured in a luminometer every 2 min for up to 1 h postinfection. Data represent the average of four independent replicates from a typical experiment ± standard error. (D) Direct quantitation of cAMP demonstrates kinetic defect of CyaA-DHFR-MavU fusion. CyaA translocation assays of CyaA-MavU and CyaA-DHFR-MavU fusion proteins were performed as described in the legend of Fig. 1 (see also Materials and Methods). Data represent the average of three replicates ± standard error from a typical experiment. RLU, relative light units.
Low-level translocation of DHFR-MavU requires IcmS.
The IcmS/W adaptor complex is necessary for translocation of a large subset of L. pneumophila effectors (63, 64). Previous work indicated that the IcmS/W complex binds to several different IDTSs, and in the case of SidG, this appears to alter the conformational state of the protein (34). Furthermore, IcmS is necessary for the stability of the T4BSS coupling protein DotL and binds directly to DotL (32, 64). Therefore, if folded DHFR precludes IcmS from binding the IDTS fusions, the folded moiety could restrict the substrate from forming a translocation-competent conformation with IcmS/W.
To investigate whether the presence of DHFR precludes IcmS from facilitating translocation, we first compared translocation of DHFR-MavU in the wild type (Lp02) to an icmS deletion mutant of L. pneumophila. The translocation efficiency of DHFR-MavU was more than 10-fold lower in the ΔicmS strain than in the wild type, similar to the magnitude of defect found with the parental MavU fusion (Fig. 7A). This indicates that the IcmS/IcmW complex is still necessary and capable of facilitating translocation of the DHFR fusion and is consistent with DHFR interfering with translocation at some level other than IcmS/W interactions.
Fig 7.
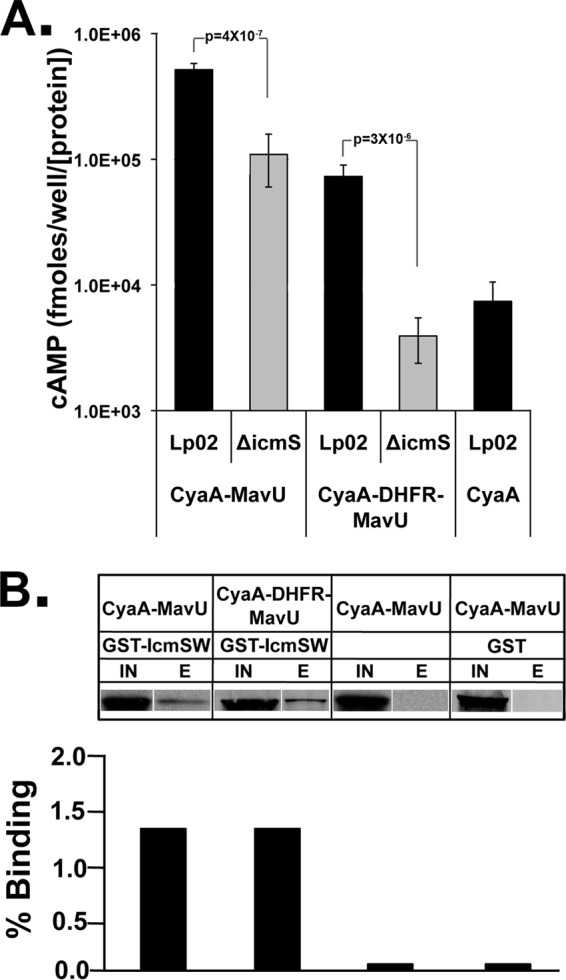
DHFR does not preclude interaction of the IcmS adapter protein with the IDTS protein fusions. (A) Lowered translocation of DHFR-MavU fusions to MavU in the absence of IcmS. CyaA-MavU and CyaA-DHFR-MavU were expressed in the wild-type strain (Lp02) and an IcmS-deficient strain (ΔicmS strain) and used to challenge U937 cells. A CyaA translocation assay was performed as described in the legend of Fig. 2. The values represent the average amount of cAMP determined by ELISA in five independent experiments (15 independent infections) normalized to the amount of the T4SS protein DotF expressed and then normalized to the amount of CyaA in the vector. (B) IcmS copurifies with MavU-DHFR fusions. Copurification of CyaA-MavU and CyaA-DHFR-MavU with GST-IcmS/W was performed. Lysates from E. coli encoding the indicated fusion with or without a second plasmid expressing GST or GST-IcmS/W were incubated with glutathione-conjugated resin for 1 h. Bound GST-IcmS and complexed proteins were eluted by reduced glutathione and concentrated by TCA precipitation. Protein samples were analyzed via Western blotting using anti-CyaA. Shown are the clarified lysate (IN) and the eluate concentrated 20-fold (E). The experiment was performed twice. The IN and E lanes from each sample were analyzed on a single blot but were loaded on noncontiguous lanes, as noted by gaps between lanes.
To further determine if DHFR prevented association of the IcmS/W complex with IDTSs, we determined the relative fraction of fusions that coassociate with the chaperone in the presence or absence of the DHFR moiety (Fig. 7B). Either CyaA-MavU or CyaA-DHFR-MavU was coexpressed in E. coli with a GST-IcmS/IcmW complex (see Materials and Methods) (Table 1). Lysates were incubated with immobilized glutathione resin, bound proteins were eluted with reduced glutathione, and CyaA-tagged proteins bound to GST-IcmS/IcmW were detected by immunoblotting. There was no significant difference in binding to GST-IcmS/W between CyaA-MavU and CyaA-DHFR-MavU (Fig. 7B). In contrast, little fusion protein was associated with the resin in the absence of coexpression of the GST-IcmS/W complex or if GST alone was coexpressed with the fusions (Fig. 7B). We thus confirm that the folded DHFR moiety does not interfere with the interaction of IcmS/W with IDTSs.
IDTS fused to folded DHFR or Ub interferes with Icm/Dot secretion channel function.
It is well documented that secretion signals fused to secretion-incompetent proteins often result in blockage of the translocation channel, resulting in a severe attenuation of channel function (36). Most relevant to this study, introduction of DHFR and Ub to type III secretion system-translocated substrates in Yersinia enterocolitica has been shown to interfere with transport of other proteins through this channel (43, 65). As the DHFR and Ub fusions interfered with translocation in the Icm/Dot T4SS, we tested whether these constructions could occlude translocation of other IDTSs or slow the rate of translocation to the extent that there would be defective intracellular growth due to a blockage the Icm/Dot system, which is known to be essential for intracellular growth.
To determine if introducing a folded moiety into RalF causes translocation inhibition, we monitored export of an endogenously expressed IDTS, SidC, in strains overproducing RalF fusions having either folded or unfolded domains inserted. U937 cells were challenged for 1.5 h with L. pneumophila that expresses either RalF or one of the RalF fusions (Fig. 8A and B), and cells were lysed with digitonin. The detergent-soluble and -insoluble fractions were then analyzed by immunoblotting to identify exported and bacterium-associated SidC, respectively (55) (see Materials and Methods). Strains expressing either the UbV76 or DHFR interfered with translocation of SidC as the translocation efficiency was less than 25% of that observed in strains harboring either RalF or the unfolded moiety-containing UbG3,13. Therefore, overproduction of the translocation-incompetent proteins interferes with function of the Icm/Dot translocon.
Fig 8.
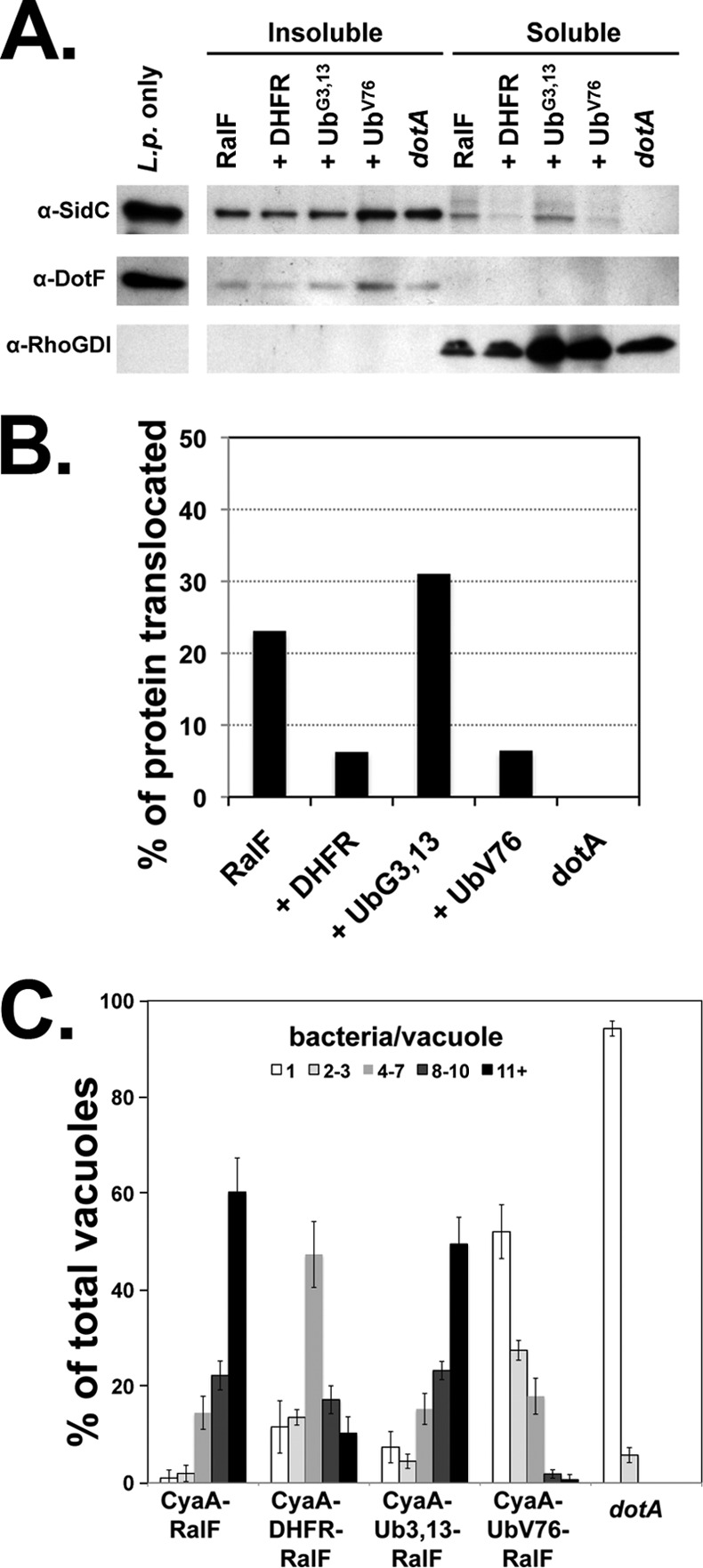
Folded protein fusions cause a partial blockage of the Icm/Dot translocon. (A) Translocation of SidC is decreased in strains harboring folded fusions. Secretion of IDTSs in strains overexpressing folded protein fusions was visualized by detergent fractionation of infected cells (see Materials and Methods). U937 cells were challenged with Legionella expressing CyaA-RalF fusions to DHFR, a folded Ub (UbV76), or the unfolded Ub (UbG3,13). After 1.5 h, cells were washed and lysed with 0.5% digitonin, and the soluble and insoluble fractions were separated by centrifugation. Translocation of the IDTS SidC into the cytoplasm of infected cells (digitonin-soluble extract) was analyzed by probing via immunoblotting. Antibodies to the bacterial T4SS protein DotF and the cytoplasmic protein RhoGDI were used to confirm that lysis does not release bacterial proteins as well as to show separation of the fractions. L.p., L. pneumophila. (B) The graph depicts the percentage of the SidC protein translocated, considered as the amount in the soluble fraction relative to the total protein in the sample, as determined by densitometry. The experiment was performed twice. Shown is a representative experiment. (C) Intracellular growth of strains harboring folded fusion proteins is inhibited. Murine bone marrow macrophages were challenged with either the wild-type strain (Lp02) or dotA strains harboring RalF fusions containing DHFR, a folded Ub (UbV76), or the unfolded Ub (UbG3,13). Growth was allowed to proceed for 14 h, at which time the infected macrophages were fixed and stained with an antibody against Legionella. The number of intracellular bacteria in individual vacuoles was determined for 100 cells each on three individual replicates for each sample. Data indicate the percentage of vacuoles with 1, 2 to 3, 4 to 7, 8 to 10, and 11 or more bacteria per vacuole ± standard error.
To determine if interference with translocation also inhibits intracellular bacterial replication, an intracellular growth assay of L. pneumophila overexpressing RalF fused to DHFR, UbG3,13, or UbV76 was performed. Monolayers of bone marrow-derived macrophages from A/J mice were challenged with wild-type L. pneumophila expressing the reporter protein fusions for 14 h, which is approximately one round of infection. Cells were fixed, and the number of bacteria in individual Legionella-containing vacuoles (LCVs) was determined by microscopy. In strains harboring RalF alone or RalF fused to the unfolded UbG3,13 variant, approximately 60% of LCVs were large vacuoles with 11 or more bacteria, consistent with efficient growth in the presence of these proteins. In the strain expressing the DHFR fusion, the presence of DHFR restricted growth, as evidenced by an increase in the percentage of vacuoles that had small numbers of bacteria (1–3), with a corresponding decrease in vacuoles having more than 11 bacteria (Fig. 8C). The growth defect was most pronounced in the strain expressing the folded ubiquitin (UbV76), with the percentage of large vacuoles of 11 or more bacteria decreasing approximately 4-fold compared to the strain expressing the unfolded Ub fusion (Fig. 8C). Furthermore, there was a significant increase in the percentage of small and medium-sized vacuoles, consistent with decreased growth in the presence of this fusion. This phenotype is indicative of a partial blockage of the translocon by folded proteins, reducing the rate of translocation of IDTSs needed for bacterial replication. Therefore, occlusion of the channel by the introduction of a folded domain interferes with the ability of L. pneumophila to grow intracellularly.
DISCUSSION
In this study, we show that translocation of substrates of the Icm/Dot T4SS into host cells is depressed by the presence of moieties that assume folded conformations. Fusion proteins of DHFR and Ub to IDTSs demonstrate a defect in translocation compared to their unfolded counterparts (Fig. 1 and 5). Furthermore, there was a small but significant increase in translocation of the IDTS RalF fused to a destabilized variant of ubiquitin (UbG3,13) relative to RalF alone, consistent with an unfolded region stimulating translocation (Fig. 5). If the unstructured moiety increases the amount of unfolding of the entire IDTS, translocation competence may be defined by the ability of the IDTS to maintain an unfolded conformation.
Many IDTSs are translated prior to infection, as indicated by the fact that protein synthesis inhibitors do not affect translocation rates (58). If we consider this in context with our data, the system would likely require one or more chaperone proteins to perpetuate an unfolded, translocation-competent state of the effectors. These may be heat shock proteins or uncharacterized chaperones but also could include the adapter protein complexes of IcmS/W and IcmS/LvgA (64). These small proteins are hypothesized to resemble the type III secretion system chaperones which act to hold proteins in a partially unfolded conformation for delivery to the translocon ATPase for subsequent unfolding and secretion (45, 46, 66). Consistent with a role for these proteins forming a pretranslocation complex, IcmS/W can associate with translocated substrates only when coexpressed in cells (34). This pretranslocation complex appears to be the form that is presented to the secretion channel as IcmS binds directly to DotL, the secretion ATPase in the Icm/Dot system (32). Although insertion of DHFR interfered with translocation, IcmS/W was still able to interact with MavU- and SidG-DHFR fusions, and IcmS was necessary to facilitate the translocation of the DHFR-MavU fusion (Fig. 7). These results indicate that the defect in translocation observed in this work is independent of IcmS/W action.
Using two different assays, we were able to demonstrate that the DHFR moiety decreases the rate of protein transport (Fig. 6). Two models can explain this result. The first possibility is that a folded domain does not cause an absolute block in translocation, either because the Icm/Dot system has some intrinsic ability to translocate folded proteins or because the secretion system is able to convert translocation-incompetent substrates into transferrable conformations. The second possibility is that there is a small subpopulation of proteins that are partially unfolded. In either model, the extent to which a protein is in a translocation-competent conformation directly correlates with the rate that it can be transported through the Icm/Dot translocon.
In addition to the translocation defect of a tightly folded protein, the effector fusions interfere with translocation of other IDTSs (Fig. 8), consistent with at least partial occlusion of the translocon. In the case of the Yersinia T3SS, the presence of folded domains in translocated substrates interferes with the assembly of specific components necessary for the function of the translocon (65). Although we cannot exclude this possibility, the fact that the DHFR fusions are very poorly translocated but cause only partial interference with intracellular growth (Fig. 8) is consistent with the Icm/Dot complex being largely intact. We propose two models for the mechanism of this blockage. First, the interfering protein fusion could disrupt transport of only a subset of translocated effectors that are critical for intracellular growth. The second model is that the fusion protein is stalled within the secretion channel, causing a global decrease in the rate of all IDTSs. The low rate of transport of the fusion proteins could generate a situation akin to a traffic jam, in which a group of slow-moving individuals causes a general reduction in the rate of movement of all individuals moving through the secretion channel.
If stalled or slow-moving substrates cause the generalized translocation defect, then there must be some explanation for why such interference occurs. Although the structure of the T4BSS has not been fully elucidated, cryo-electron microscopic and crystal structures of core components of a T4SS from plasmid pKM101 have been solved. While the Icm/Dot system constitutes a different class of secretion apparatus (T4BSS), some insight can be gained from considering the known structure. Cryo-electron microscopy of the T4SS reveals a unique structure in which the core forms a large, bulbous chamber between the inner and outer membranes (47). Importantly, the channel is constricted at the capped outer membrane face, which the crystal structure suggests undergoes a conformational change that could restrict the channel at the outer membrane, allowing opening or closing of the secretion pore (30, 47). At the base of the structure is a large entryway of 55 nm although it is believed that other components of the secretion system could fit within this entryway and make for a much narrower passage site (47). Based upon this configuration, at the very least, the transport of folded proteins through the Icm/Dot system may be hindered while exiting the translocon. This situation would likely lead to the accumulation of proteins within the secretion system core.
Blocks could also occur at the entry site of the secretion complex. In this scenario, initiation of movement into the secretion complex would result in a frustrated intermediate in which there is a physical block at the entrance. Furthermore, occlusion of IDTSs could occur at a step that is prior to entry into the complex. In the T3SS, folded effector fusions prevent translocation of other IDTSs by binding to and inhibiting the ATPase associated with the secretion system (65). Normally, chaperones carry their cognate substrates to the T3SS in a partially unfolded conformation, thus “priming” them for complete unfolding by the ATPase before translocation (45, 46). In the Icm/Dot system it has been proposed that DotL, as part of a subcomplex with the ATPases DotM and DotN, acts as a regulator of IDTS entry (67). DotL has sequence similarity to type IV secretion system coupling proteins, which are proposed to link substrates to the rest of the secretion complex (68). The fact that DotL associates with IcmS could be key to linking the substrate to the secretion system. Folded proteins may be blocked from entry by an inappropriate interaction with DotL, thus inhibiting further translocation of effectors.
These studies demonstrate that small folded domains can generate impassable substrates through the L. pneumophila T4SS, consistent with a strong selection against such moieties being present in IDTSs. Therefore, it would appear that most IDTSs are either easily unfolded, maintained with chaperones in an unfolded conformation, or have determinants that allow recognition by unfoldases associated with the Icm/Dot complex. Future work will require identifying the features of the substrates that allow them to be secretion competent and determining which bacterial proteins contribute to generating structures that maintain this conformation.
ACKNOWLEDGMENTS
R.R.I. is an investigator of HHMI, while W.M.A. and D.D. were supported by NIH training grant T32AI00742. D.D. was also supported by NIAID fellowship F31 AI098423. This work was also supported by Howard Hughes Medical Institute.
We thank Joseph Vogel for plasmid gifts as well as numerous suggestions and support, Linc Sonenshein for ICDH antibody, and Elizabeth Creasey, Sina Mohammadi, Edward Geisinger, Tamara O'Connor, and Kim Davis for review of the manuscript.
Footnotes
Published ahead of print 24 June 2013
REFERENCES
- 1.Fields BS. 1996. The molecular ecology of legionellae. Trends Microbiol. 4:286–290 [DOI] [PubMed] [Google Scholar]
- 2.Fields BS, Benson RF, Besser RE. 2002. Legionella and Legionnaires' disease: 25 years of investigation. Clin. Microbiol. Rev. 15:506–526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fields BS, Shotts EB, Jr, Feeley JC, Gorman GW, Martin WT. 1984. Proliferation of Legionella pneumophila as an intracellular parasite of the ciliated protozoan Tetrahymena pyriformis. Appl. Environ. Microbiol. 47:467–471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wadowsky RM, Butler LJ, Cook MK, Verma SM, Paul MA, Fields BS, Keleti G, Sykora JL, Yee RB. 1988. Growth-supporting activity for Legionella pneumophila in tap water cultures and implication of hartmannellid amoebae as growth factors. Appl. Environ. Microbiol. 54:2677–2682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fraser DW, Tsai TR, Orenstein W, Parkin WE, Beecham HJ, Sharrar RG, Harris J, Mallison GF, Martin SM, McDade JE, Shepard CC, Brachman PS. 1977. Legionnaires' disease: description of an epidemic of pneumonia. N. Engl. J. Med. 297:1189–1197 [DOI] [PubMed] [Google Scholar]
- 6.Horwitz MA, Silverstein SC. 1980. Legionnaires' disease bacterium (Legionella pneumophila) multiples intracellularly in human monocytes. J. Clin. Invest. 66:441–450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Segal G, Shuman HA. 1999. Legionella pneumophila utilizes the same genes to multiply within Acanthamoeba castellanii and human macrophages. Infect. Immun. 67:2117–2124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Berger KH, Isberg RR. 1993. Two distinct defects in intracellular growth complemented by a single genetic locus in Legionella pneumophila. Mol. Microbiol. 7:7–19 [DOI] [PubMed] [Google Scholar]
- 9.Segal G, Shuman HA. 1997. Characterization of a new region required for macrophage killing by Legionella pneumophila. Infect. Immun. 65:5057–5066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Altman E, Segal G. 2008. The response regulator CpxR directly regulates expression of several Legionella pneumophila icm/dot components as well as new translocated substrates. J. Bacteriol. 190:1985–1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.de Felipe KS, Glover RT, Charpentier X, Anderson OR, Reyes M, Pericone CD, Shuman HA. 2008. Legionella eukaryotic-like type IV substrates interfere with organelle trafficking. PLoS Pathog. 4:e1000117. 10.1371/journal.ppat.1000117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.de Felipe KS, Pampou S, Jovanovic OS, Pericone CD, Ye SF, Kalachikov S, Shuman HA. 2005. Evidence for acquisition of Legionella type IV secretion substrates via interdomain horizontal gene transfer. J. Bacteriol. 187:7716–7726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huang L, Boyd D, Amyot WM, Hempstead AD, Luo ZQ, O'Connor TJ, Chen C, Machner M, Montminy T, Isberg RR. 2011. The E block motif is associated with Legionella pneumophila translocated substrates. Cell Microbiol. 13:227–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Luo ZQ, Isberg RR. 2004. Multiple substrates of the Legionella pneumophila Dot/Icm system identified by interbacterial protein transfer. Proc. Natl. Acad. Sci. U. S. A. 101:841–846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nagai H, Kagan JC, Zhu X, Kahn RA, Roy CR. 2002. A bacterial guanine nucleotide exchange factor activates ARF on Legionella phagosomes. Science 295:679–682 [DOI] [PubMed] [Google Scholar]
- 16.Zusman T, Degtyar E, Segal G. 2008. Identification of a hypervariable region containing new Legionella pneumophila Icm/Dot translocated substrates by using the conserved icmQ regulatory signature. Infect. Immun. 76:4581–4591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhu W, Banga S, Tan Y, Zheng C, Stephenson R, Gately J, Luo ZQ. 2011. Comprehensive identification of protein substrates of the Dot/Icm type IV transporter of Legionella pneumophila. PLoS One 6:e17638. 10.1371/journal.pone.0017638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nagai H, Kubori T. 2011. Type IVB secretion systems of Legionella and other Gram-negative bacteria. Front. Microbiol. 2:136. 10.3389/fmicb.2011.00136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lifshitz Z, Burstein D, Peeri M, Zusman T, Schwartz K, Shuman HA, Pupko T, Segal G. 2013. Computational modeling and experimental validation of the Legionella and Coxiella virulence-related type-IVB secretion signal. Proc. Nat. Acad. Sci., U. S. A. 110:E707–E715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kagan JC, Stein MP, Pypaert M, Roy CR. 2004. Legionella subvert the functions of Rab1 and Sec22b to create a replicative organelle. J. Exp. Med. 199:1201–1211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Derre I, Isberg RR. 2004. Legionella pneumophila replication vacuole formation involves rapid recruitment of proteins of the early secretory system. Infect. Immun. 72:3048–3053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Molmeret M, Santic M, Asare R, Carabeo RA, Abu Kwaik Y. 2007. Rapid escape of the dot/icm mutants of Legionella pneumophila into the cytosol of mammalian and protozoan cells. Infect. Immun. 75:3290–3304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Christie PJ. 2001. Type IV secretion: intercellular transfer of macromolecules by systems ancestrally related to conjugation machines. Mol. Microbiol. 40:294–305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Christie PJ, Vogel JP. 2000. Bacterial type IV secretion: conjugation systems adapted to deliver effector molecules to host cells. Trends Microbiol. 8:354–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wallden K, Rivera-Calzada A, Waksman G. 2010. Type IV secretion systems: versatility and diversity in function. Cell Microbiol. 12:1203–1212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nagai H, Roy CR. 2003. Show me the substrates: modulation of host cell function by type IV secretion systems. Cell Microbiol. 5:373–383 [DOI] [PubMed] [Google Scholar]
- 27.Beare PA, Gilk SD, Larson CL, Hill J, Stead CM, Omsland A, Cockrell DC, Howe D, Voth DE, Heinzen RA. 2011. Dot/Icm type IVB secretion system requirements for Coxiella burnetii growth in human macrophages. mBio 2(4):e00175-00111. 10.1128/mBio.00175-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Leclerque A, Kleespies RG. 2008. Type IV secretion system components as phylogenetic markers of entomopathogenic bacteria of the genus Rickettsiella. FEMS Microbiol. Lett. 279:167–173 [DOI] [PubMed] [Google Scholar]
- 29.Fronzes R, Christie PJ, Waksman G. 2009. The structural biology of type IV secretion systems. Nat. Rev. Microbiol. 7:703–714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chandran V, Fronzes R, Duquerroy S, Cronin N, Navaza J, Waksman G. 2009. Structure of the outer membrane complex of a type IV secretion system. Nature 462:1011–1015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vincent CD, Friedman JR, Jeong KC, Buford EC, Miller JL, Vogel JP. 2006. Identification of the core transmembrane complex of the Legionella Dot/Icm type IV secretion system. Mol. Microbiol. 62:1278–1291 [DOI] [PubMed] [Google Scholar]
- 32.Vincent CD, Friedman JR, Jeong KC, Sutherland MC, Vogel JP. 2012. Identification of the DotL coupling protein subcomplex of the Legionella Dot/Icm type IV secretion system. Mol. Microbiol. 85:378–391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Klingenberg O, Olsnes S. 1996. Ability of methotrexate to inhibit translocation to the cytosol of dihydrofolate reductase fused to diphtheria toxin. Biochem. J. 313:647–653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cambronne ED, Roy CR. 2007. The Legionella pneumophila IcmSW complex interacts with multiple Dot/Icm effectors to facilitate type IV translocation. PLoS Pathog. 3:e188. 10.1371/journal.ppat.0030188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Burstein D, Zusman T, Degtyar E, Viner R, Segal G, Pupko T. 2009. Genome-scale identification of Legionella pneumophila effectors using a machine learning approach. PLoS Pathog. 5:e1000508. 10.1371/journal.ppat.1000508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bieker KL, Silhavy TJ. 1989. PrlA is important for the translocation of exported proteins across the cytoplasmic membrane of Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 86:968–972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Eilers M, Schatz G. 1986. Binding of a specific ligand inhibits import of a purified precursor protein into mitochondria. Nature 322:228–232 [DOI] [PubMed] [Google Scholar]
- 38.Endo T, Kawakami M, Goto A, America T, Weisbeek P, Nakai M. 1994. Chloroplast protein import. Chloroplast envelopes and thylakoids have different abilities to unfold proteins. Eur. J. Biochem. 225:403–409 [DOI] [PubMed] [Google Scholar]
- 39.Salvador N, Aguado C, Horst M, Knecht E. 2000. Import of a cytosolic protein into lysosomes by chaperone-mediated autophagy depends on its folding state. J. Biol. Chem. 275:27447–27456 [DOI] [PubMed] [Google Scholar]
- 40.McNew JA, Goodman JM. 1994. An oligomeric protein is imported into peroxisomes in vivo. J. Cell Biol. 127:1245–1257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Walton PA, Hill PE, Subramani S. 1995. Import of stably folded proteins into peroxisomes. Mol. Biol. Cell 6:675–683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wickner W, Schekman R. 2005. Protein translocation across biological membranes. Science 310:1452–1456 [DOI] [PubMed] [Google Scholar]
- 43.Lee VT, Schneewind O. 2002. Yop fusions to tightly folded protein domains and their effects on Yersinia enterocolitica type III secretion. J. Bacteriol. 184:3740–3745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sorg JA, Miller NC, Marketon MM, Schneewind O. 2005. Rejection of impassable substrates by Yersinia type III secretion machines. J. Bacteriol. 187:7090–7102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Akeda Y, Galan JE. 2005. Chaperone release and unfolding of substrates in type III secretion. Nature 437:911–915 [DOI] [PubMed] [Google Scholar]
- 46.Feldman MF, Muller S, Wuest E, Cornelis GR. 2002. SycE allows secretion of YopE-DHFR hybrids by the Yersinia enterocolitica type III Ysc system. Mol. Microbiol. 46:1183–1197 [DOI] [PubMed] [Google Scholar]
- 47.Fronzes R, Schafer E, Wang L, Saibil HR, Orlova EV, Waksman G. 2009. Structure of a type IV secretion system core complex. Science 323:266–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Feeley JC, Gibson RJ, Gorman GW, Langford NC, Rasheed JK, Mackel DC, Baine WB. 1979. Charcoal-yeast extract agar: primary isolation medium for Legionella pneumophila. J. Clin. Microbiol. 10:437–441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Berger KH, Isberg RR. 1994. Intracellular survival by Legionella. Methods Cell. Biol. 45:247–259 [DOI] [PubMed] [Google Scholar]
- 50.Coers J, Kagan JC, Matthews M, Nagai H, Zuckman DM, Roy CR. 2000. Identification of Icm protein complexes that play distinct roles in the biogenesis of an organelle permissive for Legionella pneumophila intracellular growth. Mol. Microbiol. 38:719–736 [DOI] [PubMed] [Google Scholar]
- 51.Bardill JP, Miller JL, Vogel JP. 2005. IcmS-dependent translocation of SdeA into macrophages by the Legionella pneumophila type IV secretion system. Mol. Microbiol. 56:90–103 [DOI] [PubMed] [Google Scholar]
- 52.Swanson MS, Isberg RR. 1995. Association of Legionella pneumophila with the macrophage endoplasmic reticulum. Infect. Immun. 63:3609–3620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Haug G, Wilde C, Leemhuis J, Meyer DK, Aktories K, Barth H. 2003. Cellular uptake of Clostridium botulinum C2 toxin: membrane translocation of a fusion toxin requires unfolding of its dihydrofolate reductase domain. Biochemistry 42:15284–15291 [DOI] [PubMed] [Google Scholar]
- 54.Nagai H, Cambronne ED, Kagan JC, Amor JC, Kahn RA, Roy CR. 2005. A C-terminal translocation signal required for Dot/Icm-dependent delivery of the Legionella RalF protein to host cells. Proc. Natl. Acad. Sci. U. S. A. 102:826–831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Derre I, Isberg RR. 2005. LidA, a translocated substrate of the Legionella pneumophila type IV secretion system, interferes with the early secretory pathway. Infect. Immun. 73:4370–4380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sory MP, Cornelis GR. 1994. Translocation of a hybrid YopE-adenylate cyclase from Yersinia enterocolitica into HeLa cells. Mol. Microbiol. 14:583–594 [DOI] [PubMed] [Google Scholar]
- 57.Williams MN, Poe M, Greenfield NJ, Hirshfield JM, Hoogsteen K. 1973. Methotrexate binding to dihydrofolate reductase from a methotrexate-resistant strain of Escherichia coli. J. Biol. Chem. 248:6375–6379 [PubMed] [Google Scholar]
- 58.Charpentier X, Gabay JE, Reyes M, Zhu JW, Weiss A, Shuman HA. 2009. Chemical genetics reveals bacterial and host cell functions critical for type IV effector translocation by Legionella pneumophila. PLoS Pathog. 5:e1000501. 10.1371/journal.ppat.1000501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Johnsson N, Varshavsky A. 1994. Ubiquitin-assisted dissection of protein transport across membranes. EMBO J. 13:2686–2698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Vijay-Kumar S, Bugg CE, Cook WJ. 1987. Structure of ubiquitin refined at 1.8 Å resolution. J. Mol. Biol. 194:531–544 [DOI] [PubMed] [Google Scholar]
- 61.Catic A, Fiebiger E, Korbel GA, Blom D, Galardy PJ, Ploegh HL. 2007. Screen for ISG15-crossreactive deubiquitinases. PLoS One 2:e679. 10.1371/journal.pone.0000679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Johnson ES, Bartel B, Seufert W, Varshavsky A. 1992. Ubiquitin as a degradation signal. EMBO J. 11:497–505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ninio S, Zuckman-Cholon DM, Cambronne ED, Roy CR. 2005. The Legionella IcmS-IcmW protein complex is important for Dot/Icm-mediated protein translocation. Mol. Microbiol. 55:912–926 [DOI] [PubMed] [Google Scholar]
- 64.Vincent CD, Vogel JP. 2006. The Legionella pneumophila IcmS-LvgA protein complex is important for Dot/Icm-dependent intracellular growth. Mol. Microbiol. 61:596–613 [DOI] [PubMed] [Google Scholar]
- 65.Sorg JA, Blaylock B, Schneewind O. 2006. Secretion signal recognition by YscN, the Yersinia type III secretion ATPase. Proc. Natl. Acad. Sci. U. S. A. 103:16490–16495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Stebbins CE, Galan JE. 2001. Maintenance of an unfolded polypeptide by a cognate chaperone in bacterial type III secretion. Nature 414:77–81 [DOI] [PubMed] [Google Scholar]
- 67.Sutherland MC, Nguyen TL, Tseng V, Vogel JP. 2012. The Legionella IcmSW complex directly interacts with DotL to mediate translocation of adaptor-dependent substrates. PLoS Pathog. 8:e1002910. 10.1371/journal.ppat.1002910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Buscher BA, Conover GM, Miller JL, Vogel SA, Meyers SN, Isberg RR, Vogel JP. 2005. The DotL protein, a member of the TraG-coupling protein family, is essential for Viability of Legionella pneumophila strain Lp02. J. Bacteriol. 187:2927–2938 [DOI] [PMC free article] [PubMed] [Google Scholar]