

Abstract
Macrophages are multifunctional cells that are active in TH1- and TH2-mediated responses. In this study, we demonstrate that human and mouse macrophages collaborate with neutrophils and complement to kill the parasite Strongyloides stercoralis in vitro. Infection of mice with worms resulted in the induction of alternatively activated macrophages (AAMϕ) within the peritoneal cavity. These cells killed the worms in vivo and collaborated with neutrophils and complement during the in vitro killing process. AAMϕ generated in vitro killed larvae more rapidly than naive macrophages, which killed larvae after a longer time period. In contrast, classically activated macrophages were unable to kill larvae either in vitro or in vivo. This study adds macrophages to the armamentarium of immune components that function in elimination of parasitic helminths and demonstrate a novel function by which AAMϕ control large extracellular parasites.
INTRODUCTION
Macrophages play an integral part in both pro- and anti-inflammatory responses, as well as in maintaining homeostasis. These cells exhibit a multiplicity of functions, including phagocytosis (1), antigen presentation (2), immune regulation (3), and pathogen clearance (4, 5). The plasticity of macrophages is most evident in type 1 helper T cell (TH1) responses, where classically activated macrophages (CAMϕ) are induced, and type 2 helper T cell (TH2) responses, where alternatively activated macrophages (AAMϕ) are commonly found. CAMϕ induced by microbial signals and gamma interferon (IFN-γ) are characterized by the expression of NOSII, PD-L1 (6), and other proinflammatory cytokines, such as tumor necrosis factor (TNF) and interleukin-6 (IL-6) (7). While these macrophages are highly immunomodulatory and antitumorigenic, they also function directly in the control of bacterial and protozoan pathogens (8, 9). AAMϕ, induced by IL-4/IL-13, have become the hallmark of TH2-mediated allergy, asthma, and parasite infections. These macrophages are easily distinguished by their expression of the signature markers resistin-like molecule α (Relm-α), Arginase-1 (Arg-1), and YM-1 (10), as well as the more ubiquitous costimulatory molecule PD-L2 (6). In addition to their tumor-promoting properties (11), AAMϕ exhibit several novel functions, such as proliferation (12), wound healing (13), thermogenesis (14), and metabolic regulation (15). The role of AAMϕ in response to parasitic species varies from mediating the TH2 response during helminth infections (10) to the repair of parasite-inflicted wounds (13). Despite the existence of AAMϕ in hosts with parasitic infections, the exact role these cells play during parasite killing has not been elucidated.
Reports on immunity to the parasitic nematode Strongyloides stercoralis in humans are limited. Although human macrophages and neutrophils adhere to larvae through a complement-dependent process, these cells were unable to kill the parasite (16). In mice, several components of protective primary and secondary immunity to the parasite have been described (17). Molecules from larval S. stercoralis directly recruit granulocytes to the parasite microenvironment (18–20), where they kill the parasite in collaboration with complement component C3 (21), eosinophil major basic protein, and neutrophil-derived myeloperoxidase (22, 23). The cellular composition of the parasite microenvironment in naive mice consists of high numbers of neutrophils, eosinophils, and macrophages (22). However, the role that macrophages play during the primary and secondary immune responses has not been explored.
In this study, we demonstrate that human and mouse macrophages collaborate with neutrophils and complement to kill the parasite in vitro. In mice, S. stercoralis infection resulted in the induction of AAMϕ within the peritoneal cavity, which killed the parasites in vivo and collaborated with neutrophils and complement to kill the worms in vitro. AAMϕ derived in vitro rapidly and efficiently killed larvae within 3 days, whereas naive bone marrow-derived macrophages (BMDM) killed larvae after 7 days. In contrast, CAMϕ were unable to kill larvae either in vitro or in vivo. This study adds macrophages to the armamentarium of immune components required for elimination of parasitic helminths.
MATERIALS AND METHODS
Mice.
Wild-type C57BL/6J, IL-4−/−, Arg1flox/flox, and LysMCre mice were purchased from Jackson Laboratory (Bar Harbor, ME) at 6 to 8 weeks of age. The Arg1flox/flox and LysMCre mice were crossed to generate Arg1flox/flox;LysMCre mice (8). Retnla-deficient (Relm-α−/−) mice (11) were generated at Regeneron Pharmaceuticals using VelociGene technology (24). IL-4Rα−/− mice were provided by Debroski R. Herbert (University of California–San Francisco, San Francisco, CA). Secretory IgM-deficient mice (μS−/−) were provided by Troy D. Randall (University of Rochester Medical Center, Rochester, NY). IL-4−/−, IL-4Rα−/−, Relm-α−/−, μS−/−, Arg1flox/flox, and Arg1flox/LysMCre mice all were bred at the Thomas Jefferson University Laboratory Animal Sciences facility (Philadelphia, PA). Mice were housed in ventilated cages in a specific-pathogen-free facility. All animal experiments were conducted in compliance with the guidelines set forth by the Institutional Animal Care and Use Committee at Thomas Jefferson University.
S. stercoralis larvae.
S. stercoralis larvae (L3) were obtained from 7-day-old charcoal-coprocultures made from the stool of infected laboratory dogs housed at the University of Pennsylvania School of Veterinary Medicine. The larvae were collected as previously described (25) and washed five times in a 1:1 mixture of NCTC-135 and Iscove's modified Dulbecco's medium (Sigma) supplemented with 100 U/ml penicillin plus 100 μg/ml streptomycin (Cellgro), 0.1 mg/ml gentamicin (Invitrogen), and 0.25 mg/ml levofloxacin (Levaquin; Ortho-McNeil).
Diffusion chambers, immunization, and challenge infections.
Cell-impermeable diffusion chambers were constructed as previously described (23), using 0.1-μm-pore-size membranes glued to 14-mm Lucite rings (Millipore). Mice were immunized with 5,000 larvae by subcutaneous injection, followed by a booster immunization on day 14. The mice were challenged on day 21 by surgically implanting cell-impermeable diffusion chambers containing 50 larvae and cells, as indicated, subcutaneously in the dorsal flank of naive and immunized mice. All surgical procedures were performed on mice anesthetized with isoflurane (Webster Veterinary). Diffusion chambers were recovered from the mice between 1 and 7 days postchallenge, and larval survival was assessed on the basis of larval motility and morphology. The cells recovered from diffusion chambers were counted using a hemocytometer, and cellular differentials were performed by centrifugation onto slides using a Cytospin 3 apparatus (Shandon) and staining with DiffQuik (Baxter Healthcare).
Human cell isolation.
Human neutrophil (PMN) and peripheral blood mononuclear cells (PBMC) were isolated from the blood of healthy donors. The blood was layered onto a Lympholyte-poly column (Cedarlane), and following centrifugation, the plasma, PBMC, and PMN layers were individually collected. Contaminating erythrocytes were removed from the PMN layer using lysis buffer (BD Biosciences). The purity of PMN preparations was greater than 90%, and PBMC were composed of 65% lymphocytes, 30% monocytes, and 5% PMN. Human macrophages were generated from CD34-negative stem cell fractions from granulocyte colony-stimulating factor (G-CSF)-elicited donors (26). Erythrocytes were lysed from the fractions prior to monocyte adherence for 2 h at 37°C on tissue culture-treated petri dishes (Corning). Plates were then gently washed to remove nonadherent cells, and monocytes were allowed to differentiate for 7 days in RPMI (Cellgro) medium containing human serum (Cedarlane) and 20 ng/ml of human macrophage-CSF (M-CSF; eBioscience). Macrophages were harvested for in vitro experiments using TrypLE (Invitrogen). All human studies were conducted in compliance with the guidelines set forth by the Institutional Review Board at Thomas Jefferson University.
Mouse macrophage culture and differentiation.
Macrophages were cultured by isolating bone marrow from the femurs, tibias, and humeri of naive mice. Erythrocytes were eliminated from the single-cell suspension by hypotonic lysis, and cells were cultured in Dulbecco's modified Eagle medium (DMEM) containing l-glutamine, 25% heat-inactivated fetal bovine serum (FBS) (Cellgro), 25% L929 cell-conditioned medium as a source of M-CSF, sodium pyruvate, 100 U/ml penicillin, 100 μg/ml streptomycin, and 50 μM 2-mercaptoethanol (Invitrogen). Macrophages were harvested for in vitro experiments on day 5. To induce macrophage differentiation, macrophage cultures were treated overnight on day 5 with 25 ng/ml of IL-4 (PeproTech) to induce AAMϕ or with 5 ng/ml IFN-γ (PeproTech) and 50 ng/ml of lipopolysaccharide (LPS) (Sigma) to induce CAMϕ.
Mouse neutrophil isolation.
Mouse PMN were isolated as previously described (23) by harvesting bone marrow from the femurs, tibias, and humeri of naive C57BL/6J mice. The cells were resuspended in a 45% Percoll solution and layered onto a Percoll gradient column of 80 and 62% Percoll. The cell layer at the 80 and 62% Percoll interface was then stained with anti-B220 and anti-Thy1.2 magnetically activated cell sorting (MACS) beads (Miltenyi Biotec) prior to negative selection using MACS LS columns (Miltenyi Biotec). To eliminate monocytes, the cells were placed on untreated petri dishes for 1 h at 37°C in RPMI supplemented with 10% heat-inactivated FBS. Nonadherent cells were removed, and the percentage of neutrophils was determined. The purity of the neutrophil preparations was consistently greater than 95%.
PEC isolation.
Mice were injected in the subcutaneous tissue of the neck with either 5,000 larvae or, as a control, culture medium. Peritoneal exudate cells (PEC) were collected by peritoneal lavage 7 days postinjection from these mice. The monocyte and macrophage population was further enriched by resuspending PEC in a 45% Percoll solution and placing the solution onto a gradient column of 80 and 62% Percoll. The cells at the 62 and 45% Percoll interface were collected, washed, and used for in vitro and in vivo assays. The enrichment process increased the purity of the cells from 80 to 90% monocytes and macrophages.
In vitro larval killing assay.
In vitro killing assays were performed by plating cohorts of 50 larvae into 96-well plates or 96-well Transwell plates (Millipore) with an insert containing 0.45-μm-pore-size membranes. Normal mouse serum collected from naive C57BL/6J mice via exsanguination or cardiac puncture was immediately filter sterilized prior to freezing. Serum was added to plates, as a source of complement, at a concentration of 25% of the total well volume. Macrophages (1 × 106) and PMN (4 × 106) were added to select wells in a final volume of 250 μl with RPMI (Cellgro), supplemented with 10% heat-inactivated FBS and 100 U/ml penicillin plus 100 μg/ml streptomycin (Cellgro), and incubated for 22 h. The contents of the wells were then assessed for larval survival and cell viability. The concentration and proportion of macrophages and neutrophils used in the in vitro larval killing assay were determined after cell titrations and were consistent with the cell numbers required for larval killing by neutrophils in cell-impermeable diffusion chambers in vivo (22).
ELISA.
Serum IgM and IgG levels were measured by enzyme-linked immunosorbent assay (ELISA). Maxisorp 96-well plates (Nunc) were coated with 2 μg of the deoxycholate (DOC) soluble crude larval antigen (21) overnight at 4°C. Plates were blocked with borate buffer solution (0.17 M boric acid, 0.12 M NaCl, 0.05% Tween 20, 0.025% bovine serum albumin, 1 mM EDTA, pH 8.2) at room temperature for 1 h. The samples were serially diluted in borate buffer solution overnight at 4°C. Plates were incubated with biotinylated goat anti-mouse IgG or IgM (BD Bioscience) for 1 h at room temperature. The plates were then incubated with ExtrAvidin peroxidase (Sigma) for 30 min at room temperature. The peroxidase substrate ABTS [2,2′-azino-di-(3-ethylbenzthiazoline-6-sulfonate)] (Kirkegaard & Perry Laboratories) was added, and the optical density (OD) at 405 nm was read using an iMark microplate absorbance reader (Bio-Rad).
Flow cytometry.
Surface and intracellular staining were performed on cells blocked with 20% normal mouse serum and 1 μg of TruStain FcX (93; BioLegend) for 10 min. They were stained for surface expression of F4/80 (BM8; eBioscience) using an allophycocyanin (APC)-conjugated antibody or PD-L1 (MIH5; eBioscience) and PD-L2 (TY25; BD Biosciences) using a phycoerythrin (PE)-conjugated antibody for an additional 20 min. The cells were washed and reconstituted in phosphate-buffered saline (PBS) supplemented with 1% bovine serum albumin (BSA) (Sigma). Cells were reconstituted in a final concentration of 2% paraformaldehyde for 10 min at room temperature. The cells were then fixed and permeabilized using a BD Cytofix/Cytoperm kit (BD Biosciences). Intracellular staining using primary antibodies specific to RELM-α (PeproTech) or NOSII (Polyclonal; BD Biosciences) was performed at 4°C for 45 min. The cells were washed in permeabilization wash buffer and stained with the secondary antibody anti-rabbit IgG Alexa Fluor 488-conjugated antibody (H+L; Invitrogen) for 30 min. Cells were analyzed on a FACSCalibur (BD Biosciences), and data were analyzed using FlowJo v.7 software (Tree Star).
Statistics.
In vivo experiments consisted of at least 5 mice per group unless otherwise stated, and independent experiments were performed at least twice, with each experiment having similar outcomes. Data are presented as means ± standard deviations (SD). Statistical analysis was by multivariate general linear hypothesis analysis of variance (ANOVA) using SYSTAT v.11 software (Cranes Software). Fisher's least significant difference test was performed for post hoc analysis, and P values of less than 0.05 were considered significant.
RESULTS
Human macrophages and neutrophils require complement activation for larval killing.
The capacity of human immune cells to kill S. stercoralis was investigated by culturing human neutrophils and PBMC purified from healthy donors with infective-stage larvae and normal human serum. After 48 h, larvae cultured with neutrophils or PBMC alone had greater than 90% survival. The combination of neutrophils and PBMC resulted in a 66% reduction in parasite survival (Fig. 1A). When heat-inactivated serum, which lacks active complement components, was added to cultures in lieu of normal serum, larval killing by human cells was greatly inhibited (Fig. 1A). Human neutrophils combined with macrophages, instead of PBMC, resulted in a 90% reduction in larval survival after 48 h, suggesting that macrophages are the critical cell type in PBMC (Fig. 1B).
Fig 1.
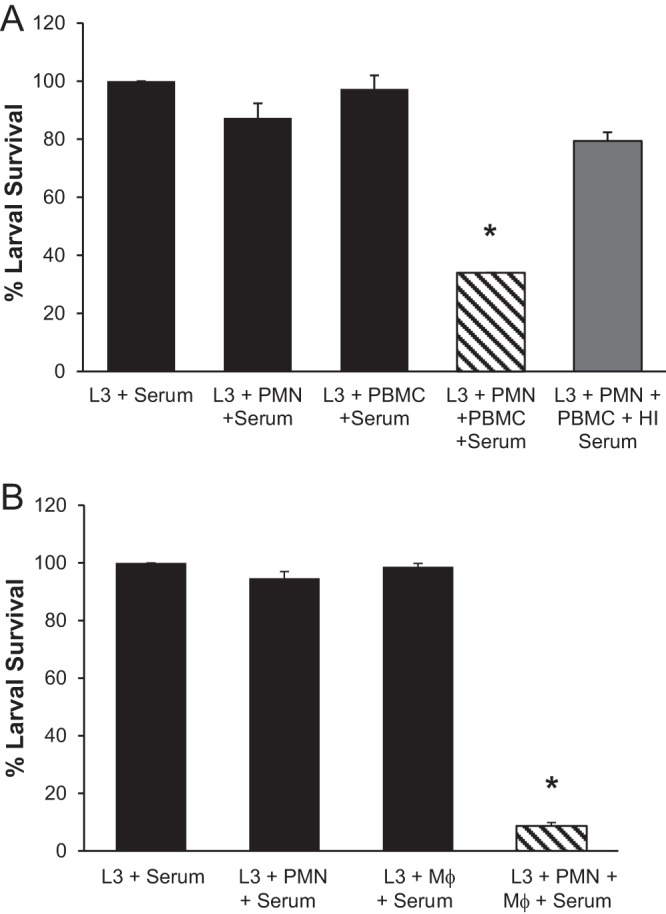
Human macrophages, neutrophils, and complement all are required for larval killing in vitro. (A) Human peripheral blood mononuclear cells (PBMC) cultured with neutrophils (PMN) and larvae (L3) in the presence of normal or heat-inactivated (HI) human serum. (B) Human macrophages (Mϕ) were cultured with PMN, larvae, and normal human serum. Larval survival was assessed after 48 h. Data are representative of at least 3 independent experiments and are depicted as means ± SD. *, P < 0.001 compared to survival of larvae with serum.
Mouse macrophages kill larvae in vitro and in vivo.
To assess the role of macrophages and neutrophils during larval killing in mice, we examined the functionality of naive mouse BMDM both in vitro and in vivo. BMDM and neutrophils were cultured with larvae and normal mouse serum for 24 h. The survival of larvae cultured with only macrophages or neutrophils was greater than 95% (Fig. 2A). Larval killing was observed when both neutrophils and macrophages were placed in culture with larvae, resulting in a 71% reduction in parasite survival. With the substitution of heat-inactivated mouse serum, larval killing was completely ablated, demonstrating that larval killing by mouse cells also depends on the presence of active complement components.
Fig 2.

Mouse macrophages kill S. stercoralis larvae during the primary and secondary immune responses. Macrophages (Mϕ) and neutrophils (PMN) isolated from C57BL/6J mice were cultured with larvae (L3) for 22 h in 96-well plates (A) or Transwell plates (B). Heat-inactivated (HI) mouse serum was added where indicated in place of normal mouse serum, which was added to all wells as a source of complement. (C) Macrophages were placed in cell-impermeable diffusion chambers containing larvae. The chambers were implanted into naive mice for 3, 5, and 7 days (D) and naive and immunized mice for 24 h. Cell viabilities were greater than 95% at the conclusion of the experiment. For panels A and B, data are representative of 4 independent experiments and are depicted as means ± SD. *, P < 0.001 compared to survival of larvae with serum. For panels C and D, data represent at least two independent experiments of five mice per group and are displayed as means ± SD. *, P < 0.01 (C) or P < 0.05 (D) compared to the no-cell control.
Transwell plates with a 0.45-μm-pore-size membrane separating the top and bottom wells was utilized to determine whether larval killing in vitro was macrophage or neutrophil mediated. As with conventional 96-well plates, a 69% reduction in larval survival occurred in Transwell plates containing macrophages, neutrophils, and complement in the bottom wells. Placement of both cell types in the top wells and larvae in the bottom wells did not result in larval killing. When neutrophils were in contact with the larvae in the bottom well and macrophages were in the top well, larval survival was reduced by 79%. Similarly, a 67% reduction of larval survival was also observed when neutrophils were in the top wells and macrophages and larvae were in the bottom wells. Therefore, both macrophages and neutrophils are capable of killing the parasite when soluble help is provided by the reciprocal cell type (Fig. 2B).
The killing capacity of macrophages in vivo was examined by implanting cell-impermeable diffusion chambers containing BMDM and larvae into naive C57BL/6J mice for 3, 5, and 7 days. BMDM killed 42% of the larvae only after 7 days (Fig. 2C). However, in immunized mice, transferred BMDM killed 60% of the larvae within 24 h (Fig. 2D). The viability of BMDM recovered from diffusion chambers was greater than 90% in both naive and immunized mice. These in vitro and in vivo findings demonstrate that BMDM are capable of killing larvae during the primary and secondary immune responses.
IgM is important for macrophage-mediated larval killing in the secondary immune response.
We examined the role of parasite-specific IgM in macrophage-mediated killing of larvae during the secondary immune response. Secretory IgM-deficient (μS−/−) and wild-type C57BL/6J mice were immunized and challenged with cell-impermeable diffusion chambers containing BMDM from wild-type mice and larvae. The challenge infection occurred at 1 week after booster immunization, a time point when IgM, but not IgG, functioned in the protective secondary immune response (27). Serum from immunized mice revealed elevated parasite-specific IgM in wild-type mice, with no detectable lgM in immunized μS−/− mice; parasite-specific IgG levels were elevated in wild-type and μS−/− mice (Fig. 3A). Diffusion chambers were recovered 24 h after implantation to assess the secondary immune response. While BMDM transferred into immunized wild-type mice were capable of larval killing, larval killing was not observed following transfer of BMDM into immunized μS−/− mice (Fig. 3B). Therefore, BMDM utilize parasite-specific IgM in the larval killing process observed during the secondary immune response.
Fig 3.

IgM is important for macrophage-mediated larval killing in the secondary immune response. (A) Serum IgM and IgG levels were measured for parasite-specific antibody following immunization of C57BL/6J (wild type) and secretory IgM-deficient (μS−/−) mice. (B) Wild-type macrophages (Mϕ) were placed in cell-impermeable diffusion chambers containing larvae and implanted in naive and immunized wild-type and μS−/− mice for 24 h. Data from at least two independent experiments of five mice per group are shown as means ± SD, where an asterisk indicates P < 0.05 compared to the naive control.
AAMϕ and not CAMϕ mediate larval killing.
The ability of macrophage subsets to kill a helminth parasite was assessed by differentiating BMDM into AAMϕ following overnight stimulation with IL-4 and CAMϕ following stimulation with IFN-γ/LPS. Intracellular staining of macrophages revealed a 53% increase in Relm-α expression following IL-4 stimulation and a 16% increase in NOSII expression following stimulation with IFN-γ/LPS (Fig. 4A). Both macrophage subset markers were absent from untreated BMDM. Further analysis of treated macrophages confirmed that PD-L1 expression coincides with NOSII and PD-L2 coincides with Relm-α as markers of subset differentiation (Fig. 4A). The macrophage subsets were cultured with neutrophils and larvae in vitro for 24 h. BMDM, AAMϕ, CAMϕ, and neutrophils did not result in larval killing when cultured independently with larvae (Fig. 4B). However, naive BMDM and AAMϕ reduced larval survival by 90% when neutrophils and complement were present. In contrast, CAMϕ failed to kill larvae both independently and when cultured with neutrophils and complement. These findings reveal a novel function for AAMϕ but not CAMϕ in cooperating with neutrophils during larval killing.
Fig 4.

AAMϕ, but not CAMϕ, kill larvae during the primary and secondary immune responses. Bone marrow-derived macrophages (BMDM) were stimulated overnight with IL-4 (25 ng/ml) to induce alternatively activated macrophages (AAMϕ) or IFN-γ (5 ng/ml) and LPS (100 μg/ml) to induce classically activated macrophages (CAMϕ). The phenotypic analysis of in vitro-generated macrophage subsets using flow cytometry was performed prior to in vitro and in vivo assays. (A) BMDM were stimulated overnight and stained for surface expression of costimulatory molecules PD-L1 and PD-L2 and intracellular marker NOSII or Relm-α. (B) Differentiated macrophages were cultured in vitro with neutrophils (PMN), normal mouse serum, and larvae (L3), and larval survival was assessed after 24 h. Macrophage subsets were placed in cell-impermeable diffusion chambers containing larvae and implanted in naive mice for 3 days (C) and in naive and immunized mice for 24 h (D). The viability of macrophages recovered from in vitro and in vivo assays was greater than 95%. For panels A and B, data are representative of at least 3 independent experiments of two to three mice per group. For panels C and D, data represent at least 2 independent experiments of five mice per group and are displayed as means ± SD, where an asterisk indicates P < 0.001 compared to naive BMDM (C) or for naive and immunized mice compared to naive mice (D).
Diffusion chambers containing larvae and either AAMϕ or CAMϕ were implanted into naive mice for 3 days to determine if the macrophage subsets kill larvae. AAMϕ displayed efficient and accelerated killing (89% reduction in larval survival) compared to that of naive BMDM (11% reduction in larval survival), which require 7 days to kill larvae in vivo (Fig. 2C). CAMϕ were unable to kill larvae when implanted with the parasites in vivo (Fig. 4C). To determine if the macrophage subsets function during the protective secondary response, diffusion chambers containing larvae and cells were implanted into naive and immunized mice for 24 h. Naive BMDM and AAMϕ were able to kill the parasites, while CAMϕ had no effect on larval survival (Fig. 4D).
AAMϕ that are systemically induced following infection with S. stercoralis in an IL-4- and IL-4Rα-dependent manner can kill the parasite.
The association of AAMϕ with S. stercoralis infection in mice was evaluated by injecting larvae subcutaneously and monitoring the systemic macrophage response in the peritoneal cavity. PEC from mice infected with larvae were composed of 90% macrophages/monocytes and 10% eosinophils. These cells were evaluated for the expression of subset-specific molecules, Relm-α for AAMϕ and NOSII for CAMϕ. Analysis of PEC 7 days after injection of larvae revealed Relm-α expression by F4/80hi macrophages and F4/80lo eosinophils, with 22% of the macrophages expressing Relm-α (Fig. 5A). Furthermore, 30% of the PEC expressed Relm-α and were F4/80hi during the secondary immune response (Fig. 5B). Despite equivalent levels of Relm-α expression in the primary and secondary responses, Relm-α was unilaterally expressed by F4/80hi macrophages during the secondary response. The frequency of NOSII-expressing peritoneal macrophages was less than 1% following both primary and secondary infections. The increase in F4/80 and Relm-α double positive cells in the peritoneal cavity of mice infected subcutaneously with larvae suggests a systemic induction of AAMϕ. The differentiation and transcription of many AAMϕ-specific genes depends on IL-4/IL-13 signaling (45). Therefore, PEC from immunized IL-4−/− and IL-4Rα−/− mice were analyzed to validate the importance of this signaling pathway during infection. The expression of Relm-α by peritoneal macrophages following subcutaneous injection of larvae was greatly diminished to 8% in IL-4−/− and further reduced to 3% in IL-4Rα−/− mice (Fig. 5C).
Fig 5.

S. stercoralis larvae systemically induced AAMϕ, which are capable of killing the parasite. (A) Primary response. Mice were infected subcutaneously with 5,000 larvae (L3) or mock infected. Peritoneal exudate cells (PEC) were isolated 7 days postinfection and stained for F4/80, classically activated macrophage (CAMϕ) marker NOSII, and alternatively activated macrophage (AAMϕ) marker Relm-α. (B) Secondary response. Mice were immunized by subcutaneous injection of 5,000 larvae, followed by a booster injection of 5,000 larvae at day 14, or were mock injected. PEC were isolated at 5 days after booster immunization and analyzed for macrophage subset-specific markers. (C) PEC from IL-4- and IL-4Rα-deficient mice were analyzed following immunization for F4/80 and Relm-α coexpression to delineate AAMϕ. Gates indicate the percentage of F4/80hi cells expressing NOSII or Relm-α. (D) Purified peritoneal macrophages (PMϕ) isolated from infected mice were cultured with neutrophils (PMN) and larvae for 24 h in the presence of normal mouse serum in vitro. (E) Purified peritoneal macrophages (1 or 3 million) were placed in cell-impermeable diffusion chambers containing larvae and implanted in naive mice for 24 h. For panels A to D, data are representative of two independent experiments. For panels A to C, cellular analysis was performed using two to five mice per group. For panels D and E, data represent a single analysis of 3 independent experiments of five mice per group, depicted as means ± SD, where an asterisk indicates P < 0.002 compared to larvae alone (D) or the no-cell control (E).
The ability of purified peritoneal macrophages isolated from infected mice to kill larvae was ascertained by performing in vitro assays. PEC (92% monocytes/macrophages, 7% eosinophils, and less than 1% basophils) or bone marrow-isolated neutrophils were unable to kill cocultured larvae independently. However, when purified peritoneal macrophages were cultured with neutrophils and complement, parasite survival was reduced by 72% within 24 h (Fig. 5D). To confirm the ability of purified peritoneal macrophages to kill in vivo, 1 or 3 million PEC and larvae were placed in cell-impermeable diffusion chambers and implanted in naive mice for 24 h. Purified peritoneal macrophages killed larvae at both concentrations, resulting in a 65% reduction in parasite survival (Fig. 5E). The viability of cells after 24 h was greater than 90% both in vitro and in vivo. Therefore, following subcutaneous infection of mice with larvae, macrophages accumulate in the peritoneal cavity, acquire an AAMϕ phenotype, and are capable of killing larvae in vivo and in conjunction with neutrophils in vitro.
Relm-α and Arg-1 are not required for PEC-mediated larval killing.
PEC were isolated from infected Relm-α−/− and Arg-1flox/LysMCre mice, which lack Arg-1 in all macrophage and granulocyte populations. Cellular differentials revealed that PEC from wild-type and Relm-α−/− mice were composed of 90% monocytes/macrophages and 10% eosinophils. A similar ratio was observed in cell differentials taken from Arg-1flox/flox and Arg-1flox/LysMCre PEC. Analysis of PEC from infected Relm-α- and Arg-1flox/LysMCre-deficient mice using flow cytometry revealed no differences in the expression of cell type-specific markers compared to that of wild-type controls (data not shown). The PEC from wild-type and Relm-α−/− mice consisted primarily of monocytes and macrophages expressing high levels of CD11b and F4/80. Low levels of Siglec-F (5% positive) and the absence of Gr-1 expression indicates the presence of a small population of eosinophils and no neutrophils. PEC from infected Arg-1flox/flox and Arg-1flox/LysMCre mice revealed a similar composition of monocytes, macrophages, and eosinophils and an absence of neutrophils following infection. Furthermore, Relm-α expression by Arg-1flox/flox and Arg-1flox/LysMCre PEC was equivalent to that of wild-type PEC, where 38% of the cells were double positive for F4/80 and Relm-α. As anticipated, Relm-α expression was undetected in Relm-α−/− mice (Fig. 6A). Wild-type and Relm-α−/− purified peritoneal macrophages derived from infected mice killed larvae at comparable rates after 24 h in vivo (Fig. 6B). Similarly, purified peritoneal macrophages from Arg-1flox/LysMCre- and Arg-1flox/flox-infected mice had equivalent larval killing capacities (Fig. 6C). These findings demonstrate that Relm-α or Arg-1 expression by peritoneal macrophages is not required for killing larval S. stercoralis.
Fig 6.

AAMϕ-specific molecules Relm-α and Arg-1 are not required for peritoneal macrophage-mediated larval killing. (A) Purified peritoneal macrophages (PMϕ) from infected wild-type, Relm-α, Arg-1flox/flox, and Arg-1flox/LysMCre mice were stained for intracellular expression of Relm-α and NOSII. (B) Cell-impermeable diffusion chambers containing larvae and purified peritoneal macrophages from infected wild-type or Relm-α−/− and (C) Arg-1flox/flox or Arg-1flox/LysMCre mice were implanted in naive mice for 24 h. Data are representative of two independent experiments, displayed as means ± SD from four to five mice per group, where an asterisk indicates P < 0.001 compared to the no-cell control (B and C).
DISCUSSION
The objective of this study was to determine the capacity of human and mouse macrophages to kill the parasitic nematode S. stercoralis. Previous studies on the primary human response to infection with S. stercoralis have shown that human peripheral blood monocytes and neutrophils adhere to S. stercoralis larvae following complement activation. Adherence of monocytes or neutrophils to larvae resulted in decreased motility but did not result in larval killing (16). In the present study, we confirm the observation that individually monocytes and neutrophils are insufficient for parasite killing. However, when human PBMC or macrophages are joined with neutrophils in vitro, the cells kill the larvae of S. stercoralis. The killing mechanism used by human cells is complement activation dependent, which is analogous to the complement-dependent killing observed in mice during the protective innate immune response (21). This study presents the first report of primary human macrophages and neutrophils from uninfected donors killing the larval stage of S. stercoralis in vitro.
BMDM from naive mice were used to corroborate the results obtained using human macrophages and neutrophils. Parasite killing was only observed when BMDM were cultured in conjunction with neutrophils and complement in vitro. The observation that complement was required in the in vitro killing mechanism is consistent with the report that complement component C3b is required for larval killing during the primary and secondary immune responses in mice (21). Two approaches were used to ascertain if mouse macrophages could kill the larvae or if they were ancillary to the function of the neutrophils. In vitro Transwell experiments revealed that macrophages killed the larvae if soluble help was provided by neutrophils. Interestingly, neutrophils could also kill the larvae if the macrophages provided soluble factors. Therefore, it appears that macrophages have two roles in killing the larvae: direct killing of the worms and providing soluble factors to support neutrophil killing of the larvae. The second approach used to determine if macrophages could kill the larvae was to implant BMDM with larvae in cell-impermeable diffusion chambers in naive and immunized mice. BMDM killed larvae in vivo during both the primary and secondary immune responses, as has been reported for neutrophils (22). This is in distinction to eosinophils, which kill the parasite only during the primary immune response (22). The membrane pore size of cell-impermeable diffusion chambers allows complement and, potentially, neutrophil soluble factors from outside the diffusion chamber to freely enter and exit. These soluble factors presumably activate macrophages within the diffusion chamber to kill the parasite.
Macrophages and neutrophils are professional phagocytes that collaborate in the immune responses to several intracellular pathogens through activation by cytokines or granular components (29, 30). Macrophages activate neutrophils following Mycobacterium avium infection through the release of IL-8 (29). Activated neutrophils secrete myeloperoxidase, which binds and activates macrophages to kill pathogens such as M. avium (29) and Candida albicans (31). Macrophages have also been shown to kill nematode larvae. Rat alveolar macrophages from Nippostrongylus brasiliensis-infected mice utilized complement for cell adherence and killing of larvae. Alveolar macrophages from naive animals did not kill the worms, suggesting that conditioning or activation of the macrophages is required for the larval killing process (4). The mechanism through which human and mouse macrophages and neutrophils cooperatively kill S. stercoralis has not been defined. Complement-dependent adherence of macrophages and neutrophils to the larval surface may stimulate the release of cytokines or soluble factors, which then activate the reciprocal cell type. Neutrophil-derived myeloperoxidase and eosinophil-derived major basic protein have both been shown to be required for the cells to kill the larvae (23). Similar toxic molecules are presumably secreted by macrophages upon activation by neutrophils.
We have previously shown that antibody is required for protective secondary immunity to S. stercoralis in mice. IgM, but not IgG, is protective at 1 week after booster immunization (27), whereas parasite-specific IgM and IgG both are protective 3 and 5 weeks after booster immunization (28). Both IgM and IgG require cells to kill the larvae, and neutrophils have been shown to collaborate with IgG and complement in the killing process (28). It was hypothesized in the present study that IgM was the mediator of larval killing by macrophages in the secondary response. Transfer of wild-type macrophages into immunized wild-type mice at 1 week after booster immunization resulted in parasite killing, whereas transfer of macrophages into secretory IgM-deficient μS−/− mice did not. Parasite-specific IgM has been shown to be bound to the surface of S. stercoralis larvae recovered from immunized mice (27). IgM-mediated killing of the parasite may require interaction between the IgM on the surface of the parasite and its receptors found on macrophages (32) activating the cells to release toxic mediators that kill the parasite. Alternatively, the capacity of IgM to activate the complement system by cleaving C3 may play a critical role in parasite killing (33). Macrophages may bind to C3 on the surface of larvae and are then induced to release the toxic molecules.
AAMϕ are associated with parasitic helminth infections, where they function in modulating the TH2 response (34, 35), augment pathology by controlling fibrosis (36), and play an integral role in wound healing following parasite-mediated tissue destruction (13, 36). There have been no reports on the role of AAMϕ in killing parasitic helminths, although they have been shown to contribute to worm expulsion during Heligmosomoides polygyrus infections (37). AAMϕ, but not CAMϕ, generated in vitro from BMDM were capable of killing S. stercoralis larvae in vivo during the primary and secondary immune responses. Optimal larval killing by AAMϕ during the primary response was observed at day 3 (Fig. 4D), a full 4 days earlier than naive BMDM (Fig. 2B), demonstrating that AAMϕ kill the parasite in an accelerated manner. Furthermore, we show that macrophage and neutrophil cooperation is restricted to naive BMDM and AAMϕ but not CAMϕ, suggesting that CAMϕ are incapable of cooperating with neutrophils or that CAMϕ inhibit parasite killing by neutrophils.
The capacity of mouse macrophages induced following S. stercoralis infection to kill larvae was examined by utilizing PEC from infected mice, which consisted predominately of macrophages and monocytes. Interestingly, the composition of PEC from infected mice is substantially different from that of naive mice, which consists primarily of neutrophils (38). Peritoneal macrophages from infected mice killed larvae in vivo and in vitro in conjunction with neutrophils. Phenotypic analysis of these cells after primary and secondary infections with S. stercoralis revealed increased expression of Relm-α by macrophages, a phenotype associated with AAMϕ. Polarization of AAMϕ in the peritoneal cavities of infected mice was dependent on IL-4 and IL-4Rα, consistent with the importance of IL-4/IL-13 signaling for the development of AAMϕ observed during other parasite infections (39–42). The presence of AAMϕ in the peritoneal cavity following a subcutaneous injection reflects a systemic immune response likely induced by the highly motile larvae of S. stercoralis (43). This systemic induction of AAMϕ and not CAMϕ following subcutaneous infection with S. stercoralis is consistent with the finding that AAMϕ are found and maintained in the peritoneal cavity of Brugia malayi-infected mice (44).
Relm-α, Arg-1, and YM-1, while not exclusively expressed by AAMϕ, are the signature markers by which AAMϕ are characterized in numerous infections (45). To evaluate the effector function of these molecules, we utilized peritoneal macrophages from Relm-α-deficient mice and Arg-1-deficient macrophages. Macrophage Relm-α does not have an essential role in the S. stercoralis larval killing process. It may have other functions, as seen with the highly homologous family member Relm-β, which significantly inhibits the chemotaxis of S. stercoralis larvae in vitro (46). Arg-1 expression by macrophages is critical in the downregulation of TH2-driven pathology, fibrosis, and T cell proliferation during chronic schistosomiasis (47). However, the expression of Arg-1 by macrophages was not required for resistance to Trichuris muris (48). Likewise, Arg-1 was not required for peritoneal macrophage-mediated killing of S. stercoralis larvae. While Relm-α and Arg-1 are not relevant for direct parasite killing of S. stercoralis, they may still play a role in modulating the TH2 response, as previously shown with Schistosoma mansoni (35, 47). Microarray and RNA-Seq studies of helminth-induced AAMϕ demonstrate a complex gene expression profile distinct from that of BMDM (49, 50). Analysis of the differentially expressed AAMϕ genes may reveal the soluble factors required for collaboration with neutrophils and the effector molecules used by AAMϕ during the larval killing process.
We conclude that human macrophages and neutrophils, like mouse cells, have the capacity to kill S. stercoralis larvae. The striking parallels between the primary immune response to S. stercoralis in humans and mice are unlike those of other infection models, where mouse responses differ drastically from those of humans (51). The multiple killing mechanisms exhibited by mice in the primary and secondary immune responses to S. stercoralis demonstrate how the immune system has evolved redundant mechanisms for pathogen elimination. The overlapping effector roles of macrophages, neutrophils, and eosinophils during the primary immune response are further amplified in the secondary response by the production of parasite-specific antibodies or through TH2 cell cytokine induction of AAMϕ, resulting in a robust protective immune response directed at the larval stage of S. stercoralis.
ACKNOWLEDGMENTS
We thank Eric B. Wong for technical assistance and Jessica Deckman for critically reviewing the manuscript. We are grateful to Judith Allen, Meera Nair, and De'Broski Herbert for their expert and enthusiastic guidance. We acknowledge the Kimmel Cancer Center Flow Cytometry Core facility at Thomas Jefferson University and the Bone Marrow Transplant Program at Thomas Jefferson University Hospital for services provided.
This work was supported by grants from the National Institutes of Health, numbers AI076345 and AI078314 (D.A.); AI82548, AI50668, and AI22662 (J.B.L.); and RR02512 (M.H.).
Footnotes
Published ahead of print 24 June 2013
REFERENCES
- 1.Cannon GJ, Swanson JA. 1992. The macrophage capacity for phagocytosis. J. Cell Sci. 101(Part 4):907–913 [DOI] [PubMed] [Google Scholar]
- 2.Gordon S. 2004. Pathogen recognition or homeostasis? APC receptor functions in innate immunity. C. R. Biol. 327:603–607 [DOI] [PubMed] [Google Scholar]
- 3.Gordon S. 1998. The role of the macrophage in immune regulation. Res. Immunol. 149:685–688 [DOI] [PubMed] [Google Scholar]
- 4.Egwang TG, Gauldie J, Befus D. 1984. Complement-dependent killing of Nippostrongylus brasiliensis infective larvae by rat alveolar macrophages. Clin. Exp. Immunol. 55:149–156 [PMC free article] [PubMed] [Google Scholar]
- 5.Mukbel RM, Patten C, Jr, Gibson K, Ghosh M, Petersen C, Jones DE. 2007. Macrophage killing of Leishmania amazonensis amastigotes requires both nitric oxide and superoxide. Am. J. Trop. Med. Hyg. 76:669–675 [PubMed] [Google Scholar]
- 6.Loke P, Allison JP. 2003. PD-L1 and PD-L2 are differentially regulated by Th1 and Th2 cells. Proc. Natl. Acad. Sci. U. S. A. 100:5336–5341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mosser DM. 2003. The many faces of macrophage activation. J. Leukoc. Biol. 73:209–212 [DOI] [PubMed] [Google Scholar]
- 8.El Kasmi KC, Qualls JE, Pesce JT, Smith AM, Thompson RW, Henao-Tamayo M, Basaraba RJ, Konig T, Schleicher U, Koo MS, Kaplan G, Fitzgerald KA, Tuomanen EI, Orme IM, Kanneganti TD, Bogdan C, Wynn TA, Murray PJ. 2008. Toll-like receptor-induced arginase 1 in macrophages thwarts effective immunity against intracellular pathogens. Nat. Immunol. 9:1399–1406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Phillips R, Svensson M, Aziz N, Maroof A, Brown N, Beattie L, Signoret N, Kaye PM. 2010. Innate killing of Leishmania donovani by macrophages of the splenic marginal zone requires IRF-7. PLoS Pathog. 6:e1000813. 10.1371/journal.ppat.1000813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jenkins SJ, Allen JE. 2010. Similarity and diversity in macrophage activation by nematodes, trematodes, and cestodes. J. Biomed. Biotechnol. 2010:262609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Munitz A, Waddell A, Seidu L, Cole ET, Ahrens R, Hogan SP, Rothenberg ME. 2008. Resistin-like molecule alpha enhances myeloid cell activation and promotes colitis. J. Allergy Clin. Immunol. 122:1200–1207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jenkins SJ, Ruckerl D, Cook PC, Jones LH, Finkelman FD, van Rooijen N, MacDonald AS, Allen JE. 2011. Local macrophage proliferation, rather than recruitment from the blood, is a signature of TH2 inflammation. Science 332:1284–1288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen F, Liu Z, Wu W, Rozo C, Bowdridge S, Millman A, Van Rooijen N, Urban JF, Jr, Wynn TA, Gause WC. 2012. An essential role for TH2-type responses in limiting acute tissue damage during experimental helminth infection. Nat. Med. 18:260–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nguyen KD, Qiu Y, Cui X, Goh YP, Mwangi J, David T, Mukundan L, Brombacher F, Locksley RM, Chawla A. 2011. Alternatively activated macrophages produce catecholamines to sustain adaptive thermogenesis. Nature 480:104–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chawla A, Nguyen KD, Goh YP. 2011. Macrophage-mediated inflammation in metabolic disease. Nat. Rev. Immunol. 11:738–749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.de Messias IJ, Genta RM, Mohren WD. 1994. Adherence of monocytes and polymorphonuclear cells to infective larvae of Strongyloides stercoralis after complement activation. J. Parasitol. 80:267–274 [PubMed] [Google Scholar]
- 17.Bonne-Annee S, Hess JA, Abraham D. 2011. Innate and adaptive immunity to the nematode Strongyloides stercoralis in a mouse model. Immunol. Res. 51: 205–214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Padigel UM, Stein L, Redding K, Lee JJ, Nolan TJ, Schad GA, Birnbaumer L, Abraham D. 2007. Signaling through Gαi2 protein is required for recruitment of neutrophils for antibody-mediated elimination of larval Strongyloides stercoralis in mice. J. Leukoc. Biol. 81:1120–1126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.O'Connell AE, Redding KM, Hess JA, Lok JB, Nolan TJ, Abraham D. 2011. Soluble extract from the nematode Strongyloides stercoralis induces CXCR2 dependent/IL-17 independent neutrophil recruitment. Microbes Infect. 13:536–544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stein LH, Redding KM, Lee JJ, Nolan TJ, Schad GA, Lok JB, Abraham D. 2009. Eosinophils utilize multiple chemokine receptors for chemotaxis to the parasitic nematode Strongyloides stercoralis. J. Innate Immun. 1:618–630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kerepesi LA, Hess JA, Nolan TJ, Schad GA, Abraham D. 2006. Complement component C3 is required for protective innate and adaptive immunity to larval Strongyloides stercoralis in mice. J. Immunol. 176:4315–4322 [DOI] [PubMed] [Google Scholar]
- 22.Galioto AM, Hess JA, Nolan TJ, Schad GA, Lee JJ, Abraham D. 2006. Role of eosinophils and neutrophils in innate and adaptive protective immunity to larval Strongyloides stercoralis in mice. Infect. Immun. 74:5730–5738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.O'Connell AE, Hess JA, Santiago GA, Nolan TJ, Lok JB, Lee JJ, Abraham D. 2011. Major basic protein from eosinophils and myeloperoxidase from neutrophils are required for protective immunity to Strongyloides stercoralis in mice. Infect. Immun. 79:2770–2778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Valenzuela DM, Murphy AJ, Frendewey D, Gale NW, Economides AN, Auerbach W, Poueymirou WT, Adams NC, Rojas J, Yasenchak J, Chernomorsky R, Boucher M, Elsasser AL, Esau L, Zheng J, Griffiths JA, Wang X, Su H, Xue Y, Dominguez MG, Noguera I, Torres R, Macdonald LE, Stewart AF, DeChiara TM, Yancopoulos GD. 2003. High-throughput engineering of the mouse genome coupled with high-resolution expression analysis. Nat. Biotechnol. 21:652–659 [DOI] [PubMed] [Google Scholar]
- 25.Abraham D, Rotman HL, Haberstroh HF, Yutanawiboonchai W, Brigandi RA, Leon O, Nolan TJ, Schad GA. 1995. Strongyloides stercoralis: protective immunity to third-stage larvae inBALB/cByJ mice. Exp. Parasitol. 80:297–307 [DOI] [PubMed] [Google Scholar]
- 26.Stroncek DF, Clay ME, Herr G, Smith J, Jaszcz WB, Ilstrup S, McCullough J. 1997. The kinetics of G-CSF mobilization of CD34+ cells in healthy people. Transfus. Med. 7:19–24 [DOI] [PubMed] [Google Scholar]
- 27.Brigandi RA, Rotman HL, Yutanawiboonchai W, Leon O, Nolan TJ, Schad GA, Abraham D. 1996. Strongyloides stercoralis: role of antibody and complement in immunity to the third stage of larvae in BALB/cByJ mice. Exp. Parasitol. 82:279–289 [DOI] [PubMed] [Google Scholar]
- 28.Ligas JA, Kerepesi LA, Galioto AM, Lustigman S, Nolan TJ, Schad GA, Abraham D. 2003. Specificity and mechanism of immunoglobulin M (IgM)- and IgG-dependent protective immunity to larval Strongyloides stercoralis in mice. Infect. Immun. 71:6835–6843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jagannath C, Pai S, Actor JK, Hunter RL., Jr 1999. CRL-1072 enhances antimycobacterial activity of human macrophages through interleukin-8. J. Interferon Cytokine Res. 19:67–76 [DOI] [PubMed] [Google Scholar]
- 30.Novais FO, Santiago RC, Bafica A, Khouri R, Afonso L, Borges VM, Brodskyn C, Barral-Netto M, Barral A, de Oliveira CI. 2009. Neutrophils and macrophages cooperate in host resistance against Leishmania braziliensis infection. J. Immunol. 183:8088–8098 [DOI] [PubMed] [Google Scholar]
- 31.Lefkowitz SS, Gelderman MP, Lefkowitz DL, Moguilevsky N, Bollen A. 1996. Phagocytosis and intracellular killing of Candida albicans by macrophages exposed to myeloperoxidase. J. Infect. Dis. 173:1202–1207 [DOI] [PubMed] [Google Scholar]
- 32.Yang L, Shen L, Shao Y, Zhao Q, Zhang W. 2009. Cytoplasmic domain of human Fcalpha/mu receptor is required for ligand internalization. Cell Immunol. 258:78–82 [DOI] [PubMed] [Google Scholar]
- 33.Heyman B, Pilstrom L, Shulman MJ. 1988. Complement activation is required for IgM-mediated enhancement of the antibody response. J. Exp. Med. 167:1999–2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhao A, Urban JF, Jr, Anthony RM, Sun R, Stiltz J, van Rooijen N, Wynn TA, Gause WC, Shea-Donohue T. 2008. Th2 cytokine-induced alterations in intestinal smooth muscle function depend on alternatively activated macrophages. Gastroenterology 135:217–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nair MG, Du Y, Perrigoue JG, Zaph C, Taylor JJ, Goldschmidt M, Swain GP, Yancopoulos GD, Valenzuela DM, Murphy A, Karow M, Stevens S, Pearce EJ, Artis D. 2009. Alternatively activated macrophage-derived RELM-α is a negative regulator of type 2 inflammation in the lung. J. Exp. Med. 206:937–952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Barron L, Wynn TA. 2011. Macrophage activation governs schistosomiasis-induced inflammation and fibrosis. Eur. J. Immunol. 41:2509–2514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Anthony RM, Urban JF, Jr, Alem F, Hamed HA, Rozo CT, Boucher JL, Van Rooijen N, Gause WC. 2006. Memory T(H)2 cells induce alternatively activated macrophages to mediate protection against nematode parasites. Nat. Med. 12:955–960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kerepesi LA, Hess JA, Leon O, Nolan TJ, Schad GA, Abraham D. 2007. Toll-like receptor 4 (TLR4) is required for protective immunity to larval Strongyloides stercoralis in mice. Microbes Infect. 9:28–34 [DOI] [PubMed] [Google Scholar]
- 39.Mohrs M, Ledermann B, Kohler G, Dorfmuller A, Gessner A, Brombacher F. 1999. Differences between IL-4- and IL-4 receptor alpha-deficient mice in chronic leishmaniasis reveal a protective role for IL-13 receptor signaling. J. Immunol. 162:7302–7308 [PubMed] [Google Scholar]
- 40.Negrao-Correa D, Pinho V, Souza D G, Pereira AT, Fernandes A, Scheuermann K, Souza AL, Teixeira MM. 2006. Expression of IL-4 receptor on non-bone marrow-derived cells is necessary for the timely elimination of Strongyloides venezuelensis in mice, but not for intestinal IL-4 production. Int. J. Parasitol. 36:1185–1195 [DOI] [PubMed] [Google Scholar]
- 41.Urban JF, Jr, Noben-Trauth N, Donaldson DD, Madden KB, Morris SC, Collins M, Finkelman FD. 1998. IL-13, IL-4Ralpha, and Stat6 are required for the expulsion of the gastrointestinal nematode parasite Nippostrongylus brasiliensis. Immunity 8:255–264 [DOI] [PubMed] [Google Scholar]
- 42.Herbert DR, Orekov T, Perkins C, Rothenberg ME, Finkelman FD. 2008. IL-4R alpha expression by bone marrow-derived cells is necessary and sufficient for host protection against acute schistosomiasis. J. Immunol. 180:4948–4955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Napier LE. 1949. Strongyloides stercoralis infection. J. Trop. Med. Hyg. 52:25. [PubMed] [Google Scholar]
- 44.Loke P, Gallagher I, Nair MG, Zang X, Brombacher F, Mohrs M, Allison JP, Allen JE. 2007. Alternative activation is an innate response to injury that requires CD4+ T cells to be sustained during chronic infection. J. Immunol. 179:3926–3936 [DOI] [PubMed] [Google Scholar]
- 45.Gordon S, Martinez FO. 2010. Alternative activation of macrophages: mechanism and functions. Immunity 32:593–604 [DOI] [PubMed] [Google Scholar]
- 46.Artis D, Wang ML, Keilbaugh SA, He W, Brenes M, Swain GP, Knight PA, Donaldson DD, Lazar MA, Miller HR, Schad GA, Scott P, Wu GD. 2004. RELMbeta/FIZZ2 is a goblet cell-specific immune-effector molecule in the gastrointestinal tract. Proc. Natl. Acad. Sci. U. S. A. 101:13596–13600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pesce JT, Ramalingam TR, Mentink-Kane MM, Wilson MS, El Kasmi KC, Smith AM, Thompson RW, Cheever AW, Murray PJ, Wynn TA. 2009. Arginase-1-expressing macrophages suppress Th2 cytokine-driven inflammation and fibrosis. PLoS Pathog. 5:e1000371. 10.1371/journal.ppat.1000371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bowcutt R, Bell LV, Little M, Wilson J, Booth C, Murray PJ, Else KJ, Cruickshank SM. 2011. Arginase-1-expressing macrophages are dispensable for resistance to infection with the gastrointestinal helminth Trichuris muris. Parasite Immunol. 33:411–420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Loke P, Nair MG, Parkinson J, Guiliano D, Blaxter M, Allen JE. 2002. IL-4 dependent alternatively-activated macrophages have a distinctive in vivo gene expression phenotype. BMC Immunol. 3:7. 10.1186/1471-2172-3-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Thomas GD, Ruckerl D, Maskrey BH, Whitfield PD, Blaxter ML, Allen JE. 2012. The biology of nematode- and IL4Ralpha-dependent murine macrophage polarization in vivo as defined by RNA-Seq and targeted lipidomics. Blood 120:e93–e104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Seok J, Warren HS, Cuenca AG, Mindrinos MN, Baker HV, Xu W, Richards DR, McDonald-Smith GP, Gao H, Hennessy L, Finnerty CC, Lopez CM, Honari S, Moore EE, Minei JP, Cuschieri J, Bankey PE, Johnson JL, Sperry J, Nathens AB, Billiar TR, West MA, Jeschke MG, Klein MB, Gamelli RL, Gibran NS, Brownstein BH, Miller-Graziano C, Calvano SE, Mason PH, Cobb JP, Rahme LG, Lowry SF, Maier RV, Moldawer LL, Herndon DN, Davis RW, Xiao W, Tompkins RG. 2013. Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc. Natl. Acad. Sci. U. S. A. 110:3507–3512 [DOI] [PMC free article] [PubMed] [Google Scholar]