

Abstract
The discovery of cancer-associated mutations in genes encoding key metabolic enzymes has provided a direct link between altered metabolism and cancer. Advances in mass spectrometry and nuclear magnetic resonance technologies have facilitated high-resolution metabolite profiling of cells and tumors and identified the accumulation of metabolites associated with specific gene defects. Here we review the potential roles of such “oncometabolites” in tumor evolution and as clinical biomarkers for the detection of cancers characterized by metabolic dysregulation.
Introduction
The emerging interest in metabolites whose abnormal accumulation causes both metabolic and nonmetabolic dysregulation and potential transformation to malignancy (herein termed “oncometabolites”) has been fueled by the identification of cancer-associated mutations in genes encoding enzymes with significant roles in cellular metabolism (1–5). Loss-of-function mutations in genes encoding the Krebs cycle enzymes fumarate hydratase (FH) and succinate dehydrogenase (SDH) cause the accumulation of fumarate and succinate, respectively (6), whereas gain-of-function isocitrate dehydrogenase (IDH) mutations increase levels of D–2-hydroxyglutarate (D-2HG) (7, 8). These metabolites have been implicated in the dysregulation of cellular processes including the competitive inhibition of α-ketoglutarate–dependent (α-KG–dependent) dioxygenase enzymes (also known as 2-oxoglutarate–dependent dioxgenases) and posttranslational modification of proteins (1, 4, 9–11). To date, several lines of biochemical and genetic evidence support roles for fumarate, succinate, and D-2HG in cellular transformation and oncogenesis (3, 12).
Production of oncometabolites in cancer
D-2HG accumulates to millimolar concentrations in tumors with monoallelic mutations in IDH1 and IDH2 (8). There are three IDH isoforms in humans: IDH1 and IDH2 are NADP+-dependent homodimers localized in the cytoplasm and mitochondria, respectively, that catalyze the reversible conversion of isocitrate to α-KG, whereas IDH3 is an NAD+-dependent heterotetramer and catalyzes the irreversible oxidative decarboxylation of isocitrate to α-KG in the Krebs cycle (13). Somatic mutations in IDH1 and IDH2 occur in multiple human cancers, including low-grade glioma and secondary glioblastoma, chondrosarcoma, cholangiocarcinoma, and acute myeloid leukemia (AML) (14–19). The most common cancer mutations map to single arginine residues in the catalytic pockets: IDH1 (R132) and IDH2 (R172 or R140) (16, 19, 20). Mutant IDH1/2 forms a dimer with the wild-type copy derived from the normal allele and displays a neomorphic activity that allows the heterodimeric enzyme to catalyze the reduction of α-KG directly to D-2HG in the presence of NADPH (refs. 7, 8, 21, 22, and Figure 1).
Figure 1. D-2HG produced by mutant IDH1/2 affects metabolism and epigenetics by modulating activities of α-KG–dependent oxygenases.
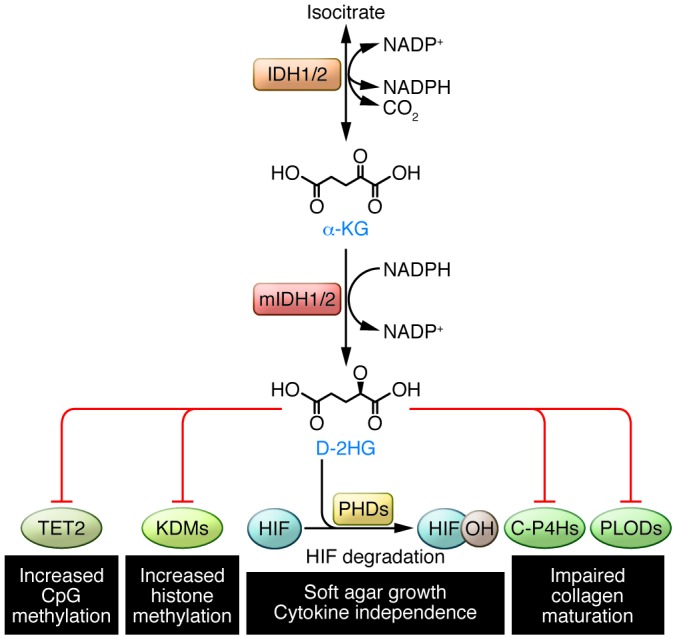
Wild-type IDH1 and IDH2 catalyze the NADP+-dependent reversible conversion of isocitrate to α-KG, whereas cancer-associated gain-of-function mutations enable mutant IDH1/2 (mIDH1/2) to catalyze the oxidation of α-KG to D-2HG, using NADPH as a cofactor. Because D-2HG is structurally similar to α-KG, its accumulation can modulate the activities of α-KG–utilizing dioxygenases. Inhibition of 5mC hydroxylase TET2 and the KDMs results in increased CpG island methylation and increased histone methylation marks, respectively, thus blocking lineage-specific cell differentiation. Inhibition of collagen prolyl and lysyl hydroxylases (C-P4Hs and PLODs, respectively) leads to impaired collagen maturation and disrupted basement membrane formation. D-2HG can also stimulate the activities of HIF PHDs, leading to enhanced HIF degradation and a diminished HIF response, which are associated with increased soft agar growth of human astrocytes and growth factor independence of leukemic cells. Together these processes exert pleiotrophic effects on cell signaling and gene expression that probably contribute to the malignancy of IDH1/2-mutant cells.
Succinate and fumarate accumulation occur in tumors driven by inactivating mutations in SDH and FH, the gene products of which catalyze sequential steps in the Krebs cycle. SDH mutations are commonly found in hereditary paraganglioma (PGL) and pheochromocytoma (PCC), while FH mutations are associated with hereditary leiomyomatosis and renal cell cancer (HLRCC) (23). Affected individuals inherit a loss-of-function mutation in one allele, with their tumors displaying loss of heterozygosity in the other allele, usually through somatic deletion or chromosomal loss; thus both genes follow the hereditary pattern of typical tumor suppressors (24). SDH is a highly conserved heterotetrameric protein, with SDHA and SDHB as catalytic subunits and SDHC and SDHD as ubiquinone-binding and membrane-anchorage subunits. In addition to its role in the Krebs cycle, SDH also functions as complex II of the electron transport chain (ETC), catalyzing the oxidation of succinate to fumarate in a reaction that generates FADH2, and donates electrons to the ETC. Mutations in genes encoding SDH subunits as well as the SDH assembly factor 2 occur frequently in PGL/PCC, but have also been identified in other types of tumors such as gastrointestinal stromal tumors (GISTs), renal tumors, thyroid tumors, testicular seminomas, and neuroblastomas (25). FH exists as a homotetrameric enzyme that catalyzes the stereospecific and reversible hydration of fumarate to malate. Though characterized by renal cancer, renal cysts, and skin and uterine leiomyomas (26, 27), evidence suggests that FH mutations may also be involved in the pathogenesis of breast, bladder, and Leydig cell tumors (28, 29). Both SDH and FH mutations significantly reduce their enzymatic activities, leading to accumulation of high levels of succinate and fumarate, respectively (refs. 6, 30–32, and Figure 2).
Figure 2. Candidate oncogenic mechanisms of succinate and fumarate accumulation.

SDH and FH are Krebs cycle enzymes and tumor suppressors. Loss-of-function mutations in SDH and FH result in abnormal accumulation of Krebs cycle metabolites succinate (Succ) and fumarate (Fum), respectively, both of which can inhibit the activities of α-KG–dependent oxygenases. Inhibition of HIF PHDs leads to activation of HIF-mediated pseudohypoxic response, whereas inhibition of KDMs and TET family of 5mC hydroxylases causes epigenetic alterations. Fumarate is electrophilic and can also irreversibly modify cysteine residues in proteins by succination. Succination of KEAP1 in FH deficiency results in the constitutive activation of the antioxidant defense pathway mediated by NRF2, conferring a reductive milieu that promotes cell proliferation. Succination of the Krebs cycle enzyme Aco2 impairs aconitase activity in Fh1-deficient MEFs. Fumarate accumulation may also affect cytosolic pathways by inhibiting the reactions involved in the biosynthesis of arginine and purine. AcCoA, acetyl CoA; Mal, malate; OAA, oxaloacetate; Succ-CA, succinyl CoA.
Epigenetic alterations
A common oncogenic mechanism linking D-2HG, succinate, and fumarate is the inhibition of α-KG–dependent dioxygenases (9), particularly of the Jumonji C domain–containing histone lysine demethylases (KDMs) and the ten eleven translocation (TET) family of 5-methylcytosine (5mC) hydroxylases, which results in epigenetic alterations that affect the expression of genes involved in cell differentiation and the acquisition of malignant features (Figures 1 And 2). Turcan et al. showed that D-2HG accumulation in IDH1-mutant gliomas substantially remodeled the DNA methylome and established a distinct CpG island methylator phenotype, which can be phenocopied by the stable expression of IDH1 R132H in primary human astrocytes (33). Elevated DNA methylation is also observed in AML samples harboring either 2HG-producing mutations in IDH1/2 or inactivating mutations in the α-KG–dependent TET2 enzyme (34, 35), and expression of mutant IDH1/2 or treatment with D-2HG is sufficient to inhibit TET2 function and impair differentiation in erythroleukemia cells (36). Expression of IDH mutants also suppressed histone demethylation, which repressed the expression of genes involved in lineage-specific differentiation and impaired adipocyte differentiation in culture (37). Taken together, these observations suggest that D-2HG acts as an oncogenic driver via epigenetic reprogramming in IDH-associated cancers.
Succinate and fumarate have been shown to inhibit KDM and TET enzymes in vitro and in cultured cells, leading to enhanced histone methylation marks and decreased global 5-hydroxymethylcytosine (5hmC) levels (38). Recently, two independent studies both demonstrated that succinate could also remodel the cancer epigenome. Killian et al. studied the methylation profiles of GIST tumors driven by mutations in either SDH or KIT and uncovered substantial DNA hypermethylation in the SDH-mutant subgroup (39). The authors also examined PGL/PCC tumors and identified hypermethylation in those harboring SDH mutations (39). In parallel, Letouzé and colleagues classified the methylation status of a large PGL/PCC cohort and identified a distinct epigenetic subcluster characterized by a hypermethylator phenotype (40). Strikingly, SDH mutations were discovered in 16 of 17 samples from the hypermethylated subgroup, in which the expression of 191 genes was downregulated concordantly with levels of CpG promoter hypermethylation. Two of the most significantly epigenetically silenced genes, PNMT and KRT19, are involved in neuroendocrine differentiation and epithelial-mesenchymal transition, respectively, which potentially explains the undifferentiated phenotype and aggressive nature of SDH-related PGL/PCC. Furthermore, Sdhb-deficient mouse chromaffin cells exhibit higher 5mC/5hmC ratios compared with wild-type cells, which can be reversed by the addition of exogenous α-KG, supporting the hypothesis that succinate inhibits TET-catalyzed DNA modification. Interestingly, the only tumor in the hypermethylated PGL/PCC subgroup with wild-type SDH harbored germline and somatic FH mutations, strongly suggesting that fumarate may also play a role in epigenetic rewiring in HLRCC (40). Finally, both groups analyzed developmentally distinct tumors containing SDH and IDH mutations and discovered an overlap in their hypermethylation patterns, potentially implicating a shared role for D-2HG, succinate, and fumarate in reprogramming of the epigenetic landscape in cancer.
Pseudohypoxia
In SDH- and FH-deficient cells and tumors, the activation of a HIF-orchestrated “pseudohypoxic” response has been reported (41–44), which could at least in part be attributed to the allosteric inhibition of HIF prolyl hydroxylases (PHDs) by elevated levels of succinate or fumarate (Figure 2). Although generally regarded as a major player in human cancer by transcriptionally regulating pathways that facilitate tumor growth and progression, the exact role of HIF in tumorigenesis is unclear (45). At least in the context of FH deficiency, a causal role for HIF in cancer initiation is debatable, as indicated by the observation that combined inactivation of Fh1 and Hif1a in mouse renal tubular cells exacerbated the cystic phenotype associated with Fh1 inactivation alone (46). Similarly, there have been contradictory observations of HIF levels in IDH-mutant models. Mice bearing the Idh1 R132H mutant accumulated Hif-1α and upregulated the expression of Hif target genes (47, 48). Ectopic expression of IDH1 R132H also increased HIF-1α levels in HEK293T and U-87MG glioblastoma cells (49, 50). In contrast, Koivunen et al. showed that, whereas L-2HG inhibits PHD activity, D-2HG promotes it by acting as a cofactor for these enzymes at pathophysiological concentrations, leading to diminished HIF levels, which confers a proliferative advantage for human astrocytes in soft agar growth (ref. 51 and Figure 1).
D-2HG is present at low levels in normal cells and can be metabolized by the enantiomer-specific D-2HG dehydrogenase (D2HGDH) to α-KG (52). Accumulation of either D- or L-2HG has been detected in the body fluids of patients with rare metabolic disorders that result from mutations in the corresponding dehydrogenase D2HGDH or L2HGDH (53, 54). Compared to D-2HG, L-2HG is a more potent inhibitor of the α-KG–dependent dioxygenases tested to date (49, 51). Interestingly, there is an increased risk for pediatric glioma in systemic L2HGDH deficiency, but not in patients with D2HGDH deficiency (53, 55), suggesting that the tumorigenic effect of D-2HG in adult tumors may be context specific or dose dependent. Furthermore, gliomas harboring IDH1/2 mutations are associated with better prognosis (49, 51). Kaelin and colleagues (36) proposed that the seemingly paradoxical observations could potentially be explained by the differential effect of D-2HG on HIF PHDs and histone/DNA demethylases. The authors showed that IDH1 R132H expression promoted growth factor independence and impaired differentiation of erythroleukemia cells, whereas loss of PHD2 activity blocked the leukemic transformation induced by IDH1 R132H expression or TET2 suppression. At tumor-relevant concentrations, D-2HG exerts an inhibitory effect on TET2 and causes epigenetic reprogramming while acting as an agonist for PHDs to promote HIF degradation. Indeed, HIF may have a tumor-suppressive effect at least in this setting, supported by the observations that HIF can inhibit hematopoietic stem cell and leukemic cell proliferation (56, 57).
Collagen maturation
Interestingly, recent work employing a small-molecule inhibitor against IDH1 R132 suggested that D-2HG might promote cancer growth through factors other than its epigenetic effects (58). Potential explanations may include modulation of HIF activity as discussed above, and dysregulation of other α-KG–dependent dioxygenases and corresponding cellular programs. Sasaki et al. showed that mice bearing Idh1 R132H mutations exhibit defects in collagen protein maturation and disrupted basement membrane formation, which could be attributed to D-2HG–mediated inhibition of collagen prolyl and lysyl hydroxylases and may contribute to abnormal tissue morphology and, potentially, glioma progression (47, 48). RNA expression array analyses of mutant IDH1 xenograft tumors also identified alterations in type IV collagen expression (58). Recently, the range of reactions catalyzed by α-KG–dependent dioxygenases has been further extended to ribosome protein modifications (59), and it remains to be determined whether translational processes are also sensitive to D-2HG targeting.
Succination and succinylation
Fumarate is an electrophilic metabolite that can react spontaneously with free sulfhydryl groups in cysteine residues by a Michael addition reaction to produce S-(2-succino)-cysteine (2SC), a process termed succination (60, 61). Succination can impair protein function, notably that of Kelch-like ECH-associated protein 1 (KEAP1), which is a major cellular electrophile sensor and negative regulator of the transcription factor nuclear factor E2-related factor 2 (NRF2) (62). Under normal physiological conditions, KEAP1 interacts with NRF2, promoting its ubiquitylation and proteasomal-mediated degradation. In the presence of oxidative stress or electrophiles, the KEAP1-NRF2 interaction is disrupted, resulting in the nuclear translocation of NRF2 and activation of an assortment of genes involved in antioxidant defense (63, 64). Work from ourselves and others demonstrated that in FH-deficient cells, KEAP1 is succinated on two critical cysteine residues (Cys155 and Cys288), which abrogates its interaction with NRF2 and elicits the constitutive expression of NRF2 target genes (refs. 46, 65, and Figure 2). Several lines of evidence suggest that activation of the NRF2-mediated antioxidant defense pathway may promote tumorigenesis by enhanced ROS detoxification as well as conferring a more reduced intracellular environment that can promote cell survival and proliferation (66, 67). This model is also supported by the observations that inactivating KEAP1 mutations and activating NRF2 mutations are found in various human cancers (68) and that NRF2 and its target genes are overexpressed in many cancer cell lines and human tumors (69–71). The identification of the NRF2 target gene heme oxygenase (decycling) 1 (HMOX1), which encodes a protein involved in heme degradation, as synthetically lethal to FH deletion, further supports the hypothesis that NRF2 can promote survival of FH-deficient cells (72), as does the recent discovery of NRF2-activating somatic mutations in sirtuin 1 (SIRT1), cullin 3 (CUL3), and NRF2 in sporadic papillary renal cell carcinomas (translocation associated) type 2 (PRCC2) (73).
We recently conducted a proteomic-based screen for novel 2SC targets and identified succination in more than 90 proteins in Fh1-deficient mouse embryonic fibroblasts (MEFs) and tissues. Nearly half of the 2SC targets thus identified are involved in metabolic processes, among which we identified the succination of the Krebs cycle enzyme aconitase 2 (ACO2) at three cysteine residues that are required for iron-sulfur cluster binding (ref. 74 and Figure 2). Fumarate-mediated succination of ACO2 exerted a dose-dependent inhibition on its enzymatic activity in vitro, and Fh1-deficient MEFs displayed reduced aconitase activity compared with wild-type counterparts. In contrast to other cell lines with compromised mitochondrial function, through either mutation or hypoxic stress, Fh1-deficient MEFs did not utilize the reductive carboxylation pathway for citrate synthesis, possibly due to succination of ACO2 (74–77). Taken together with previous work demonstrating succination and inhibition of GAPDH activity (78), it is plausible that succination is widespread in FH deficiency and that fumarate may cause dysregulated metabolism via succination of key metabolic proteins, potentially contributing to the oncogenesis of HLRCC.
Succination is a distinct process from succinylation, which typically occurs on lysine residues and is considered to be mediated by succinyl coenzyme A, producing a thioester derivative protein product (79). Recently, increases in protein succinylation have also been shown to occur under conditions where succinate levels are elevated and the activity of the desuccinylase SIRT5 is suppressed. Succinylated proteins targets identified include malate dehydrogenase, GAPDH, glutamate carrier 1, L-lactate dehydrogenase A chain and transaldolase (80). Protein succinylation levels and functional consequences in SDH-mutant tumors are interesting topics for future investigation.
ROS
Dysregulation of mitochondrial function characterized by Krebs cycle defects has been associated with overproduction of ROS, which may participate in oncogenic signaling and tumor progression by irreversible modification of DNA and oxidation of proteins (81, 82). Mouse fibroblasts transfected with an SDHC mutant displayed sustained ROS production and elevated DNA mutation frequency (81). A separate study using hamster fibroblasts expressing a truncated form of SDHC also showed increased steady-state levels of ROS and genomic instability (83). Guzy and colleagues further proposed that ROS-induced inactivation of PHDs triggers the HIF response in SDH-deficient cells, thus providing an alternative model for HIF activation in SDH deficiency (84).
Cells harboring 2HG-producing IDH mutations manifest depleted cellular glutamate levels, possibly because the metabolite is shunted to produce α-KG and subsequently converted to D-2HG, and elevated NADP+/NADPH ratios as a result of impaired generation but increased consumption of NADPH (3). Both events could suppress glutathione synthesis and regeneration in IDH mutant cells (85), potentially conferring an oxidative intracellular status and affecting biosynthesis. Surprisingly, Idh1 R132H–expressing mouse brain cells displayed attenuated ROS levels despite an increased NADP+/NADPH ratio (47).
Very recently, Sullivan et al. showed that succination of the antioxidant glutathione by fumarate in FH-deficient cells results in the accumulation of succinated glutathione, which acts as an alternative substrate to glutathione reductase, leading to decreased NADPH levels, enhanced mitochondrial ROS, and HIF-1α stabilization (86). Interestingly, the authors showed that fumarate-dependent succination, rather than ROS-induced oxidation, of KEAP1, is the dominant mechanism for NRF2 activation in these cells. The NRF2-mediated antioxidant pathways in turn serve to mitigate enhanced ROS levels, thus reflecting the intricate adaptive mechanisms of FH-deficient cells in combating redox stress.
Fumarate-dependent dysregulation of the urea cycle
The identification of KEAP1 succination suggests that fumarate accumulation occurs in subcellular compartments other than mitochondria in FH-deficient cells. In addition to its role in the Krebs cycle, fumarate also participates in the urea cycle and the purine biosynthesis pathway (refs. 87–90 and Figure 2). Recently it has been reported that fumarate accumulation can cause reversal of the argininosuccinate lyase–catalyzed reaction in the urea cycle, resulting in accumulation of fumarate-derived argininosuccinate and rendering the FH-deficient cells auxotrophic for arginine and sensitive to pharmacological arginine depletion from the growth media compared with controls (91, 92). Argininosuccinate accumulation was also detected in kidney and urine samples of Fh1-deficient mice, raising its potential as a biomarker for HLRCC (91, 92). Further, re-expression of cytosolic FH in vivo ameliorated both renal cyst development and urea cycle defects observed in mice with renal-specific Fh1 deletion, implicating a potential role for extra-mitochondrial metabolic pathways in FH-associated oncogenesis (92).
Biomarkers
IDH mutations correlate with better survival of glioma patients (93), hence assessment of IDH mutation status carries significant diagnostic and prognostic value. Techniques for routine neuropathological detection of IDH mutations include DNA-based sequencing approaches and immunohistochemistry (IHC) staining using mutation-specific antibodies (94–97). However, there are at least five reproducible cancer-associated mutations that can result in 2HG production, and further, conventional gene sequencing methods may lead to false positives due to genetic polymorphism and sequencing artifacts (98). In comparison, screening for elevated 2HG levels is a sensitive and specific approach to detect IDH mutations in tumors. Whereas patient sera/plasma can be assessed in the case of AML (7, 8, 21, 99), exciting advances with proton magnetic resonance spectroscopy (MRS) have been made in the noninvasive detection of 2HG in patients with gliomas (100–103). Using MRS sequence optimization and spectral fitting techniques, Maher and colleagues examined 30 patients with glioma and showed that the detection of 2HG correlated 100% with the presence of IDH1 or IDH2 mutations (102). Andronesi et al. further demonstrated that two-dimensional correlation spectroscopy could effectively distinguish 2HG from chemically similar metabolites present in the brain (103).
Negative IHC staining for SDHB correlates with the presence of SDH mutations, whether in SDHB, SDHC, or SDHD (104). This finding is most likely explained by the fact that mutations in any of the four subunits of SDH can destabilize the entire enzyme complex. PGLs/PCCs associated with an SDHA mutation show negative staining for SDHA as well as SDHB (105). Therefore, IHC staining for SDHB is a useful diagnostic tool to triage patients for genetic testing of any SDH mutation, and subsequent staining for the other subunits may further narrow the selection of genes to be tested. In contrast, detection of FH protein is often evident in HLRCC tumors due to retention of the nonfunctional mutant allele (106). However, staining of cysts and tumors for 2SC immunoreactivity reveals a striking correlation between FH inactivation and the presence of 2SC-modified protein (2SCP), which is absent in non-HLRCC tumors and normal tissue controls (106). IHC staining for 2SCP thus provides a robust diagnostic biomarker for FH deficiency (107).
Therapeutic targeting
Because D-2HG is a product of neomorphic enzyme activities, curtailing the D-2HG supply by specifically inhibiting the mutant IDH enzymes provides an elegant approach to target IDH-mutant cancers. Indeed, recent reports of small-molecule inhibitors against mutant forms of IDH1 and IDH2 demonstrated the feasibility of this method. An inhibitor against IDH2 R140Q was shown to reduce both intracellular and extracellular levels of D-2HG, suppress cell growth, and increase differentiation of primary human AML cells (108). Similarly, small-molecule inhibition of IDH1 R132H suppressed colony formation and increased tumor cell differentiation in a xenograft model for IDH1 R132H glioma (58). The inhibitors exhibited a cytostatic rather than cytotoxic effect, and therefore their therapeutic efficacy over longer time periods may need further assessment (109). Letouzé et al. showed that the DNA methytransferase inhibitor decitabine could repress the migration capacities of SDHB-mutant cells (40). However, for SDH- and FH-associated cancers, a synthetic lethality approach is worth exploring because of the pleiotrophic effects associated with succinate and fumarate accumulation.
Outlook
The application of next-generation sequencing technologies in the field of cancer genomics has substantially increased our understanding of cancer biology. Detection of germline and somatic mutations in specific tumor types not only expands the current repertoire of driver mutations and downstream effectors in tumorigenesis, but also sheds light on how oncometabolites may exert their oncogenic roles. For example, the identification of mutually exclusive mutations in IDH1 and TET2 in AML led to the characterization of TET2 as a major pathological target of D-2HG (34, 110). Additionally, the discovery of somatic CUL3, SIRT1, and NRF2 mutations in sporadic PRCC2 converges with FH mutation in HLRCC, in which NRF2 activation is a consequence of fumarate-mediated succination of KEAP1, indicating the functional prominence of the NRF2 pathway in PRCC2 (73). In light of this, the identification of somatic mutations in genes encoding the chromatin-modifying enzymes histone H3K36 methyltransferase (SETD2), histone H3K4 demethylase JARID1C (KDM5C), histone H3K27 demethylase UTX (KDM6A), and the SWI/SNF chromatin remodelling complex gene PBRM1 in clear cell renal cell carcinoma (111–113) highlights the importance of epigenetic modulation in human cancer and raises the potential for systematic testing in other types of tumors such as those associated with FH mutations.
Technological advances such as those in gas and liquid-chromatography mass spectrometry (114, 115) and nuclear magnetic resonance imaging (102) have greatly improved the ability to measure low-molecular-weight metabolites in tumor samples with high resolution (116). Combined with metabolic flux analyses employing isotope tracers and mathematical modeling, modern-era metabolomic approaches can provide direct pathophysiological insights into tumor metabolism and serve as an excellent tool for biomarker discovery. Using a data-driven approach, Jain and colleagues constructed the metabolic profiles of 60 cancer cell lines and discovered glycine consumption as a key metabolic event in rapidly proliferating cancer cells (117), thus demonstrating the power of metabolomic analyses and the relevance to future cancer research and therapeutics.
Acknowledgments
The Cancer Biology and Metabolism Group is funded by Cancer Research UK and the European Research Council under the European Community’s Seventh Framework Programme (FP7/2007-2013)/ERC grant agreement no. 310837 to Dr. Pollard. Professor Soga receives funding from a Grant-in-Aid for scientific research on Innovative Areas, Japan (no. 22134007), and the Yamagata Prefectural Government and City of Tsuruoka.
Footnotes
Conflict of interest: Tomoyoshi Soga is a founder of Human Metabolome Technologies.
Citation for this article: J Clin Invest. 2013;123(9):3652–3658. doi:10.1172/JCI67228.
References
- 1.Yang M, Soga T, Pollard PJ, Adam J. The emerging role of fumarate as an oncometabolite. Front Oncol. 2012;2:85. doi: 10.3389/fonc.2012.00085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ward PS, Thompson CB. Metabolic reprogramming: a cancer hallmark even warburg did not anticipate. Cancer Cell. 2012;21(3):297–308. doi: 10.1016/j.ccr.2012.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324(5930):1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Thompson CB. Metabolic enzymes as oncogenes or tumor suppressors. N Engl J Med. 2009;360(8):813–815. doi: 10.1056/NEJMe0810213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schulze A, Harris AL. How cancer metabolism is tuned for proliferation and vulnerable to disruption. Nature. 2012;491(7424):364–373. doi: 10.1038/nature11706. [DOI] [PubMed] [Google Scholar]
- 6.Pollard PJ, et al. Accumulation of Krebs cycle intermediates and over-expression of HIF1alpha in tumours which result from germline FH and SDH mutations. Hum Mol Genet. 2005;14(15):2231–2239. doi: 10.1093/hmg/ddi227. [DOI] [PubMed] [Google Scholar]
- 7.Ward PS, et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell. 2010;17(3):225–234. doi: 10.1016/j.ccr.2010.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dang L, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2010;465(7300):966. doi: 10.1038/nature09132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kaelin WG. Cancer and altered metabolism: potential importance of hypoxia-inducible factor and 2-oxoglutarate-dependent dioxygenases. Cold Spring Harb Symp Quant Biol. 2011;76:335–345. doi: 10.1101/sqb.2011.76.010975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.DeBerardinis RJ, Thompson CB. Cellular metabolism and disease: what do metabolic outliers teach us? Cell. 2012;148(6):1132–1144. doi: 10.1016/j.cell.2012.02.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang Z, Tan M, Xie Z, Dai L, Chen Y, Zhao Y. Identification of lysine succinylation as a new post-translational modification. Nat Chem Biol. 2010;7(1):58–63. doi: 10.1038/nchembio.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mullen AR, DeBerardinis RJ. Genetically-defined metabolic reprogramming in cancer. Trends Endocrinol Metab. 2012;23(11):552–559. doi: 10.1016/j.tem.2012.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Frezza C, Pollard PJ, Gottlieb E. Inborn and acquired metabolic defects in cancer. J Mol Med (Berl). 2011;89(3):213–220. doi: 10.1007/s00109-011-0728-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Paschka P, et al. IDH1 and IDH2 mutations are frequent genetic alterations in acute myeloid leukemia and confer adverse prognosis in cytogenetically normal acute myeloid leukemia with NPM1 mutation without FLT3 internal tandem duplication. J Clin Oncol. 2010;28(22):3636–3643. doi: 10.1200/JCO.2010.28.3762. [DOI] [PubMed] [Google Scholar]
- 15.Abbas S, et al. Acquired mutations in the genes encoding IDH1 and IDH2 both are recurrent aberrations in acute myeloid leukemia: prevalence and prognostic value. Blood. 2010;116(12):2122–2126. doi: 10.1182/blood-2009-11-250878. [DOI] [PubMed] [Google Scholar]
- 16.Yan H, et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med. 2009;360(8):765–773. doi: 10.1056/NEJMoa0808710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Amary MF, et al. IDH1 and IDH2 mutations are frequent events in central chondrosarcoma and central and periosteal chondromas but not in other mesenchymal tumours. J Pathol. 2011;224(3):334–343. doi: 10.1002/path.2913. [DOI] [PubMed] [Google Scholar]
- 18.Borger DR, et al. Frequent mutation of isocitrate dehydrogenase (IDH)1 and IDH2 in cholangiocarcinoma identified through broad-based tumor genotyping. Oncologist. 2012;17(1):72–79. doi: 10.1634/theoncologist.2011-0386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mardis ER, et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med. 2009;361(11):1058–1066. doi: 10.1056/NEJMoa0903840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Parsons DW, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321(5897):1807–1812. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gross S, et al. Cancer-associated metabolite 2-hydroxyglutarate accumulates in acute myelogenous leukemia with isocitrate dehydrogenase 1 and 2 mutations. J Exp Med. 2010;207(2):339–344. doi: 10.1084/jem.20092506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Leonardi R, Subramanian C, Jackowski S, Rock CO. Cancer-associated isocitrate dehydrogenase mutations inactivate NADPH-dependent reductive carboxylation. J Biol Chem. 2012;287(18):14615–14620. doi: 10.1074/jbc.C112.353946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bayley JP, Devilee P. Warburg tumours and the mechanisms of mitochondrial tumour suppressor genes. Barking up the right tree? Curr Opin Genet Dev. 2010;20(3):324–329. doi: 10.1016/j.gde.2010.02.008. [DOI] [PubMed] [Google Scholar]
- 24.Gottlieb E, Tomlinson IP. Mitochondrial tumour suppressors: a genetic and biochemical update. Nat Rev Cancer. 2005;5(11):857–866. doi: 10.1038/nrc1737. [DOI] [PubMed] [Google Scholar]
- 25.Bardella C, Pollard PJ, Tomlinson I. SDH mutations in cancer. Biochim Biophys Acta. 2011;1807(11):1432–1443. doi: 10.1016/j.bbabio.2011.07.003. [DOI] [PubMed] [Google Scholar]
- 26.Tomlinson IP, et al. Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer. Nat Genet. 2002;30(4):406–410. doi: 10.1038/ng849. [DOI] [PubMed] [Google Scholar]
- 27.Launonen V, et al. Inherited susceptibility to uterine leiomyomas and renal cell cancer. Proc Natl Acad Sci U S A. 2001;98(6):3387–3392. doi: 10.1073/pnas.051633798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lehtonen HJ, et al. Increased risk of cancer in patients with fumarate hydratase germline mutation. J Med Genet. 2006;43(6):523–526. doi: 10.1136/jmg.2005.036400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Carvajal-Carmona LG, et al. Adult leydig cell tumors of the testis caused by germline fumarate hydratase mutations. J Clin Endocrinol Metab. 2006;91(8):3071–3075. doi: 10.1210/jc.2006-0183. [DOI] [PubMed] [Google Scholar]
- 30.Gimenez-Roqueplo AP, et al. Functional consequences of a SDHB gene mutation in an apparently sporadic pheochromocytoma. J Clin Endocrinol Metab. 2002;87(10):4771–4774. doi: 10.1210/jc.2002-020525. [DOI] [PubMed] [Google Scholar]
- 31.Gimenez-Roqueplo AP, et al. The R22X mutation of the SDHD gene in hereditary paraganglioma abolishes the enzymatic activity of complex II in the mitochondrial respiratory chain and activates the hypoxia pathway. Am J Hum Genet. 2001;69(6):1186–1197. doi: 10.1086/324413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Alam NA, et al. Genetic and functional analyses of FH mutations in multiple cutaneous and uterine leiomyomatosis, hereditary leiomyomatosis and renal cancer, and fumarate hydratase deficiency. Hum Mol Genet. 2003;12(11):1241–1252. doi: 10.1093/hmg/ddg148. [DOI] [PubMed] [Google Scholar]
- 33.Turcan S, et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature. 2012;483(7390):479–483. doi: 10.1038/nature10866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Figueroa ME, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell. 2010;18(6):553–567. doi: 10.1016/j.ccr.2010.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Delhommeau F, et al. Mutation in TET2 in myeloid cancers. N Engl J Med. 2009;360(22):2289–2301. doi: 10.1056/NEJMoa0810069. [DOI] [PubMed] [Google Scholar]
- 36.Losman JA, et al. (R)-2-hydroxyglutarate is sufficient to promote leukemogenesis and its effects are reversible. Science. 2013;339(6127):1621–1625. doi: 10.1126/science.1231677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lu C, et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature. 2012;483(7390):474–478. doi: 10.1038/nature10860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xiao M, et al. Inhibition of alpha-KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes Dev. 2012;26(12):1326–1338. doi: 10.1101/gad.191056.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Killian JK, et al. Succinate dehydrogenase mutation underlies global epigenomic divergence in gastrointestinal stromal tumor. Cancer Discov. 2013;3(6):648–657. doi: 10.1158/2159-8290.CD-13-0092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Letouzé E, et al. SDH mutations establish a hypermethylator phenotype in paraganglioma. Cancer Cell. 2013;23(6):739–752. doi: 10.1016/j.ccr.2013.04.018. [DOI] [PubMed] [Google Scholar]
- 41.Selak MA, et al. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-α prolyl hydroxylase. Cancer Cell. 2005;7(1):77–85. doi: 10.1016/j.ccr.2004.11.022. [DOI] [PubMed] [Google Scholar]
- 42.Isaacs JS, et al. HIF overexpression correlates with biallelic loss of fumarate hydratase in renal cancer: novel role of fumarate in regulation of HIF stability. Cancer Cell. 2005;8(2):143–153. doi: 10.1016/j.ccr.2005.06.017. [DOI] [PubMed] [Google Scholar]
- 43.O’Flaherty L, et al. Dysregulation of hypoxia pathways in fumarate hydratase-deficient cells is independent of defective mitochondrial metabolism. Hum Mol Genet. 2010;19(19):3844–3851. doi: 10.1093/hmg/ddq305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Briere JJ, et al. Mitochondrial succinate is instrumental for HIF1alpha nuclear translocation in SDHA-mutant fibroblasts under normoxic conditions. Hum Mol Genet. 2005;14(21):3263–3269. doi: 10.1093/hmg/ddi359. [DOI] [PubMed] [Google Scholar]
- 45.Young RM, Simon MC. Untuning the tumor metabolic machine: HIF-alpha: pro- and antitumorigenic? Nat Med. 2012;18(7):1024–1025. doi: 10.1038/nm.2865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Adam J, et al. Renal cyst formation in Fh1-deficient mice is independent of the Hif/Phd pathway: roles for fumarate in KEAP1 succination and Nrf2 signaling. Cancer Cell. 2011;20(4):524–537. doi: 10.1016/j.ccr.2011.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sasaki M, et al. D-2-hydroxyglutarate produced by mutant IDH1 perturbs collagen maturation and basement membrane function. Genes Dev. 2012;26(18):2038–2049. doi: 10.1101/gad.198200.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sasaki M, et al. IDH1(R132H) mutation increases murine haematopoietic progenitors and alters epigenetics. Nature. 2012;488(7413):656–659. doi: 10.1038/nature11323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xu W, et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell. 2011;19(1):17–30. doi: 10.1016/j.ccr.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhao S, et al. Glioma-derived mutations in IDH1 dominantly inhibit IDH1 catalytic activity and induce HIF-1alpha. Science. 2009;324(5924):261–265. doi: 10.1126/science.1170944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Koivunen P, et al. Transformation by the (R)-enantiomer of 2-hydroxyglutarate linked to EGLN activation. Nature. 2012;483(7390):484–488. doi: 10.1038/nature10898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wanders RJ, Mooyer P. D-2-hydroxyglutaric acidaemia: identification of a new enzyme, D-2-hydroxyglutarate dehydrogenase, localized in mitochondria. J Inherit Metab Dis. 1995;18(2):194–196. doi: 10.1007/BF00711764. [DOI] [PubMed] [Google Scholar]
- 53.Struys EA. D-2-Hydroxyglutaric aciduria: unravelling the biochemical pathway and the genetic defect. J Inherit Metab Dis. 2006;29(1):21–29. doi: 10.1007/s10545-006-0317-9. [DOI] [PubMed] [Google Scholar]
- 54.Kranendijk M, Struys EA, Salomons GS, Van der Knaap MS, Jakobs C. Progress in understanding 2-hydroxyglutaric acidurias. J Inherit Metab Dis. 2012;35(4):571–587. doi: 10.1007/s10545-012-9462-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kranendijk M, et al. IDH2 mutations in patients with D-2-hydroxyglutaric aciduria. Science. 2010;330(6002):336. doi: 10.1126/science.1192632. [DOI] [PubMed] [Google Scholar]
- 56.Pollard PJ, Kranc KR. Hypoxia signaling in hematopoietic stem cells: a double-edged sword. Cell Stem Cell. 2010;7(3):276–278. doi: 10.1016/j.stem.2010.08.006. [DOI] [PubMed] [Google Scholar]
- 57.Suda T, Takubo K, Semenza GL. Metabolic regulation of hematopoietic stem cells in the hypoxic niche. Cell Stem Cell. 2011;9(4):298–310. doi: 10.1016/j.stem.2011.09.010. [DOI] [PubMed] [Google Scholar]
- 58.Rohle D, et al. An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science. 2013;340(6132):626–630. doi: 10.1126/science.1236062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ge W, et al. Oxygenase-catalyzed ribosome hydroxylation occurs in prokaryotes and humans. Nat Chem Biol. 2012;8(12):960–962. doi: 10.1038/nchembio.1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nagai R, et al. Succination of protein thiols during adipocyte maturation: a biomarker of mitochondrial stress. J Biol Chem. 2007;282(47):34219–34228. doi: 10.1074/jbc.M703551200. [DOI] [PubMed] [Google Scholar]
- 61.Alderson NL, et al. S-(2-Succinyl)cysteine: a novel chemical modification of tissue proteins by a Krebs cycle intermediate. Arch Biochem Biophys. 2006;450(1):1–8. doi: 10.1016/j.abb.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 62.Sun Z, Zhang S, Chan JY, Zhang DD. Keap1 controls postinduction repression of the Nrf2-mediated antioxidant response by escorting nuclear export of Nrf2. Mol Cell Biol. 2007;27(18):6334–6349. doi: 10.1128/MCB.00630-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Itoh K, et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem Biophys Res Commun. 1997;236(2):313–322. doi: 10.1006/bbrc.1997.6943. [DOI] [PubMed] [Google Scholar]
- 64.Itoh K, et al. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999;13(1):76–86. doi: 10.1101/gad.13.1.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ooi A, et al. An antioxidant response phenotype shared between hereditary and sporadic type 2 papillary renal cell carcinoma. Cancer Cell. 2011;20(4):511–523. doi: 10.1016/j.ccr.2011.08.024. [DOI] [PubMed] [Google Scholar]
- 66.DeNicola GM, et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature. 2011;475(7354):106–109. doi: 10.1038/nature10189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mitsuishi Y, et al. Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell. 2012;22(1):66–79. doi: 10.1016/j.ccr.2012.05.016. [DOI] [PubMed] [Google Scholar]
- 68.Hayes JD, McMahon M. NRF2 and KEAP1 mutations: permanent activation of an adaptive response in cancer. Trends Biochem Sci. 2009;34(4):176–188. doi: 10.1016/j.tibs.2008.12.008. [DOI] [PubMed] [Google Scholar]
- 69.Lau A, Villeneuve NF, Sun Z, Wong PK, Zhang DD. Dual roles of Nrf2 in cancer. Pharmacol Res. 2008;58(5–6):262–270. doi: 10.1016/j.phrs.2008.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Inami Y, et al. Persistent activation of Nrf2 through p62 in hepatocellular carcinoma cells. J Cell Biol. 2011;193(2):275–284. doi: 10.1083/jcb.201102031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Network TCGAR. Comprehensive genomic characterization of squamous cell lung cancers. Nature. 2012;489(7417):519–525. doi: 10.1038/nature11404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Frezza C, et al. Haem oxygenase is synthetically lethal with the tumour suppressor fumarate hydratase. Nature. 2011;477:225–228. doi: 10.1038/nature10363. [DOI] [PubMed] [Google Scholar]
- 73.Ooi A, et al. CUL3 and NRF2 mutations confer an NRF2 activation phenotype in a sporadic form of papillary renal cell carcinoma. Cancer Res. 2013;73(7):2044–2051. doi: 10.1158/0008-5472.CAN-12-3227. [DOI] [PubMed] [Google Scholar]
- 74.Ternette N, et al. Inhibition of mitochondrial aconitase by succination in fumarate hydratase deficiency. Cell Rep. 2013;3(3):689–700. doi: 10.1016/j.celrep.2013.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Metallo CM, et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature. 2012;481(7381):380–384. doi: 10.1038/nature10602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mullen AR, et al. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature. 2012;481(7381):385–388. doi: 10.1038/nature10642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wise DR, et al. Hypoxia promotes isocitrate dehydrogenase-dependent carboxylation of α-ketoglutarate to citrate to support cell growth and viability. Proc Natl Acad Sci U S A. 2011;108(49):19611–19616. doi: 10.1073/pnas.1117773108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Blatnik M, Frizzell N, Thorpe SR, Baynes JW. Inactivation of glyceraldehyde-3-phosphate dehydrogenase by fumarate in diabetes: formation of S-(2-succinyl)cysteine, a novel chemical modification of protein and possible biomarker of mitochondrial stress. Diabetes. 2008;57(1):41–49. doi: 10.2337/db07-0838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Frizzell N, Lima M, Baynes JW. Succination of proteins in diabetes. Free Radic Res. 2011;45(1):101–109. doi: 10.3109/10715762.2010.524643. [DOI] [PubMed] [Google Scholar]
- 80.Tannahill GM, et al. Succinate is an inflammatory signal that induces IL-1beta through HIF-1α. Nature. 2013;496(7444):238–242. doi: 10.1038/nature11986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ishii T, Yasuda K, Akatsuka A, Hino O, Hartman PS, Ishii N. A mutation in the SDHC gene of complex II increases oxidative stress, resulting in apoptosis and tumorigenesis. Cancer Res. 2005;65(1):203–209. [PubMed] [Google Scholar]
- 82.Ishii N, et al. A mutation in succinate dehydrogenase cytochrome b causes oxidative stress and ageing in nematodes. Nature. 1998;394(6694):694–697. doi: 10.1038/29331. [DOI] [PubMed] [Google Scholar]
- 83.Slane BG, et al. Mutation of succinate dehydrogenase subunit C results in increased O2.-, oxidative stress, and genomic instability. Cancer Res. 2006;66(15):7615–7620. doi: 10.1158/0008-5472.CAN-06-0833. [DOI] [PubMed] [Google Scholar]
- 84.Guzy RD, Sharma B, Bell E, Chandel NS, Schumacker PT. Loss of the SdhB, but Not the SdhA, subunit of complex II triggers reactive oxygen species-dependent hypoxia-inducible factor activation and tumorigenesis. Mol Cell Biol. 2008;28(2):718–731. doi: 10.1128/MCB.01338-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Reitman ZJ, et al. Profiling the effects of isocitrate dehydrogenase 1 and 2 mutations on the cellular metabolome. Proc Natl Acad Sci U S A. 2011;108(8):3270–3275. doi: 10.1073/pnas.1019393108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sullivan LB, et al. The Proto-oncometabolite fumarate binds glutathione to amplify ROS-dependent signaling. Mol Cell. 2013;51(2):236–248. doi: 10.1016/j.molcel.2013.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Shambaugh GE. Urea biosynthesis I. The urea cycle and relationships to the citric acid cycle. Am J Clin Nutr. 1977;30(12):2083–2087. doi: 10.1093/ajcn/30.12.2083. [DOI] [PubMed] [Google Scholar]
- 88.Brosnan ME, Brosnan JT. Renal arginine metabolism. J Nutr. 2004;134(10 suppl):2791S–2795S. doi: 10.1093/jn/134.10.2791S. [DOI] [PubMed] [Google Scholar]
- 89.Stepinski J, Bizon D, Piec G, Angielski S. The purine nucleotide cycle activity in renal cortex and medulla. Am J Kidney Dis. 1989;14(4):307–309. doi: 10.1016/s0272-6386(89)80209-4. [DOI] [PubMed] [Google Scholar]
- 90.Salway JG. Metabolism At A Glance. Oxford, United Kingdom: Blackwell Publishing; 1999. [Google Scholar]
- 91.Zheng L, et al. Reversed argininosuccinate lyase activity in fumarate hydratase-deficient cancer cells. Cancer Metab. 2013;1:12. doi: 10.1186/2049-3002-1-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Adam J, et al. A role for cytosolic fumarate hydratase in urea cycle metabolism and renal neoplasia. Cell Rep. 2013;3(5):1440–1448. doi: 10.1016/j.celrep.2013.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hartmann C, et al. Type and frequency of IDH1 and IDH2 mutations are related to astrocytic and oligodendroglial differentiation and age: a study of 1,010 diffuse gliomas. Acta Neuropathol. 2009;118(4):469–474. doi: 10.1007/s00401-009-0561-9. [DOI] [PubMed] [Google Scholar]
- 94.De Witte O, Lefranc F, Levivier M, Salmon I, Brotchi J, Goldman S. FDG-PET as a prognostic factor in high-grade astrocytoma. J Neurooncol. 2000;49(2):157–163. doi: 10.1023/A:1026518002800. [DOI] [PubMed] [Google Scholar]
- 95.Preusser M, Capper D, Hartmann C. IDH testing in diagnostic neuropathology: review and practical guideline article invited by the Euro-CNS research committee. Clin Neuropathol. 2011;30(5):217–230. doi: 10.5414/np300422. [DOI] [PubMed] [Google Scholar]
- 96.Preusser M, Wohrer A, Stary S, Hoftberger R, Streubel B, Hainfellner JA. Value and limitations of immunohistochemistry and gene sequencing for detection of the IDH1-R132H mutation in diffuse glioma biopsy specimens. J Neuropathol Exp Neurol. 2011;70(8):715–723. doi: 10.1097/NEN.0b013e31822713f0. [DOI] [PubMed] [Google Scholar]
- 97.Metellus P, et al. IDH mutation status impact on in vivo hypoxia biomarkers expression: new insights from a clinical, nuclear imaging and immunohistochemical study in 33 glioma patients. J Neurooncol. 2011;105(3):591–600. doi: 10.1007/s11060-011-0625-2. [DOI] [PubMed] [Google Scholar]
- 98.Ward PS, et al. Identification of additional IDH mutations associated with oncometabolite R(-)-2-hydroxyglutarate production. Oncogene. 2012;31(19):2491–2498. doi: 10.1038/onc.2011.416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Pollyea DA, et al. 2-Hydroxyglutarate in IDH mutant acute myeloid leukemia: predicting patient responses, minimal residual disease and correlations with methylcytosine and hydroxymethylcytosine levels. Leuk Lymphoma. 2013;54(2):408–410. doi: 10.3109/10428194.2012.701009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Pope WB, et al. Non-invasive detection of 2-hydroxyglutarate and other metabolites in IDH1 mutant glioma patients using magnetic resonance spectroscopy. J Neurooncol. 2012;107(1):197–205. doi: 10.1007/s11060-011-0737-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Elkhaled A, et al. Magnetic resonance of 2-hydroxyglutarate in IDH1-mutated low-grade gliomas. Sci Transl Med. 2012;4(116):116ra115. doi: 10.1126/scitranslmed.3002796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Choi C, et al. 2-hydroxyglutarate detection by magnetic resonance spectroscopy in IDH-mutated patients with gliomas. Nat Med. 2012;18(4):624–629. doi: 10.1038/nm.2682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Andronesi OC, et al. Detection of 2-hydroxyglutarate in IDH-mutated glioma patients by in vivo spectral-editing and 2D correlation magnetic resonance spectroscopy. Sci Transl Med. 2012;4(116):116ra114. doi: 10.1126/scitranslmed.3002693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.van Nederveen FH, et al. An immunohistochemical procedure to detect patients with paraganglioma and phaeochromocytoma with germline SDHB, SDHC, or SDHD gene mutations: a retrospective and prospective analysis. Lancet Oncol. 2009;10(8):764–771. doi: 10.1016/S1470-2045(09)70164-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Korpershoek E, et al. SDHA immunohistochemistry detects germline SDHA gene mutations in apparently sporadic paragangliomas and pheochromocytomas. J Clin Endocrinol Metab. 2011;96(9):E1472–E1476. doi: 10.1210/jc.2011-1043. [DOI] [PubMed] [Google Scholar]
- 106.Bardella C, et al. Aberrant succination of proteins in fumarate hydratase-deficient mice and HLRCC patients is a robust biomarker of mutation status. J Pathol. 2011;225(1):4–11. doi: 10.1002/path.2932. [DOI] [PubMed] [Google Scholar]
- 107.Maxwell PH. Seeing the smoking gun: a sensitive and specific method to visualize loss of the tumour suppressor, fumarate hydratase, in human tissues. J Pathol. 2011;225(1):1–3. doi: 10.1002/path.2950. [DOI] [PubMed] [Google Scholar]
- 108.Wang F, et al. Targeted inhibition of mutant IDH2 in leukemia cells induces cellular differentiation. Science. 2013;340(6132):622–626. doi: 10.1126/science.1234769. [DOI] [PubMed] [Google Scholar]
- 109.Kim J, DeBerardinis RJ. Cancer. Silencing a metabolic oncogene. Science. 2013;340(6132):558–559. doi: 10.1126/science.1238523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Gaidzik VI, et al. TET2 mutations in acute myeloid leukemia (AML): results from a comprehensive genetic and clinical analysis of the AML study group. J Clin Oncol. 2012;30(12):1350–1357. doi: 10.1200/JCO.2011.39.2886. [DOI] [PubMed] [Google Scholar]
- 111.Varela I, et al. Exome sequencing identifies frequent mutation of the SWI/SNF complex gene PBRM1 in renal carcinoma. Nature. 2011;469(7331):539–542. doi: 10.1038/nature09639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Dalgliesh GL, et al. Systematic sequencing of renal carcinoma reveals inactivation of histone modifying genes. Nature. 2010;463(7279):360–363. doi: 10.1038/nature08672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.van Haaften G, et al. Somatic mutations of the histone H3K27 demethylase gene UTX in human cancer. Nat Genet. 2009;41(5):521–523. doi: 10.1038/ng.349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Soga T, Ohashi Y, Ueno Y, Naraoka H, Tomita M, Nishioka T. Quantitative metabolome analysis using capillary electrophoresis mass spectrometry. J Proteome Res. 2003;2(5):488–494. doi: 10.1021/pr034020m. [DOI] [PubMed] [Google Scholar]
- 115.Soga T, et al. Differential metabolomics reveals ophthalmic acid as an oxidative stress biomarker indicating hepatic glutathione consumption. J Biol Chem. 2006;281(24):16768–16776. doi: 10.1074/jbc.M601876200. [DOI] [PubMed] [Google Scholar]
- 116.Tomita M, Kami K. Cancer. Systems biology, metabolomics, and cancer metabolism. Science. 2012;336(6084):990–991. doi: 10.1126/science.1223066. [DOI] [PubMed] [Google Scholar]
- 117.Jain M, et al. Metabolite profiling identifies a key role for glycine in rapid cancer cell proliferation. Science. 2012;336(6084):1040–1044. doi: 10.1126/science.1218595. [DOI] [PMC free article] [PubMed] [Google Scholar]