

Abstract
The opportunistic pathogen Pneumocystis jirovecii is a significant cause of disease in HIV-infected patients and others with immunosuppressive conditions. Pneumocystis can also cause complications in treatment following antiretroviral therapy or reversal of immunosuppressive therapy, as the newly reconstituted immune system can develop a pathological inflammatory response to remaining antigens or a previously undetected infection. To target β-(1,3)-glucan, a structural component of the Pneumocystis cell wall with immune-stimulating properties, we have developed immunoadhesins consisting of the carbohydrate binding domain of Dectin-1 fused to the Fc regions of the 4 subtypes of murine IgG (mIgG). These immunoadhesins bind β-glucan with high affinity, and precoating the surface of zymosan with Dectin-1:Fc can reduce cytokine production by macrophages in an in vitro stimulation assay. All Dectin-1:Fc variants showed specificity of binding to the asci of Pneumocystis murina, but effector activity of the fusion molecules varied depending on Fc subtype. Dectin-1:mIgG2a Fc was able to reduce the viability of P. murina in culture through a complement-dependent mechanism, whereas previous studies have shown the mIgG1 Fc fusion to increase macrophage-dependent killing. In an in vivo challenge model, systemic expression of Dectin-1:mIgG1 Fc significantly reduced ascus burden in the lung. When administered postinfection in a model of immune reconstitution inflammatory syndrome (IRIS), both Dectin-1:mIgG1 and Dectin-1:mIgG2a Fc reduced hypoxemia despite minimal effects on fungal burden in the lung. Taken together, these data indicate that molecules targeting β-glucan may provide a mechanism for treatment of fungal infection and for modulation of the inflammatory response to Pneumocystis and other pathogens.
INTRODUCTION
Pneumocystis is an opportunistic fungal pathogen that causes pneumonia in immunocompromised hosts, including those with human immunodeficiency virus (HIV) and immunosuppression secondary to chemotherapy or organ transplantation. Despite a significant decline in the incidence of Pneumocystis pneumonia (PCP) following the introduction of PCP chemoprophylaxis and potent combination antiretroviral therapy (ART), PCP remains a leading opportunistic infection in HIV-positive adults and children worldwide (1–3). In addition, as more potent immunotherapies are developed and the number of patients receiving immunosuppressive therapy and antitumor chemotherapeutic agents increases, the prevalence of PCP in non-HIV-infected patients continues to grow (4, 5).
The depletion or dysfunction of CD4+ T cells is the primary risk factor for host susceptibility to Pneumocystis infection, and HIV-positive patients with CD4+ T cell counts below 200 cells/μl are highly susceptible to PCP if not receiving preventive therapy (6, 7). Animal models have also demonstrated the fundamental role of CD4+ T cells in protective immunity against Pneumocystis. Selective depletion of CD4+ T cells can render mice susceptible to persistent pulmonary infection (8, 9), and SCID, Rag1−/−, and Rag2−/− mice which lack functional B and T cells are also highly susceptible to PCP (10, 11). In HIV-infected patients, the goal of ART is to increase the population of CD4+ T cells and restore protective immunity against pathogens such as Pneumocystis, but this restoration may be associated with a paradoxical clinical deterioration and the development of a severe inflammatory response against specific foreign or self-antigens (12–14). This immune reconstitution inflammatory syndrome (IRIS), also termed immune reconstitution disease (IRD) or immune reconstitution syndrome (IRS), has become a major challenge in the clinical treatment of HIV, affecting from 8 to 33% of ART responders (15–18). Development of IRIS has also been reported in non-HIV patient populations following withdrawal of immunosuppressive therapy, such as transplant recipients (19) and patients receiving tumor necrosis factor antagonists (20).
IRIS in the context of Pneumocystis infection has been described both in HIV-infected patients (21–24) and for those with non-HIV immunosuppressive conditions (25, 26). PCP-associated IRIS has been modeled in murine hosts using selective transfer of lymphocyte subsets (27), and depletion of regulatory T cells from these cell populations can exacerbate lung injury (28). In further support of effector T cells mediating lung injury in Pneumocystis IRIS, it has also been shown that anti-CD3 antibodies (Abs) can ameliorate lung injury in the adoptive transfer model (29).
Carbohydrate antigens such as β-glucans in the Pneumocystis cell wall are believed to play a significant role in both protective immunity and the pathological inflammatory response of IRIS (30, 31). Signaling of the pattern recognition receptor Dectin-1 in response to β-glucan can regulate expression of innate response genes, linking innate to adaptive immunity (32). Dectin-1 signaling can promote activation of dendritic cells, rendering them competent to prime Th1 and Th17 responses as well as cytotoxic T lymphocyte responses (33, 34). Prior studies in the lab demonstrated that administration of a fusion protein that carries the extracellular domain of the β-glucan receptor Dectin-1 fused to a murine IgG (mIgG) Fc can opsonize Pneumocystis organisms and can reduce organism burden in the lungs of Pneumocystis murina-infected SCID mice (35). In this study, we investigated the role of the IgG Fc fusion isotype in killing of Pneumocystis in vitro as well as reduction of organism burden in vivo. Moreover, we examined the effect of these molecules on Dectin-1 signaling and the ability of Dectin-1:Fc to ameliorate lung injury in the IRIS adoptive transfer model.
MATERIALS AND METHODS
Mice.
Six- to 8-week-old, wild-type (WT) C57BL/6J mice and immunodeficient B6.129S7-Rag1tm1Mom/J (Rag1−/−) mice were obtained from The Jackson Laboratory (Bar Harbor, ME). Immunodeficient B10;B6-Rag2tm1Fwa II2rgtm1Wjl (Rag2−/−Il2rg−/−) mice were obtained from Taconic (Hudson, NY). Animals were housed in a pathogen-free environment and given food and water ad libitum. All experiments were approved by the University of Pittsburgh Institutional Animal Care and Use Committee.
MAbs and Fc fusion proteins.
Anti-Dectin-1 monoclonal antibody (MAb), clone 2A11, rat IgG2b (AbD Serotec, Raleigh, NC), was used as previously described (36). Construction of recombinant Dectin-1:mIgG1 Fc was previously described (35). Additional Dectin-1 fusion vectors containing an IgK leader sequence to facilitate secretion, the extracellular domain of the Dectin-1 receptor (amino acids 69 to 244), and a hexapeptide linker with a thrombin cleavage site cloned in frame with the Fc regions of murine IgG2a, IgG2b, and IgG3 were constructed using the parent Dectin-1:mIgG1 Fc vector and the vectors pFUSE-mIgG2a-Fc, pFUSE-mIgG2b-Fc, and pFUSE-mIgG3-Fc, respectively (Invivogen, San Diego, CA).
Molecular interaction analysis.
Real-time surface plasmon resonance experiments were performed on a Biacore 3000 instrument with CM5 sensor chips (GE Healthcare Life Sciences, Piscataway, NJ) at 25°C. Amine coupling with N-hydroxysuccinimide (NHS)–1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC) was used to covalently immobilize rabbit anti-mouse capture antibodies onto the chip surface (mouse antibody capture kit; GE Healthcare Life Sciences). Affinity-purified Dectin-1:Fc was captured by injection at a flow rate of 5 μl/min (total, 35 μl). Laminarin (Sigma-Aldrich, St. Louis, MO) was dissolved in phosphate-buffered saline (PBS) and diluted in running buffer (10 mM HEPES [pH 8.0], 150 mM NaCl, 0.002% Tween 20), injected by Kinject, and allowed to reach equilibrium, after which only running buffer was applied. Immobilized rabbit anti-mouse Abs or an irrelevant IgG2a MAb served as a control surface, and nonspecific binding of laminarin was subtracted from the signal in the active flow cell. The association rate constant (ka) and dissociation rate constant (kd) were calculated, and the equilibrium association (KA) and dissociation (KD) constants were determined using BIAevaluation 4.1 software.
In vitro macrophage stimulation.
Zymosan (Invivogen) was depleted of TLR-activating ligands by being boiled in hot alkali as previously described (37) and optionally pretreated with 100 μg/ml Dectin-1:Fc for 30 min at 37°C. RAW 264.7 cells cultured in Dulbecco's modified Eagle's medium (DMEM) plus 10% fetal bovine serum (FBS) were plated into 96-well flat-bottom plates at a density of 1 × 105 cells per well and stimulated with 50 μg/ml zymosan. A control group of RAW cells were also pretreated for 30 min with 5 μg/ml of MAb 2A11 to block Dectin-1 binding to zymosan. Cell culture supernatants were harvested 16 h after stimulation and assayed for granulocyte colony-stimulating factor (G-CSF) and tumor necrosis factor alpha (TNF-α) production using a custom multiplex bead assay (EMD Millipore, Billerica, MA).
Pneumocystis isolation and inoculum.
Pneumocystis murina organisms were isolated from the lung tissue of Rag2−/−Il2rg−/− mice previously inoculated with P. murina. The inoculum for infectious challenge was prepared by differential centrifugation as previously described (38). Briefly, Rag2−/−Il2rg−/− mice with Pneumocystis pneumonia were sacrificed, and the lungs were aseptically removed and frozen in 1 ml of sterile phosphate-buffered saline (PBS) at −80°C. Frozen lungs were thawed, homogenized through a 70-μm filter, and pelleted by centrifugation at 800 × g for 10 min at 4°C. The pellet was resuspended in 1 ml PBS, and a 1:10 dilution was stained with Hema-3 modified Wright-Giemsa stain (Fisher Scientific, Pittsburgh, PA). The number of P. murina asci (cyst forms) was quantified microscopically (8), and the inoculum concentration was adjusted to 2 × 106 asci per ml. One hundred microliters of this inoculum, corresponding to 2 × 105 asci per mouse, was given by oropharyngeal aspiration using the tongue-pull technique. Briefly, mice were lightly anesthetized using 2 to 3% isoflurane and suspended by their front incisors, and the tongue was gently extended using forceps. The inoculum was pipetted into the trachea, and the tongue was held until two breaths were completed. For ascus-enriched preparations of P. murina, asci were isolated from trophic forms using sucrose gradient density centrifugation, according to a method previously described (39).
Pneumocystis viability assay.
P. murina (1 × 104 asci per well, estimated 1:10 ascus-to-trophic-form ratio) was cultured in 96-well round-bottom plates in DMEM plus 10% fetal bovine serum (FBS). Serum was treated by heat inactivation for 30 min at 56°C to deplete complement activity (HI FBS) or left untreated (non-HI FBS). P. murina was treated with affinity-purified Dectin-1:Fc at various concentrations and cultured for 24 h. A viability control of P. murina incubated with control medium was included. Following incubation, the contents of the wells were collected and total RNA was isolated using TRIzol-LS reagent (Life Technologies, Carlsbad, CA). The viability of P. murina was analyzed with real-time PCR measurement of rRNA copy number as described below.
Flow cytometry.
For comparison of different Fc isotypes, P. murina organisms were stained with affinity-purified Dectin-1:Fc and fluorescein isothiocyanate (FITC)-conjugated Fcγ fragment-specific (subclass 1 + 2a + 2b + 3) secondary antibody (Jackson ImmunoResearch, West Grove, PA) and analyzed using an LSRII flow cytometer (BD Biosciences, San Jose, CA). To assess the specificity of Dectin-1:Fc to specific forms of Pneumocystis, ascus-enriched or total preparations of P. murina were stained with Dectin-1:Fc directly conjugated to peridinin chlorophyll protein (PerCP)-Cy5.5 (Abcam, Cambridge, MA) and analyzed with an LSRII flow cytometer. Secondary analysis was performed using FlowJo software (Tree Star, Ashland, OR).
Immunofluorescence.
Pneumocystis samples were heat fixed onto slides and further fixed and permeabilized by immersion in ice-cold methanol. Slides were washed with PBS and blocked using 5% mouse serum and 1% bovine serum albumin in PBS. Samples were stained with Alexa Fluor 555-conjugated Dectin-1:mIgG1 and Dectin-1:mIgG2a Fc (Apex antibody labeling kit; Invitrogen). DAPI (4′,6-diamidino-2-phenylindole)-containing mounting medium (Vector Laboratories, Burlingame, CA) was used to coverslip samples, which were visualized at an ×60 magnification.
Hydrodynamic injection of plasmid DNA.
Expression of Dectin-1 Fc fusion proteins was achieved in vivo by systemic administration of plasmid DNA according to the method of Liu et al. (40). Mice were injected with 10 μg endotoxin-free plasmid DNA in isotonic saline (0.9% [wt/vol] NaCl) or Ringer's solution (0.9% NaCl, 0.03% KCl, and 0.016% CaCl2). A total injection volume of 1 ml per 10 g mouse body weight was injected via tail vein within 5 to 10 s.
Purification of T cell subsets and adoptive transfer.
Spleens from naive C57BL/6J mice were collected, teased apart, and filtered through a 70-μm cell strainer under sterile conditions. CD4+ CD25− cells for adoptive transfer were purified by magnetic bead separation using only the negative selection step of a CD4+ CD62L+ T cell isolation kit to deplete non-CD4 cells (Miltenyi Biotec, Auburn, CA). To induce immune reconstitution syndrome, 3 × 105 purified CD4+ CD25− cells were injected intravenously via tail vein to wild-type (WT) or Rag1−/− mice, 25 days after infection with 2 × 105 P. murina asci and 3 days after hydrodynamic injection. Reconstituted mice were sacrificed 11 days after adoptive transfer.
BALF, serum, and lung tissue collection.
At time of sacrifice, mice were anesthetized with ketamine-xylazine and serum was collected from the posterior vena cava. Tracheas were cannulated, and 1 ml of PBS without calcium and magnesium (PBS-free) was instilled through the cannula and aspirated. Bronchoalveolar lavage fluid (BALF) cells were pelleted by centrifugation at 500 × g, and the supernatant was stored at −80°C for further analysis. Following the collection of lavage fluid, lungs were harvested for RNA isolation, cytokine analysis, and histology. After the right bronchus was tied off, one lobe of the right lung was excised and placed into 1 ml TRIzol (Life Technologies), homogenized, and stored at −80°C until RNA isolation. The remaining lobes of the right lung were placed into 1 ml PBS-free containing Complete protease inhibitor (Roche, Indianapolis, IN) and then homogenized and centrifuged at 12,000 × g for 15 min at 4°C. The supernatant was stored at −80°C for later cytokine analysis. For histological study, the left lung was inflated with buffered zinc formalin (Z-fix; Anatech, Battle Creek, MI) and placed into fixative. Paraffin-embedded sections were stained with Gomori methenamine silver (GMS) and scored blindly for intensity of infection as previously described using a semiquantitative scale, ranging from 1 (rare asci per high-power field) to 4 (asci throughout most alveoli with foamy exudate) (38).
RNA isolation and TaqMan probes and primers for Pneumocystis rRNA.
The assay for determination of P. murina copy number per whole lung has been previously described (38). Briefly, cDNA was synthesized with iScript reverse transcription reagents (Bio-Rad, Hercules, CA), and real-time PCR was performed using primers for the P. murina large-subunit rRNA gene with SsoFast Probes Supermix (Bio-Rad). The threshold cycle values were converted to rRNA copy number by using a standard curve of known copy number of Pneumocystis rRNA as previously described (41).
Pulse oximetry and parameters of lung injury.
Blood oxygen saturation was measured using a MouseOx pulse oximeter with a tail sensor (Starr Life Sciences, Oakmont, PA). Following anesthesia with 100 mg intraperitoneal ketamine/kg of body weight, the tail sensor was placed at the base of the tail and measurements were recorded using MouseOx software. One-minute readings were taken from each mouse, and average values of 10 to 20 stable, error-free measurements over this interval are reported. Total protein in the BALF was assayed with a bicinchoninic acid (BCA) protein assay kit (Pierce, Rockford, IL) per the manufacturer's instructions. Lactate dehydrogenase (LDH) levels in the BALF were analyzed by an LDH activity assay kit (BioVision, Milpitas, CA) per the manufacturer's instructions. Expression levels of Dectin-1:Fc proteins in serum and BALF of Rag1−/− mice were measured using isotype-specific IgG enzyme-linked immunosorbent assay (ELISA) quantitation sets (Bethyl Laboratories, Montgomery, TX).
Cytokine analysis.
BALF and lung homogenate samples were analyzed for protein levels of G-CSF, granulocyte-macrophage colony-stimulating factor (GM-CSF), gamma interferon (IFN-γ), interleukin-10 (IL-10), IL-12(p40), IL-12(p70), IL-13, IL-15, IL-17, IL-1α, IL-1β, IL-2, IL-4, IL-5, IL-6, IL-7, IL-9, IP-10, KC, monocyte chemoattractant protein 1 (MCP-1), MIP-1α, MIP-1β, MIP-2, RANTES, and TNF-α using a Milliplex Map mouse cytokine/chemokine magnetic bead panel (EMD Millipore) on a Bio-Plex 200 instrument (Bio-Rad). The data were analyzed using Bio-Plex Manager software (Bio-Rad). Data are reported as means ± standard errors of the means (SEM).
Statistical analysis.
GraphPad Prism (GraphPad Software, La Jolla, CA) was used to calculate P values using one-way analysis of variance (ANOVA) with a Holm-Sidak multiple-comparison posttest. For testing of nonparametric data with three or more groups, such as P. murina ascus burden, the Kruskal-Wallis test was performed with a Dunn multiple-comparison posttest. For comparison of HI and non-HI FBS in Dectin-1:Fc treatment on P. murina viability, multiple t test comparisons were performed with a Holm-Sidak correction. A P value of ≤0.05 was considered statistically significant.
RESULTS
Characterization of Dectin-1:Fc protein reactivity and structure.
A recombinant Dectin-1 receptor Fc fusion protein containing the extracellular domain of the murine β-glucan receptor Dectin-1 (amino acids 69 to 244), a thrombin-sensitive hexapeptide linker, and the murine IgG1 hinge and CH2 through CH3 domains was previously shown to increase macrophage-dependent killing of P. murina in vitro and enhance host recognition and clearance of P. murina in immunodeficient SCID mice (35). As these findings suggested that FcγR-based targeting of P. murina via cell wall carbohydrate recognition can promote resistance against P. murina pneumonia in the immunodeficient host, we sought to understand the relative contribution of antibody-dependent cell-mediated cytotoxicity (ADCC) and complement-dependent cytotoxicity (CDC) by creating Dectin-1 fusions containing the Fc regions of murine IgG2a, IgG2b, and IgG3. The resulting vectors Dectin-1:mIgG2a, Dectin-1:mIgG2b, and Dectin-1:mIgG3 were used to produce recombinant proteins in transfected HEK293T cells, and we assessed the binding of these Fc fusion proteins to β-(1,3)-d-glucan using surface plasmon resonance measurements (Fig. 1).
Fig 1.
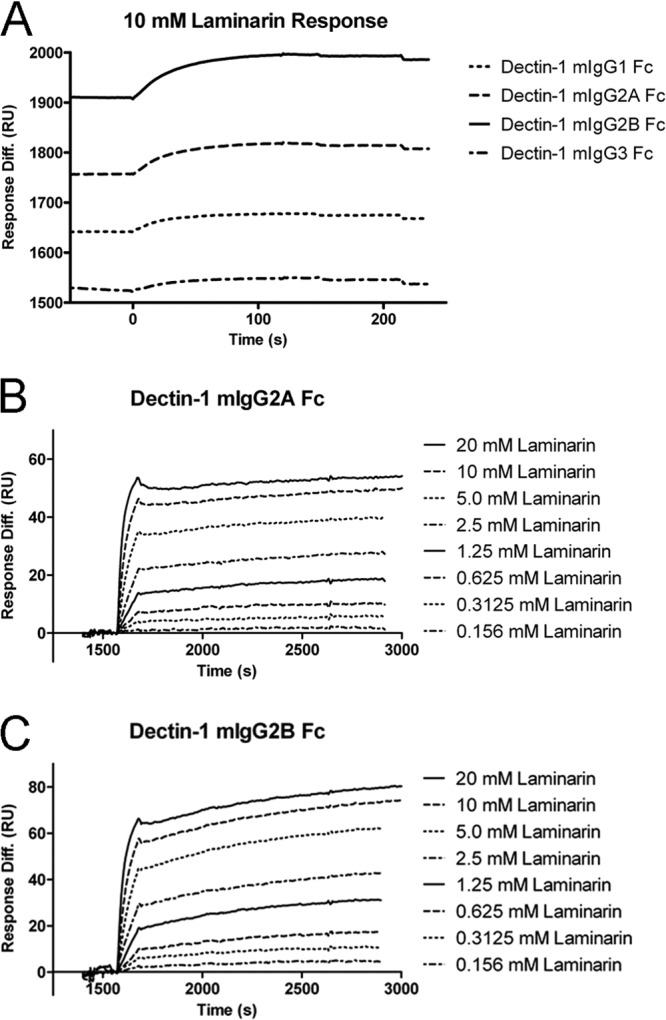
Surface plasmon resonance analysis of the interaction between Dectin-1:Fc and β-glucan. Binding of laminarin to immobilized Dectin-1:Fc was studied in real time using a Biacore 3000 instrument. Rabbit anti-mouse antibodies were chemically immobilized on CM5 chips and used as a capture surface for Dectin-1:Fc. Nonspecific binding was measured using a control flow cell consisting of immobilized capture Ab alone or with an irrelevant IgG2a MAb and was subtracted from each laminarin binding curve. (A) Dectin-1:mIgG1, Dectin-1:mIgG2a, Dectin-1:mIgG2b, and Dectin-1:mIgG3 were captured at various concentrations and measured for response to 10 mM laminarin. Response units (RU) for laminarin were proportional to RU for Dectin-1:Fc loaded for each Ig subtype, indicating similar β-glucan binding capacities (Table 1). (B and C) Detailed kinetic and affinity analysis was performed for Dectin-1:mIgG2a (B) and Dectin-1:mIgG2b (C) using a 2-fold dilution series of laminarin ranging from 20 mM to 0.156 mM. The rate of association (ka), the rate of dissociation (kd), and the equilibrium constants of association (KA) and dissociation (KD) were calculated (Table 2).
Initially, various amounts of each affinity-purified Dectin-1 Fc protein were captured with immobilized anti-mouse antibodies on a CM5 sensor chip (GE Healthcare Life Sciences, Piscataway, NJ) and measured for response to laminarin, a glucan molecule consisting of primarily β-(1,3)-linked glucan. The ratios of laminarin binding response units (RU) per Dectin-1:Fc protein RU loaded were similar for all Fc subtypes, indicating that none of the Fc subtypes grossly interfered with the carbohydrate recognition domain of Dectin-1 (Fig. 1A and Table 1). We further characterized the binding of Dectin-1:mIgG2a and Dectin-1:mIgG2b by performing kinetic and affinity analyses with laminarin in 2-fold dilution series from 20 mM to 0.156 mM (Fig. 1B and C). The rate of association (ka), the rate of dissociation (kd), and the equilibrium constants of association (KA) and dissociation (KD) were calculated (Table 2), demonstrating high-affinity binding of Dectin-Fc proteins to laminarin, similar in quality to our previous results with Dectin-1:mIgG1 Fc (35) and to that of various carbohydrate-directed Abs or lectin receptors for their ligands. In addition, these affinity analyses demonstrate the generation of a stable interaction between laminarin and immobilized Dectin-1:Fc over the course of the kinetic analysis. This binding phenomenon may affect the calculation of the equilibrium constants, as the multiple glucan subunit interactions on each laminarin chain stabilize each other and prevent dissociation of laminarin from the Dectin-1 complex on the surface of the chip.
Table 1.
Dectin-1:Fc binding of different isotypes
| Protein | RU loaded | RU for laminarin | Response ratio |
|---|---|---|---|
| Dectin-1:mIgG1 | 549.9 | 34.3 | 16.03 |
| Dectin-1:mIgG2a | 1,011.8 | 59.1 | 17.12 |
| Dectin-1:mIgG2b | 1,460.4 | 85.4 | 17.1 |
| Dectin-1:mIgG3 | 485.2 | 24.1 | 20.1 |
Table 2.
Rate and affinity constants of Dectin-1:Fc
| Protein | ka (M−1 s−1) | kd (s−1) | KA (M−1) | KD (M) |
|---|---|---|---|---|
| Dectin-1:mIgG2a | 2.10E+03 | 4.40E−06 | 4.80E+08 | 2.10E−09 |
| Dectin-1:mIgG2b | 2.10E+03 | 1.10E−06 | 1.90E+09 | 5.40E−10 |
Dectin-1:Fc masking of β-(1,3)-glucan residues reduces zymosan-induced stimulation of macrophages.
To address the ability of Dectin-1:Fc to modify recognition of particles containing β-(1,3)-d-glucan, such as the cell wall surface of Pneumocystis and other fungi, we evaluated whether Dectin-1:mIgG1 and Dectin-1:mIgG2a could affect RAW 264.7 macrophage stimulation by particulate zymosan. Zymosan is an insoluble cellular wall polysaccharide derived from Saccharomyces cerevisiae, composed mostly of β-glucan and mannan (42). To ensure the specificity of stimulation to Dectin-1 in the assay, we removed all TLR2-activating components of zymosan by treating zymosan with hot alkali to make depleted zymosan (37). RAW cells were stimulated with depleted zymosan (Zym) with or without precoating with a molar excess of Dectin-1:Fc and compared to cells pretreated with the Dectin-1 receptor-blocking monoclonal antibody 2A11 (43). After 18 h of stimulation, production of granulocyte colony-stimulating factor (G-CSF) and tumor necrosis factor alpha (TNF-α) in the supernatant was measured (Fig. 2A and B). The results indicate decreased stimulation by zymosan in the Dectin-1:Fc-treated groups, with significantly lower production of G-CSF than in those treated with both zymosan alone and zymosan plus 2A11 MAb. Dectin-1:Fc treatment of zymosan also significantly reduced macrophage production of TNF-α.
Fig 2.

Effect of Dectin-1:Fc pretreatment of zymosan on stimulation of macrophages. RAW 264.7 cells were stimulated with 50 μg/ml untreated TLR agonist-depleted zymosan (Zym) or with depleted zymosan pretreated with 10 μg/ml Dectin-1:Fc. RAW cells were also pretreated with 5 μg/ml of the Dectin-1-blocking MAb 2A11 as an inhibition control. Cell culture supernatant was assayed at 24 h for the production of granulocyte colony-stimulating factor (A) and tumor necrosis factor (B). Data are expressed as mean pg/ml ± SEM for n = 3 per group. *, P < 0.05; **, P < 0.01; ***, P < 0.001. Using one-way ANOVA with a Holm-Sidak multiple-comparison test.
Dectin-1:mIgG2a Fc binds Pneumocystis organisms and enhances macrophage-independent killing.
Our previous studies using fluorescent deconvolution microscopy and flow cytometry have demonstrated that Dectin-1:mIgG1 Fc binds specifically to the surface of Pneumocystis asci (cyst forms) (35). In this study, we tested and confirmed the binding of the novel Dectin-1:mIgG2a, Dectin-1:mIgG2b, and Dectin-1:mIgG3 Fc fusion proteins to P. murina organisms by flow cytometry (Fig. 3A). When subpopulations of asci and trophic forms were analyzed using flow cytometry, Dectin-1 Fc proteins showed an increased binding to P. murina asci, indicating a greater availability of β-(1,3)-d-glucan in the cell wall of the ascus (Fig. 3B and C). Immunofluorescence staining of whole P. murina with fluorochrome-labeled Dectin-1 Fc also showed specific binding to the ascus (Fig. 3D and E).
Fig 3.

Dectin-1:Fc binds the cell wall of Pneumocystis asci. (A) P. murina organisms were stained with Dectin-1:Fc fusion proteins and a FITC-conjugated anti-mouse IgG secondary antibody and measured using flow cytometry. Staining of each Fc subtype fusion protein is shown as labeled compared to unstained P. murina (underlying histogram). Nonspecific staining of isotype controls was minimal for each subtype (data not shown). (B and C) Binding affinities of Dectin-1:mIgG1 (B) and Dectin-1:mIgG2a (C) for P. murina (PC) asci were also determined by flow cytometry. Populations of ascus-enriched P. murina (black line) and whole P. murina (gray dashed line) were prepared by rate-zonal centrifugation over a sucrose gradient and stained with Dectin-1:Fc directly conjugated to PerCP-Cy5.5. Unstained whole P. murina (gray-shaded histogram) was indistinguishable from unstained ascus-enriched P. murina (not shown). (D and E) Immunofluorescence staining was performed on whole P. murina using Alexa Fluor 555-conjugated Dectin-1:mIgG1 (D) and Dectin-1:mIgG2a (E) and DAPI. Dectin-1:Fc (red) stains the P. murina asci (white arrows), which contain 8 individual DAPI-positive ascospore nuclei (blue). Trophic forms (white arrowheads) are represented by lone nuclei without Dectin-1:Fc staining.
The Dectin-1:mIgG1 Fc protein was previously shown to enhance the macrophage-dependent killing of P. murina in vitro (35). Preopsonization of P. murina organisms with Dectin-1:mIgG1 Fc diminished overall copy numbers by 3-fold in studies with thioglycolate-elicited peritoneal macrophages and by up to 10-fold with alveolar macrophages (35). We sought to determine if Dectin-1 Fc proteins could enhance P. murina killing in vitro without macrophages through a complement-dependent cytotoxic mechanism. We studied decreases in absolute quantities of P. murina mitochondrial large-subunit rRNA (PC mtLSU rRNA) copy numbers as a correlate of in vitro P. murina killing, a methodology validated by previous work (41, 44). P. murina organisms were isolated from infected murine lung homogenates and were treated with Dectin-1:Fc in medium supplemented with fetal bovine serum that was untreated or heat inactivated for 30 min at 56°C to remove complement activity (HI). Dectin-1:mIgG1 Fc did not reduce P. murina rRNA copies in vitro in the absence of macrophages (Fig. 4A). In contrast, Dectin-1:mIgG2a exhibited potent P. murina killing activity at 35 and 14 μg/ml, but only in medium supplemented with non-heat-inactivated serum, indicating a role for complement fixation in the killing process (Fig. 4B). These results are consistent with previous observations that mouse IgG2a is the most efficient IgG at fixing complement and exhibits the most CDC (45).
Fig 4.

Pneumocystis-killing activity of Dectin-1:Fc fusion proteins in vitro. Dectin-1:mIgG1 (A) and Dectin-1:mIgG2a (B) Fc fusion proteins were tested for P. murina (PC) killing in medium containing 10% heat-inactivated (HI) or untreated (Non-HI) fetal bovine serum (FBS). P. murina asci (2 × 105 per well) were cultured in medium alone (PC only) or with various amounts of Dectin-1:Fc. After 24 h, total RNA was harvested and viability was assessed through real-time RT-PCR measurement of P. murina mitochondrial large-subunit (mtLSU) rRNA copy number. All data are reported as means ± SEM for n = 3 per group. *, P < 0.05; **, P < 0.01. Using multiple t tests with a Holm-Sidak correction.
Sustained expression of Dectin-1:Fc via hydrodynamic injection can reduce P. murina asci in B and T cell-deficient Rag1−/− mice.
Following our previous reports of increased macrophage-dependent killing of P. murina in vitro and protection in vivo in a SCID model with Dectin-1:mIgG1 Fc (35) and our current observations with macrophage-independent in vitro killing of P. murina by Dectin-1:mIgG2a Fc, we sought to compare the effects of these molecules in an immunodeficient mouse model of P. murina infection. For this model, we used Rag1−/− mice, which lack B and T cells, and chose to express the Dectin-1:Fc fusion proteins through hydrodynamic injection of plasmid DNA via tail vein. Groups of Rag1−/− mice were injected with Dectin-1:mIgG1 Fc, Dectin-1:mIgG2a Fc, or buffer control on day 0 and inoculated with P. murina on day 1. Blood samples were collected at baseline and at 4, 7, and 10 days to assess protein production, and all mice were sacrificed on day 15 to assess P. murina lung and ascus burden (Fig. 5A). Hydrodynamic injection of 10 μg plasmid DNA resulted in sustained high-level expression of Dectin-1:Fc proteins during the course of infection (Fig. 5B). At 14 days postinfection, total P. murina lung burdens assessed by real-time reverse transcription-PCR (RT-PCR) for P. murina mtLSU rRNA were not significantly different between any groups (Fig. 5C). However, this assay does not discriminate between asci and trophic forms of the organism. In contrast, quantification of asci by histological scoring of Gomori methenamine silver (GMS)-stained lung sections revealed a significant decrease in the P. murina ascus burden in Dectin-1:mIgG1 Fc-injected mice (Fig. 5D).
Fig 5.

Pneumocystis infection in Rag1−/− mice expressing Dectin-1:mIgG1 and Dectin-1:mIgG2a Fc fusion proteins. (A) Rag1−/− mice were treated with a hydrodynamic injection of 10 μg Dectin-1:Fc plasmid DNA (Dectin-1:mIgG1,2a) or buffer control (Rag1−/−) on day 0, infected with P. murina (PC) on day 1, and sacrificed at day 15 to assess P. murina burden. (B) Dectin-1:Fc protein levels in serum of Rag1−/− mice at 4, 7, and 10 days after hydrodynamic injection of 10 μg plasmid DNA. Data are reported as median values plus errors. (C) P. murina organism burden at 2 weeks postinfection, determined by real-time RT-PCR and reported as mtLSU copy number/μg lung RNA. Data are reported as means ± SEM. (D) P. murina ascus burden, determined by qualitative scoring of GMS-stained histological sections of lung tissue. Horizontal lines denote median values. (E) Representative micrographs of lung sections stained with GMS. n = 5 per group. *, P < 0.05; **, P < 0.01; ***, P < 0.001. Using a nonparametric Kruskal-Wallis test with a Dunn multiple-comparison posttest (D).
Dectin-1:Fc decreases markers of inflammation in an immune reconstitution inflammatory syndrome model of P. murina infection.
As fungal β-(1,3)-glucan is known to trigger potent inflammatory responses, altering the glucan surface content of an infectious organism may have strong potential to modulate the host immune response. In the case of Pneumocystis, we have demonstrated that Dectin-1:Fc has an increased binding affinity for the P. murina ascus, which contains a greater proportion of β-(1,3)-glucan in the cell wall (46). We have also shown that macrophages exhibit decreased production of inflammatory cytokines when these glucan residues are masked by soluble Dectin-1:Fc in an in vitro stimulation assay. We hypothesized that these properties along with the increased P. murina-killing activity shown by Dectin-1:mIgG1 and Dectin-1:mIgG2a Fc via macrophage- and complement-dependent mechanisms may provide a therapeutic benefit in a mouse model of immune reconstitution inflammatory syndrome by reducing the generation of damaging inflammatory responses in the lung.
To test this hypothesis, WT or Rag1−/− mice were inoculated with P. murina at day 0 and allowed to progress for 3 weeks of infection, followed by hydrodynamic injection treatment at day 22. WT mice received buffer control only, and Rag1−/− mice were treated with either buffer control, IgG1 Fc control, IgG2a Fc control, Dectin-1:mIgG1 Fc, or Dectin-1:mIgG2a Fc (Fig. 6A). Dectin-1:Fc protein expression was assayed 3 days later, with expression levels in serum averaging 22 and 14 μg/ml for Dectin-1:mIgG1 and Dectin-1:mIgG2a, respectively (Fig. 6B). At this time point, 3 × 105 purified CD4+ CD25− T cells were adoptively transferred to induce immune reconstitution syndrome as previously described (28). At 11 days posttransfer, mice were sacrificed and assessed for markers of inflammation and P. murina burden. Oxygen saturation was measured under anesthesia at the time of sacrifice using a rodent pulse oximeter (Starr Life Sciences, Holliston, MA). Data are reported as percent oxygen saturation relative to uninfected WT controls (Fig. 6C). Reconstituted Rag1−/− mice in the control injection group showed significantly lower oxygen saturation levels at the time of sacrifice than did reconstituted Dectin-1-treated Rag1−/− mice and WT mice that underwent the same infection and adoptive transfer procedure, indicating more severe inflammation and lung pathology (Fig. 6C). To further assess lung injury, total protein content and lactate dehydrogenase (LDH) activity were measured in the bronchoalveolar lavage fluid (BALF). Levels of total protein and LDH were significantly higher in all Rag1−/− groups regardless of treatment with Dectin-1:Fc (Fig. 6D and E). BALF levels of Dectin-1:Fc proteins were also measured at 7 and 14 days after hydrodynamic injection and ranged from 10 to 20 ng/ml. We further assessed lung inflammation by measuring inflammatory cytokine and chemokine levels in the BALF using a bead-based multiplex assay. Following adoptive transfer, immunocompetent WT mice expressed the lowest levels of total protein, LDH, and inflammatory cytokine levels in the BALF. Total lung P. murina burdens and histological P. murina ascus burdens were not significantly different among Rag1−/− groups (Fig. 6F and G) regardless of Dectin-1 Fc treatment. However, several inflammatory cytokines and chemokines were significantly reduced by administration of Dectin-1 Fc in immune-reconstituted Rag1−/− mice, including IL-12(p40) in Dectin-1:mIgG1 treatment, and reduction of KC and MIP-2 for both Dectin-1:Fc subtypes (Fig. 6H). MCP-1 also showed a trend toward reduction with Dectin-1:mIgG1 treatment, but levels of TNF-α were not reduced for either construct (Fig. 6H). We also assessed immune cell numbers and phenotype in the lung during IRIS using intracellular staining of phorbol myristate acetate (PMA)-ionomycin-stimulated lung cells collected at day 11 after adoptive transfer. The predominant T cell response after stimulation appeared to be Th1, with stimulated T cells showing an increase in the percentage and total number of IFN-γ-positive cells but not IL-17 or IL-13 (see Fig. S1A in the supplemental material). Dectin-1:Fc treatment did not result in a significant decrease of this IFN-γ+ population.
Fig 6.

Effect of systemic Dectin-1:mIgG1 and Dectin-1:mIgG2a Fc expression in a model of Pneumocystis (PC)-associated immune reconstitution inflammatory syndrome. (A) WT C57BL/6 and Rag1−/− mice were infected with P. murina on day 0 and treated on day 22 with hydrodynamic injection. WT mice received buffer control, and Rag1−/− mice received buffer control or a 10-μg plasmid injection. Plasmid groups included mIgG1 Fc control, mIgG2a Fc control, Dectin-1:mIgG1 Fc, and Dectin-1:mIgG2a Fc. Immune reconstitution syndrome was induced by adoptive transfer of 3 × 105 purified CD4+ CD25− naive T cells on day 25, and mice were sacrificed at day 36 to assess P. murina burden and markers of inflammation and lung damage. (B) Levels of Dectin-1:Fc protein expression in serum at day 25 (3 days after hydrodynamic injection). (C) Oxygen saturation levels in anesthetized mice, measured by pulse oximeter at time of sacrifice on day 36 and reported as a percentage of the uninfected control. (D) Total protein concentration in bronchoalveolar lavage fluid (BALF) measured by BCA assay. (E) Lactate dehydrogenase (LDH) activity levels in BALF. (F) P. murina organism burden at 36 days postinfection, determined by real-time RT-PCR and reported as mtLSU copy number/μg lung RNA. (G) P. murina ascus burden determined by qualitative scoring of GMS-stained histological sections of lung tissue. (H) Protein levels of KC, MCP-1, IL-12(p40), MIP-2, and TNF-α in BALF measured by multiplex bead array. Data are reported as means ± SEM (B to F and H) or median values (G) for n = 4 to 5 per group. *, P < 0.05; **, P < 0.01; ***, P < 0.001. Using a nonparametric Kruskal-Wallis test with a Dunn multiple-comparison posttest (G) or one-way ANOVA with a Holm-Sidak multiple-comparison test (C to E and H).
DISCUSSION
The β-(1,3)-glucan receptor Dectin-1 plays a pivotal role in recognition and clearance of fungal pathogens such as Pneumocystis (41, 47), Candida albicans (48, 49), Aspergillus fumigatus (36, 50), and Coccidioides species (51, 52). β-Glucans are major structural components of the fungal cell wall, but exposure of β-glucan to the immune system can be masked by mannoproteins and cell wall compositions can differ depending on the life cycle stage of the organism (53). Dectin-1 has been shown to specifically bind to swollen conidia and early germlings of Aspergillus and to the yeast form of Candida albicans (36, 48). In the case of Pneumocystis, the ascus or cyst form has been shown to exhibit a thicker cell wall with higher β-glucan content and greater availability of β-glucan to host binding by Dectin-1 (35, 46). Previous rodent studies have indicated that a purified Pneumocystis ascus preparation induces inflammation and a fulminant PCP infection (54), whereas echinocandin treatment can selectively ablate the cyst population and prevent transmission of infection (55). Another recent publication by Linke et al. has shown that murine hosts mount unique immune responses to the different life forms of P. murina and that the ascus can contribute to the detrimental inflammatory response associated with infection and immune reconstitution syndrome (56). As β-glucan recognition is a critical component of fungal immunity, we have sought to test the effect of systemic expression of recombinant Dectin-1:Fc fusion proteins on host defense and inflammatory response in a murine model of P. murina infection.
To investigate the contributions of different IgG Fc subtypes on efficacy of protection in our model, we constructed fusion proteins of the Dectin-1 carbohydrate recognition domain with the Fc regions of murine IgG1, IgG2a, IgG2b, and IgG3 to form antibody-like molecules that specifically bind β-glucan. We then compared the abilities of the different Fc fusions in their ability to bind laminarin by surface plasmon resonance and found that Fc subtype did not affect carbohydrate binding for any of the constructs (Fig. 1). Next, we tested the ability of Dectin-1:Fc to modify the stimulation of macrophages by zymosan, a β-glucan-rich, particulate cell wall preparation. Precoating TLR-agonist-depleted zymosan with a molar excess of Dectin-1:Fc decreased production of G-CSF and TNF-α upon exposure to RAW cells (Fig. 2). This reduction may result from competition of Dectin-1:Fc with macrophage Dectin-1 for binding of available β-glucan on the surface of zymosan. This competition could not completely abrogate cytokine production, however, as Fcγ receptor-mediated recognition of Dectin-1:Fc-opsonized zymosan can activate macrophages. Previous studies in our lab indicate that blockade of macrophage FcγRII and FcγRIII results in diminished binding and recognition of Dectin-1:Fc-coated particulates (35). Other Dectin-1-independent mechanisms may also contribute to stimulation, as treatment with Dectin-1-blocking MAb 2A11 did not completely reduce cytokine production by unopsonized zymosan. These results are consistent with observations of normal cytokine production in Dectin-1-deficient mice infected with Pneumocystis (47).
Our previous studies in have shown that Dectin-1:mIgG1 Fc can specifically bind to the cell wall of Pneumocystis and increase killing of P. murina by macrophages in vitro (35). In our current study, we found that all Dectin-1:Fc fusion isotypes bound Pneumocystis equally, and increased binding of Dectin-1:Fc to the ascus-enriched population indicated specific recognition of the ascus, which contains a significant portion of β-(1,3)-d-glucan in its cell wall (Fig. 3). Other studies have shown that Dectin-1-deficient mice infected with P. murina harbor a greater ascus burden in the lung early in infection, particularly under immunocompromised conditions, indicating the importance of carbohydrate recognition to killing of the ascus (47).
In a direct killing assay, a Dectin-1:mIgG1 Fc fusion did not mediate killing in the absence of macrophages, whereas Dectin-1:mIgG2a Fc could reduce the viability of P. murina in culture through a complement-dependent process (Fig. 4). Our P. murina preparation assumes a 1:10 ascus-to-trophic-form ratio, but there may be some variation in the total number of trophic forms (57). In addition, asci contain 8 ascospores with separate nuclei and mitochondria, and potentially, reduction of 1 ascus can reduce mitochondrial RNA subunit transcript levels as much as can the reduction of 8 trophic forms. We have tested RNA from ascus-enriched and trophic-form-enriched populations and found their mitochondrial RNA transcript levels to be roughly equivalent for the same amount of total input RNA. This indicates that the assay has the potential to report killing of both life forms but that it may be weighted more toward a reduction of asci. In addition, trophic forms may contain low levels of β-glucan in their cell walls, although at a level that is much reduced from that in asci and less accessible to Dectin-1 binding. The process of culturing P. murina during the killing assay may result in more exposure of β-glucan by trophic forms, rendering them more accessible to killing by Dectin-1 immunoadhesins. The macrophage-independent process of P. murina killing by Dectin-1:mIgG2a is dependent on an intact complement system, as heat inactivation of complement proteins in the fetal bovine serum used for the killing assay abrogates killing. However, the membrane attack complex formed by the complement proteins C6 through C9 has not been shown to directly cause lysis of Pneumocystis and other fungi with large structural cell walls, such as Cryptococcus and Candida species. It may be possible that the membrane attack complex or another component of the complement system, such as C3a, is able to sufficiently perforate or perturb the membrane integrity of Pneumocystis and induce killing, but it is unlikely that the ascus will be completely lysed. Trophic forms may be more susceptible to complement-mediated lysis, which we are continuing to investigate.
In contrast to the in vitro studies, the IgG1 isotype fusion of Dectin-1 was more effective in reducing ascus burden in vivo, with the IgG2a isotype showing a trend toward a greater reduction in total lung burden as measured by P. murina rRNA copy number (Fig. 5). The Dectin-1:mIgG2a Fc construct showed a greater in vitro killing efficacy, perhaps in part due to differences in concentration of the target molecule. Previous studies have shown that systemic administration of a recombinant Fc-conjugated TNF inhibitor can result in functional levels of protein in the lung (58). We have tested blood and BALF samples at the time of sacrifice in the IRIS experiment (14 days after hydrodynamic injection), and levels of both Dectin-1:mIgG1 and Dectin-1:mIgG2a Fc were waning in the blood and BALF. At an earlier time point of 7 days after hydrodynamic injection, there are higher levels of Dectin-1:Fc in the alveolar lumen (10 to 20 ng/ml), but protein availability through the course of a severe infection may contribute to lack of efficacy in vivo. Complement-dependent killing induced by Dectin-1:mIgG2a Fc may require a higher threshold of expression in vivo, which may be overcome through an alternate method of therapeutic delivery.
We also observed a greater reduction in ascus burden by the IgG1 isotype when it was administered prechallenge than when it was administered after established infection. This may be due to better efficacy of the molecule when ascus burden is low in the lung. Both constructs were effective in reducing hypoxemia in the IRIS model despite minimal effects on fungal burden in the lung (Fig. 6). These data indicate that β-glucan- and Dectin-1-mediated signals contribute to hypoxemia during Pneumocystis IRIS, consistent with recent observations implicating the β-glucan-rich ascus in producing a proinflammatory response (56). Possible mechanisms of inflammatory exacerbation include induction of IL-8 secretion and activation of the IL-23/IL-17 axis by Pneumocystis β-glucan (59, 60), as well as the effects of Dectin-1 signaling on Th1 and Th17 differentiation of CD4+ T cells (33, 56, 61, 62), which have been shown to contribute to the risk for IRIS (63, 64). Although intracellular staining of PMA- and ionomycin-stimulated lung cells did not reveal a strong Th17 signature (see Fig. S1A in the supplemental material), antigen-specific stimulation of lung or draining lymph node cells may exhibit greater induction of IL-17. Another potential method for Dectin-1:Fc to exert anti-inflammatory activity is through a recently discovered interaction between FcγRIIB and Dectin-1, in which galactosylated IgG1 immune complexes suppress complement receptor-mediated inflammation (65). However, this mechanism has not been described in antibodies of the IgG2a isotype. Administration of control Fc-only vectors did not improve hypoxemia in our IRIS model, indicating that observed differences with the administration of Dectin-1:Fc were specific to the Dectin-1 moiety (Fig. 6C). Despite improved hypoxemia, the Dectin immunoadhesins did not affect markers of cell death (LDH) or vascular leakage (total protein). There are several possible explanations for these data. One possibility is that these markers of lung injury are Dectin-1 and β-glucan independent. Second, since we did not achieve a significant reduction of P. murina asci before reconstitution in this model, the retained ascus burden may be sufficient to mediate lung injury. Lastly, as these molecules do not affect the burden of the trophic form in the lung, this life form may also contribute to lung injury in this model. In summary, these data provide evidence that immunoadhesins targeting β-glucan may provide a mechanism for treatment of fungal infection and for modulation of the inflammatory response to Pneumocystis and other pathogens.
Supplementary Material
Footnotes
Published ahead of print 8 July 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.00136-13.
REFERENCES
- 1.Morris A, Lundgren JD, Masur H, Walzer PD, Hanson DL, Frederick T, Huang L, Beard CB, Kaplan JE. 2004. Current epidemiology of Pneumocystis pneumonia. Emerg. Infect. Dis. 10:1713–1720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kelley CF, Checkley W, Mannino DM, Franco-Paredes C, Del Rio C, Holguin F. 2009. Trends in hospitalizations for AIDS-associated Pneumocystis jirovecii pneumonia in the United States (1986 to 2005). Chest 136:190–197 [DOI] [PubMed] [Google Scholar]
- 3.Sudjaritruk T, Oberdorfer P, Puthanakit T, Sirisanthana T, Sirisanthana V. 2012. Causes of first hospitalization among 1121 HIV-infected children: comparison of the pre-Pneumocystis jiroveci pneumonia prophylaxis, pre-antiretroviral therapy and antiretroviral therapy periods. Int. J. STD AIDS 23:335–339 [DOI] [PubMed] [Google Scholar]
- 4.Mansharamani NG, Garland R, Delaney D, Koziel H. 2000. Management and outcome patterns for adult Pneumocystis carinii pneumonia, 1985 to 1995: comparison of HIV-associated cases to other immunocompromised states. Chest 118:704–711 [DOI] [PubMed] [Google Scholar]
- 5.McKinnell JA, Cannella AP, Kunz DF, Hook EW, Moser SA, Miller LG, Baddley JW, Pappas PG. 2012. Pneumocystis pneumonia in hospitalized patients: a detailed examination of symptoms, management, and outcomes in human immunodeficiency virus (HIV)-infected and HIV-uninfected persons. Transpl. Infect. Dis. 14:510–518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Phair J, Muñoz A, Detels R, Kaslow R, Rinaldo C, Saah A. 1990. The risk of Pneumocystis carinii pneumonia among men infected with human immunodeficiency virus type 1. Multicenter AIDS Cohort Study Group. N. Engl. J. Med. 322:161–165 [DOI] [PubMed] [Google Scholar]
- 7.Stansell JD, Osmond DH, Charlebois E, LaVange L, Wallace JM, Alexander BV, Glassroth J, Kvale PA, Rosen MJ, Reichman LB, Turner JR, Hopewell PC. 1997. Predictors of Pneumocystis carinii pneumonia in HIV-infected persons. Pulmonary Complications of HIV Infection Study Group. Am. J. Respir. Crit. Care Med. 155:60–66 [DOI] [PubMed] [Google Scholar]
- 8.Shellito J, Suzara VV, Blumenfeld W, Beck JM, Steger HJ, Ermak TH. 1990. A new model of Pneumocystis carinii infection in mice selectively depleted of helper T lymphocytes. J. Clin. Invest. 85:1686–1693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Harmsen AG, Stankiewicz M. 1990. Requirement for CD4+ cells in resistance to Pneumocystis carinii pneumonia in mice. J. Exp. Med. 172:937–945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Roths JB, Marshall JD, Allen RD, Carlson GA, Sidman CL. 1990. Spontaneous Pneumocystis carinii pneumonia in immunodeficient mutant scid mice. Natural history and pathobiology. Am. J. Pathol. 136:1173–1186 [PMC free article] [PubMed] [Google Scholar]
- 11.Hanano R, Reifenberg K, Kaufmann SH. 1996. Naturally acquired Pneumocystis carinii pneumonia in gene disruption mutant mice: roles of distinct T-cell populations in infection. Infect. Immun. 64:3201–3209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.French MA, Lenzo N, John M, Mallal SA, McKinnon EJ, James IR, Price P, Flexman JP, Tay-Kearney ML. 2000. Immune restoration disease after the treatment of immunodeficient HIV-infected patients with highly active antiretroviral therapy. HIV Med. 1:107–115 [DOI] [PubMed] [Google Scholar]
- 13.Cheng VC, Yuen KY, Chan WM, Wong SS, Ma ES, Chan RM. 2000. Immunorestitution disease involving the innate and adaptive response. Clin. Infect. Dis. 30:882–892 [DOI] [PubMed] [Google Scholar]
- 14.Shelburne SA, Hamill RJ, Rodriguez-Barradas MC, Greenberg SB, Atmar RL, Musher DW, Gathe JC, Visnegarwala F, Trautner BW. 2002. Immune reconstitution inflammatory syndrome: emergence of a unique syndrome during highly active antiretroviral therapy. Medicine 81:213–227 [DOI] [PubMed] [Google Scholar]
- 15.Shelburne SA, Visnegarwala F, Darcourt J, Graviss EA, Giordano TP, White AC, Hamill RJ. 2005. Incidence and risk factors for immune reconstitution inflammatory syndrome during highly active antiretroviral therapy. AIDS 19:399–406 [DOI] [PubMed] [Google Scholar]
- 16.Zolopa A, Andersen J, Powderly W, Sanchez A, Sanne I, Suckow C, Hogg E, Komarow L. 2009. Early antiretroviral therapy reduces AIDS progression/death in individuals with acute opportunistic infections: a multicenter randomized strategy trial. PLoS One 4:e5575. 10.1371/journal.pone.0005575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Grant PM, Komarow L, Andersen J, Sereti I, Pahwa S, Lederman MM, Eron J, Sanne I, Powderly W, Hogg E, Suckow C, Zolopa A. 2010. Risk factor analyses for immune reconstitution inflammatory syndrome in a randomized study of early vs. deferred ART during an opportunistic infection. PLoS One 5:e11416. 10.1371/journal.pone.0011416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Achenbach CJ, Harrington RD, Dhanireddy S, Crane HM, Casper C, Kitahata MM. 2012. Paradoxical immune reconstitution inflammatory syndrome in HIV-infected patients treated with combination antiretroviral therapy after AIDS-defining opportunistic infection. Clin. Infect. Dis. 54:424–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sun H-Y, Singh N. 2011. Opportunistic infection-associated immune reconstitution syndrome in transplant recipients. Clin. Infect. Dis. 53:168–176 [DOI] [PubMed] [Google Scholar]
- 20.Sun H-Y, Singh N. 2009. Immune reconstitution inflammatory syndrome in non-HIV immunocompromised patients. Curr. Opin. Infect. Dis. 22:394–402 [DOI] [PubMed] [Google Scholar]
- 21.Barry SM, Lipman MCI, Deery AR, Johnson MA, Janossy G. 2002. Immune reconstitution pneumonitis following Pneumocystis carinii pneumonia in HIV-infected subjects. HIV Med. 3:207–211 [DOI] [PubMed] [Google Scholar]
- 22.Koval CE, Gigliotti F, Nevins D, Demeter LM. 2002. Immune reconstitution syndrome after successful treatment of Pneumocystis carinii pneumonia in a man with human immunodeficiency virus type 1 infection. Clin. Infect. Dis. 35:491–493 [DOI] [PubMed] [Google Scholar]
- 23.Takahashi T, Nakamura T, Iwamoto A. 2002. Reconstitution of immune responses to Pneumocystis carinii pneumonia in patients with HIV infection who receive highly active antiretroviral therapy. Res. Commun. Mol. Pathol. Pharmacol. 112:59–67 [PubMed] [Google Scholar]
- 24.Jagannathan P, Davis E, Jacobson M, Huang L. 2009. Life-threatening immune reconstitution inflammatory syndrome after Pneumocystis pneumonia: a cautionary case series. AIDS 23:1794–1796 [DOI] [PubMed] [Google Scholar]
- 25.Wu AKL, Cheng VCC, Tang BSF, Hung IFN, Lee RA, Hui DS, Yuen KY. 2004. The unmasking of Pneumocystis jiroveci pneumonia during reversal of immunosuppression: case reports and literature review. BMC Infect. Dis. 4:57. 10.1186/1471-2334-4-57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Otahbachi M, Nugent K, Buscemi D. 2007. Granulomatous Pneumocystis jiroveci pneumonia in a patient with chronic lymphocytic leukemia: a literature review and hypothesis on pathogenesis. Am. J. Med. Sci. 333:131–135 [DOI] [PubMed] [Google Scholar]
- 27.Wright TW, Gigliotti F, Finkelstein JN, McBride JT, An CL, Harmsen AG. 1999. Immune-mediated inflammation directly impairs pulmonary function, contributing to the pathogenesis of Pneumocystis carinii pneumonia. J. Clin. Invest. 104:1307–1317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McKinley L, Logar AJ, McAllister F, Zheng M, Steele C, Kolls JK. 2006. Regulatory T cells dampen pulmonary inflammation and lung injury in an animal model of pneumocystis pneumonia. J. Immunol. 177:6215–6226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bhagwat SP, Wright TW, Gigliotti F. 2010. Anti-CD3 antibody decreases inflammation and improves outcome in a murine model of Pneumocystis pneumonia. J. Immunol. 184:497–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Drummond RA, Brown GD. 2011. The role of Dectin-1 in the host defence against fungal infections. Curr. Opin. Microbiol. 14:392–399 [DOI] [PubMed] [Google Scholar]
- 31.Singh N. 2008. Novel immune regulatory pathways and their role in immune reconstitution syndrome in organ transplant recipients with invasive mycoses. Eur. J. Clin. Microbiol. Infect. Dis. 27:403–408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Qi C, Cai Y, Gunn L, Ding C, Li B, Kloecker G, Qian K, Vasilakos J, Saijo S, Iwakura Y, Yannelli JR, Yan J. 2011. Differential pathways regulating innate and adaptive antitumor immune responses by particulate and soluble yeast-derived β-glucans. Blood 117:6825–6836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.LeibundGut-Landmann S, Gross O, Robinson MJ, Osorio F, Slack EC, Tsoni SV, Schweighoffer E, Tybulewicz V, Brown GD, Ruland J, Reis e Sousa C. 2007. Syk- and CARD9-dependent coupling of innate immunity to the induction of T helper cells that produce interleukin 17. Nat. Immunol. 8:630–638 [DOI] [PubMed] [Google Scholar]
- 34.Leibundgut-Landmann S, Osorio F, Brown GD, Reis e Sousa C. 2008. Stimulation of dendritic cells via the Dectin-1/Syk pathway allows priming of cytotoxic T-cell responses. Blood 112:4971–4980 [DOI] [PubMed] [Google Scholar]
- 35.Rapaka RR, Goetzman ES, Zheng M, Vockley J, McKinley L, Kolls JK, Steele C. 2007. Enhanced defense against Pneumocystis carinii mediated by a novel Dectin-1 receptor Fc fusion protein. J. Immunol. 178:3702–3712 [DOI] [PubMed] [Google Scholar]
- 36.Steele C, Rapaka RR, Metz A, Pop SM, Williams DL, Gordon S, Kolls JK, Brown GD. 2005. The beta-glucan receptor Dectin-1 recognizes specific morphologies of Aspergillus fumigatus. PLoS Pathog. 1:e42. 10.1371/journal.ppat.0010042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gantner BN, Simmons RM, Canavera SJ, Akira S, Underhill DM. 2003. Collaborative induction of inflammatory responses by Dectin-1 and Toll-like receptor 2. J. Exp. Med. 197:1107–1117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zheng M, Shellito JE, Marrero L, Zhong Q, Julian S, Ye P, Wallace V, Schwarzenberger P, Kolls JK. 2001. CD4+ T cell-independent vaccination against Pneumocystis carinii in mice. J. Clin. Invest. 108:1469–1474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lim SK, Eveland WC, Porter RJ. 1973. Development and evaluation of a direct fluorescent antibody method for the diagnosis of Pneumocystis carinii infections in experimental animals. Appl. Microbiol. 26:666–671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu F, Song Y, Liu D. 1999. Hydrodynamics-based transfection in animals by systemic administration of plasmid DNA. Gene Ther. 6:1258–1266 [DOI] [PubMed] [Google Scholar]
- 41.Steele C, Marrero L, Swain S, Harmsen AG, Zheng M, Brown GD, Gordon S, Shellito JE, Kolls JK. 2003. Alveolar macrophage-mediated killing of Pneumocystis carinii f. sp. muris involves molecular recognition by the Dectin-1 beta-glucan receptor. J. Exp. Med. 198:1677–1688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Di Carlo FJ, Fiore JV. 1958. On the composition of zymosan. Science 127:756–757 [DOI] [PubMed] [Google Scholar]
- 43.Brown GD, Taylor PR, Reid DM, Willment JA, Williams DL, Martinez-Pomares L, Wong SYC, Gordon S. 2002. Dectin-1 is a major beta-glucan receptor on macrophages. J. Exp. Med. 196:407–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McAllister F, Steele C, Zheng M, Shellito JE, Kolls JK. 2005. In vitro effector activity of Pneumocystis murina-specific T-cytotoxic-1 CD8+ T cells: role of granulocyte-macrophage colony-stimulating factor. Infect. Immun. 73:7450–7457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Leatherbarrow RJ, Dwek RA. 1984. Binding of complement subcomponent C1q to mouse IgG1, IgG2a and IgG2b: a novel C1q binding assay. Mol. Immunol. 21:321–327 [DOI] [PubMed] [Google Scholar]
- 46.Matsumoto Y, Matsuda S, Tegoshi T. 1989. Yeast glucan in the cyst wall of Pneumocystis carinii. J. Protozool. 36:21S–22S [DOI] [PubMed] [Google Scholar]
- 47.Saijo S, Fujikado N, Furuta T, Chung S, Kotaki H, Seki K, Sudo K, Akira S, Adachi Y, Ohno N, Kinjo T, Nakamura K, Kawakami K, Iwakura Y. 2007. Dectin-1 is required for host defense against Pneumocystis carinii but not against Candida albicans. Nat. Immunol. 8:39–46 [DOI] [PubMed] [Google Scholar]
- 48.Gantner BN, Simmons RM, Underhill DM. 2005. Dectin-1 mediates macrophage recognition of Candida albicans yeast but not filaments. EMBO J. 24:1277–1286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Netea MG, Gow NAR, Munro CA, Bates S, Collins C, Ferwerda G, Hobson RP, Bertram G, Hughes HB, Jansen T, Jacobs L, Buurman ET, Gijzen K, Williams DL, Torensma R, McKinnon A, MacCallum DM, Odds FC, Van der Meer JWM, Brown AJP, Kullberg BJ. 2006. Immune sensing of Candida albicans requires cooperative recognition of mannans and glucans by lectin and Toll-like receptors. J. Clin. Invest. 116:1642–1650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hohl TM, Van Epps HL, Rivera A, Morgan LA, Chen PL, Feldmesser M, Pamer EG. 2005. Aspergillus fumigatus triggers inflammatory responses by stage-specific beta-glucan display. PLoS Pathog. 1:e30. 10.1371/journal.ppat.0010030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Viriyakosol S, Fierer J, Brown GD, Kirkland TN. 2005. Innate immunity to the pathogenic fungus Coccidioides posadasii is dependent on Toll-like receptor 2 and Dectin-1. Infect. Immun. 73:1553–1560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Del Pilar Jiménez-A M, Viriyakosol S, Walls L, Datta SK, Kirkland T, Heinsbroek SEM, Brown G, Fierer J. 2008. Susceptibility to Coccidioides species in C57BL/6 mice is associated with expression of a truncated splice variant of Dectin-1 (Clec7a). Genes Immun. 9:338–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wheeler RT, Fink GR. 2006. A drug-sensitive genetic network masks fungi from the immune system. PLoS Pathog. 2:e35. 10.1371/journal.ppat.0020035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lasbury ME, Durant PJ, Lee C-H. 2003. Pneumocystis carinii cyst is associated with inflammation in the host. J. Eukaryot. Microbiol. 50(Suppl):639–640 [DOI] [PubMed] [Google Scholar]
- 55.Cushion MT, Linke MJ, Ashbaugh A, Sesterhenn T, Collins MS, Lynch K, Brubaker R, Walzer PD. 2010. Echinocandin treatment of pneumocystis pneumonia in rodent models depletes cysts leaving trophic burdens that cannot transmit the infection. PLoS One 5:e8524. 10.1371/journal.pone.0008524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Linke MJ, Ashbaugh A, Collins MS, Lynch K, Cushion MT. 2013. Characterization of a distinct host response profile to Pneumocystis murina asci during clearance of pneumocystis pneumonia. Infect. Immun. 81:984–995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang J, Gigliotti F, Maggirwar S, Johnston C, Finkelstein JN, Wright TW. 2005. Pneumocystis carinii activates the NF-kappaB signaling pathway in alveolar epithelial cells. Infect. Immun. 73:2766–2777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kolls JK, Lei D, Nelson S, Summer WR, Greenberg S, Beutler B. 1995. Adenovirus-mediated blockade of tumor necrosis factor in mice protects against endotoxic shock yet impairs pulmonary host defense. J. Infect. Dis. 171:570–575 [DOI] [PubMed] [Google Scholar]
- 59.Carmona EM, Lamont JD, Xue A, Wylam M, Limper AH. 2010. Pneumocystis cell wall beta-glucan stimulates calcium-dependent signaling of IL-8 secretion by human airway epithelial cells. Respir. Res. 11:95. 10.1186/1465-9921-11-95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Carmona EM, Kottom TJ, Hebrink DM, Moua T, Singh R-D, Pagano RE, Limper AH. 2012. Glycosphingolipids mediate pneumocystis cell wall β-glucan activation of the IL-23/IL-17 axis in human dendritic cells. Am. J. Respir. Cell Mol. Biol. 47:50–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gringhuis SI, Den Dunnen J, Litjens M, Van der Vlist M, Wevers B, Bruijns SCM, Geijtenbeek TBH. 2009. Dectin-1 directs T helper cell differentiation by controlling noncanonical NF-kappaB activation through Raf-1 and Syk. Nat. Immunol. 10:203–213 [DOI] [PubMed] [Google Scholar]
- 62.Espinosa V, Rivera A. 2012. Cytokines and the regulation of fungus-specific CD4 T cell differentiation. Cytokine 58:100–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Antonelli LRV, Mahnke Y, Hodge JN, Porter BO, Barber DL, DerSimonian R, Greenwald JH, Roby G, Mican J, Sher A, Roederer M, Sereti I. 2010. Elevated frequencies of highly activated CD4+ T cells in HIV+ patients developing immune reconstitution inflammatory syndrome. Blood 116:3818–3827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Grant PM, Komarow L, Lederman MM, Pahwa S, Zolopa AR, Andersen J, Asmuth DM, Devaraj S, Pollard RB, Richterman A, Kanthikeel S, Sereti I. 2012. Elevated interleukin 8 and T-helper 1 and T-helper 17 cytokine levels prior to antiretroviral therapy in participants who developed immune reconstitution inflammatory syndrome during ACTG A5164. J. Infect. Dis. 206:1715–1723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Karsten CM, Pandey MK, Figge J, Kilchenstein R, Taylor PR, Rosas M, McDonald JU, Orr SJ, Berger M, Petzold D, Blanchard V, Winkler A, Hess C, Reid DM, Majoul IV, Strait RT, Harris NL, Köhl G, Wex E, Ludwig R, Zillikens D, Nimmerjahn F, Finkelman FD, Brown GD, Ehlers M, Köhl J. 2012. Anti-inflammatory activity of IgG1 mediated by Fc galactosylation and association of FcγRIIB and Dectin-1. Nat. Med. 10.1038/nm.2862 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.