

Abstract
Recent genome-wide discovery studies have identified a spectrum of mutations in different malignancies and have led to the elucidation of novel pathways that contribute to oncogenic transformation. The discovery of mutations in the genes encoding isocitrate dehydrogenase (IDH) has uncovered a critical role for altered metabolism in oncogenesis, and the neomorphic, oncogenic function of IDH mutations affects several epigenetic and gene regulatory pathways. Here we discuss the relevance of IDH mutations to leukemia pathogenesis, therapy, and outcome and how mutations in IDH1 and IDH2 affect the leukemia epigenome, hematopoietic differentiation, and clinical outcome.
Introduction
Mutations in isocitrate dehydrogenase (IDH) have been identified in a spectrum of human malignancies. Mutations in IDH1 were first identified in an exome resequencing analysis of patients with colorectal cancer (1). Shortly thereafter, recurrent IDH1 and IDH2 mutations were found in patients with glioma, most commonly in patients who present with lower-grade gliomas (2). IDH1 mutations were subsequently discovered in patients with acute myeloid leukemia (AML) through whole genome sequencing (3), which was followed by the identification of somatic IDH2 mutations in patients with AML (4–6). Further studies revealed that IDH mutations induce a neomorphic function to produce the oncometabolite 2-hydroxyglutarate (2HG) (7, 8), which can inhibit many cellular processes (9, 10). In particular, the ability of 2HG to alter the epigenetic landscape makes IDH a prototypical target for prognostic studies and drug targeting in leukemias.
Neomorphic function leads to oncometabolite production
IDH proteins catalyze the oxidative decarboxylation of isocitrate to α-ketoglutarate (αKG, also known as 2-oxoglutarate). IDH3 primarily functions as the allosterically regulated, rate-limiting enzymatic step in the TCA cycle, while the other two isoforms, which are mutated in cancer, utilize this catalytic process in additional contexts including metabolism and glucose sensing (IDH1) and regulation of oxidative respiration (IDH2) (11, 12). Loss-of-function mutations in other TCA cycle components have previously been identified in other types of cancer, specifically in mutations in fumarate hydratase (FH) and succinate dehydrogenase (SDH). As such, many hypothesized that IDH1/2 mutations would result in loss of metabolic activity, and indeed, enzymatic studies confirmed that the mutant protein’s ability to perform its native function is markedly attenuated, as measured by reduced production of αKG or NADPH (13, 14).
However, the genetic data relating to these mutations were more consistent with gain-of-function mutation: all of the observed alterations are somatic, heterozygous mutations that occur at highly conserved positions, which appear to be functionally equivalent between different isoforms. This discrepancy was resolved when metabolic profiling showed that the IDH1 mutant protein catalyzes a neomorphic reaction that converts αKG to 2HG. 2HG can be detected at high levels in gliomas harboring these mutations (4), and the accumulation of 2HG was further found to be common to oncogenic IDH mutations (8). This finding indicated that 2HG may serve as a potential functional biomarker of IDH mutation, and later, metabolomics analysis of 2HG content in patient samples led to the identification of IDH2 mutations in leukemias (6). IDH mutant proteins have been proposed to form a heterodimer with the remaining wild-type IDH isoform (7, 8, 14), which is consistent with genetic data showing retention of the wild-type allele in IDH-mutant cancers.
2HG and tet family enzymes in leukemogenesis
The discovery of the neomorphic function of IDH opened the doors for true investigation into the implications of these mutations and the resultant intracellular accumulation of 2HG. 2HG is thought to competitively inhibit the activity of a broad spectrum of αKG-dependent enzymes with known and postulated roles in oncogenic transformation. Some targets, such as the prolyl 4-hydroxylases, have unclear implications in leukemia pathogenesis. However, the recent demonstration that alterations in epigenetic factors occur in the majority of acute leukemias led to investigations of the effects of 2HG on the jumonji C domain histone-modifying enzymes and the newly characterized tet methylcytosine dioxygenase (TET) family of methylcytosine hydroxylases. Importantly, expression of IDH or exposure to chemically modified, cell-permeable 2HG affects hematopoietic differentiation, likely due to changes in epigenetic regulation that induce reversible alterations in differentiation states (15).
TET1 was initially discovered as a binding partner of mixed-lineage leukemia (MLL) in patients with MLL-translocated AML (16, 17). However, the function of the TET gene family and its role in leukemogenesis remained unknown until TET1 was shown to catalyze αKG-dependent addition of a hydroxyl group to methylated cytosines (18), which precedes DNA demethylation and results in altered epigenetic control (10, 18–24). TET enzymes have further been shown to catalyze conversion of 5-methylcytosine (5mC) to 5-formylcytosine (5fC) or 5-carboxylcytosine (5cC) (25, 26). These data suggest that loss of TET2 enzymatic function can lead to aberrant cytosine methylation and epigenetic silencing in malignant settings. TET2 mutations were initially found in array-comparative genomic hybridization and genome-wide SNP arrays, which identified microdeletions containing this gene in a patient with myeloproliferative neoplasm (MPN) and myelodysplastic syndrome (MDS) (27). This discovery was followed by the identification of somatic missense, nonsense, and frameshift TET2 mutations in patients with MDS, MPN, AML, and other myeloid malignancies (27–30). Most TET2 alleles result in nonsense/frameshift mutations, which result in loss of TET2 catalytic function (31), consistent with a tumor suppressor function in myeloid malignancies.
When 2HG was hypothesized to affect specific enzymatic processes in oncogenesis, AML patients were observed to harbor IDH1/2 and TET mutations in a mutually exclusive manner (9). Of note, exploration into the functional relationship between these mutant IDH proteins and the function of TET2 ultimately suggested a role for 2HG in inhibiting TET enzymatic function. IDH- or TET2-mutant patient samples are characterized by increased global hypermethylation of DNA and transcriptional silencing of genes with hypermethylated promoters. Expression of these IDH-mutant alleles in experimental models was further observed to result in increased methylation, reduced hydroxymethylation, and impaired TET2 function (9). Finally, in biochemical assays, 2HG was shown to directly inhibit TET2 as well as other αKG-dependent enzymes (10). These data demonstrate that a key feature of IDH1/2 mutations in hematopoietic cells is to impair TET2 function and disrupt DNA methylation (Figure 1).
Figure 1. Normal IDH functions to convert isocitrate to αKG in the Krebs cycle.

Oncogenic mutations in IDH induce neomorphic function to produce the oncometabolite 2HG. In leukemias, 2HG affects the TET family of proteins, which results in impaired hydroxymethylation of DNA and disrupted epigenetic control. Inhibitors of mutated IDH have shown promise in preclinical testing as well as in mechanistic studies of this system.
Models of leukemia employing IDH/2HG and TET2
Conditional loss of Tet2 expression in mice results in a chronic myelomonocytic leukemia (CMML) phenotype and in increased hematopoietic self-renewal in vivo (32). Of note, in vitro systems have shown that TET2 silencing and expression of IDH1/2 mutant alleles leads to impaired hematopoietic differentiation and expansion of stem/progenitor cells (9). More recently, IDH1 (R132H) conditional knockin mice with hematopoietic-specific recombination were analyzed and found to have myeloid expansion, although they did not develop overt AML. This suggests that IDH mutations by themselves cannot promote overt transformation, and that additional genetic, epigenetic, and/or microenvironmental factors are needed to cooperate with mutant IDH alleles to promote hematologic malignancies. The hematopoietic defects included increased numbers of hematopoietic stem cells and myeloid progenitor cells, and a DNA methylation signature that was similar to observed patterns in primary AML patients with IDH1 mutations (33). While many models of IDH-mutant leukemia have shown potential, future models that incorporate the complexity seen in human patients are needed, as discussed below. More recently, the effects of IDH1/2 mutations on hematopoietic cell lines were replicated using exogenously applied 2HG, which was rendered permeable to the cell membrane by esterification. The Kaelin group used this system to dissect the role of 2HG in the αKG-dependent pathways that may be affected in IDH mutation, and to show that the effects are reversible (34). Tools such as these will help advance our understanding of the biology of IDH mutations and, by extension, the potential therapies that may affect mutant IDH and the downstream pathways. Indeed, given the recent description of mutant-selective IDH1/2 inhibitors (34–37), the development of genetically accurate models of IDH mutant–mediated leukemogenesis will be critical to evaluate the effects of targeted therapies in mice with AML and subsequently in the clinical context.
Prevalence of IDH mutations in leukemia patients
IDH mutations are most common in patients with cytogenetically normal AML (CN-AML), and the prevalence of IDH1 and IDH2 missense mutations among patients with AML is between 5% and 20% (Table 1). In comparison with other cancers in which IDH1 mutations are more common than IDH2 mutations, IDH2 mutations are relatively more common in AML, such that the frequency of IDH1 and IDH2 are comparable in AML patients (38). IDH1 and IDH2 mutations are mutually exclusive from one another in AML.
Table 1.
Prevalence of IDH hotspot mutations in leukemia subtypes
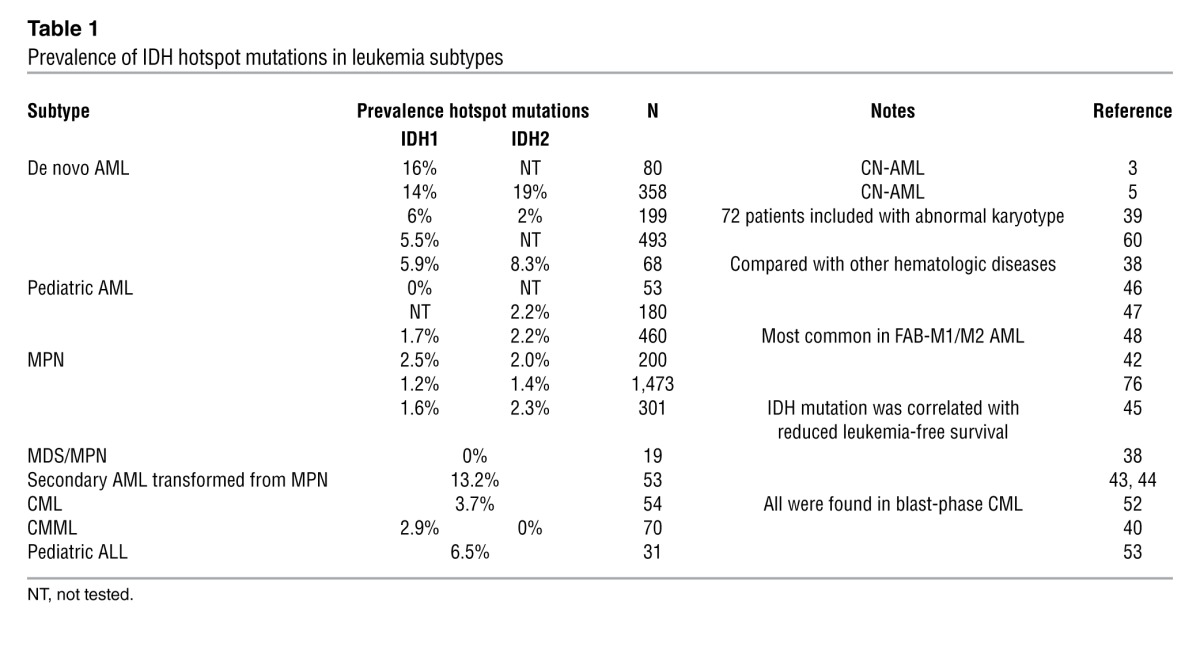
Different subtypes of AML have been postulated to have different biases toward IDH mutation. For instance, some studies suggest that IDH1 mutations are more common in patients with abnormal karyotype, while IDH2 mutations are primarily found in patients with CN-AML (39). In addition, 3% of patients with CMML were observed to have IDH1 mutations (40).
Patients can present with de novo AML, or alternatively can progress to AML from chronic-phase MPN and MDS. IDH mutations are associated with trisomy 8 in MDS and AML patients, but this association has been thought to imply a leukemogenic advantage for IDH mutant clones, rather than cooperation between these two events (41). In MPN, IDH mutations have been observed in 5%–20% of patients whose disease transforms to secondary AML (42). In pairwise comparisons of MPN and resultant post-MPN AML from the same patients, the acquisition of IDH mutations was observed during the process of transformation, implying an important role for IDH mutation in the progression to post-MPN secondary leukemia (43, 44). In addition, IDH mutations in chronic-phase MPN patients predicts for an increased risk of subsequent transformation to AML, suggesting IDH mutations may serve as a marker of incipient transformation in patients in advance of pathologic evidence of transformation (Figure 2 and ref. 45).
Figure 2. Many mutations have been observed in conjunction with IDH1/2 mutations in different types of leukemia.
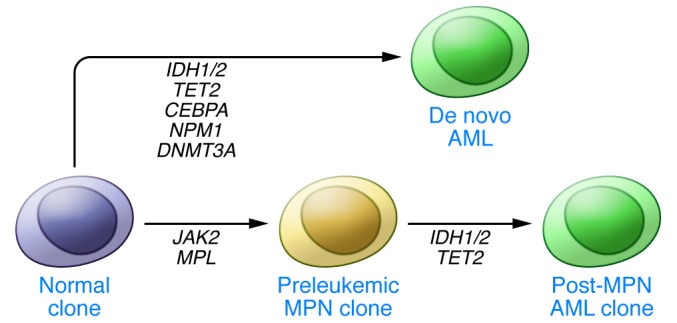
In de novo adult AML, these mutations should be observed in the context of other prognostic indicators such as CEBPA, NPM1, and DNMT3A mutation. In AML that progresses from MPN, IDH1/2 mutations can be examined separately from the mutations responsible for MPN (such as JAK2 or MPL mutations) using paired pre- and post-transformation samples. Evidence supports a role for IDH1/2 hotspot mutations in leukemic transformation.
IDH mutations are less commonly observed in childhood AML. In a study of 531 patients with childhood AML, IDH1 hotspot mutations were not found, though they were observed in adults with AML (46). Further investigation by the same group indicated that IDH2 mutations were also rare in this population (47). Another study examining patients from the Berlin-Frankfurt-Munster AML (AML-BFM) and Children’s Oncology Group (COG) study cohorts identified IDH mutations in 4% of 460 pediatric AML cases, most commonly in patients with AML classified according to the French-American-British classification system as M1/M2 (48), which is defined as immature or granulocytic differentiation by flow cytometry. In a study of children with a spectrum of malignancies, IDH1/2 mutations were found in only one of 115 AML patients (49). Recent studies have corroborated this prevalence and classification (50). In contrast, examination of AML in young adults (<60 years) showed that 6.6% of all patients harbored an IDH1 mutation (51), suggesting that IDH mutations are a consequence of age-related oncogenesis. Similar results have been observed for TET2 and DNMT3a mutations in pediatric AML (47, 50), suggesting that mutations in epigenetic modifiers are uncommon in children with AML and increase in frequency with increasing age. Alternatively, it is possible that IDH mutations occur early in leukemogenesis, and that there is a latent period between acquisition of IDH mutations, accumulation of 2HG, and leukemic transformation.
IDH mutations are not as broadly characterized in other hematopoietic disorders. In chronic myeloid leukemia (CML), mutant IDH has been suggested to play a role in progression to blast phase of advanced disease (52). Examination of acute lymphoblastic leukemia (ALL) has also shown limited prevalence of IDH mutation, including 3%–5% of adult ALL and 0% of childhood ALL (53, 54).
Prognosis for patients with IDH mutations
The prognostic significance of IDH mutations is of great interest in leukemia, particularly given that IDH mutations are associated with relatively favorable outcome in gliomas. In AML, patients with the R132H IDH1 mutation have lower overall survival (55, 56). Patients with R172K IDH2 mutation have lower rates of compete remission (5), and patients with this mutation have a worse prognosis than those with R140Q IDH2 mutations (57). However, the relevance of IDH mutations to outcome in AML is more complex than it first appears. In one study of newly diagnosed patients, IDH mutations were associated with older age and higher platelet levels, but were not independent predictors of survival (58). IDH2 mutations have no influence on treatment outcome in CN-AML (59), and IDH1 mutations appear to remain stable over the course of disease, are not acquired at relapse, and do not have any independent impact on survival (60). In childhood AML, IDH mutation was actually associated with increased overall survival, but this covariate was not found to be an independent predictor of survival (48). Of note, two recent studies have suggested that R140Q IDH2 mutations are associated with favorable outcome in younger adults treated with dose-intensive therapy (57, 61), suggesting that the prognostic relevance of IDH mutations may depend on the specific allele, patient age, or treatment regimen. By contrast, IDH mutations, particularly R172K IDH2 mutations, are associated with adverse outcome in older adults with AML, suggesting an age-dependent relevance of IDH mutations to outcome in AML (5).
Although the independent prognostic value of IDH mutations themselves varies in different studies, IDH has been defined as part of prognostic subsets in combination with other mutations in AML. IDH mutation has often been observed to cooccur with mutations in the nucleophosmin (NPM1) protein in adults (57, 58, 62) as well as children (48, 50). In an effort to determine if a larger set of mutant disease alleles can inform outcome in AML, Patel and colleagues performed mutational profiling of 502 AML patients younger than 60 years of age (63). Of note, NPM1/IDH-mutant patients without the FLT3 internal tandem duplication were found to compose a favorable subset of patients with outcome superior to patients with core-binding factor translocations, which historically have been observed to be associated with the most favorable outcome in AML (64). This observation may be specific to the specific mutation: one study suggested that NPM1 mutations coincident with IDH2 mutations had a lower risk of relapse, which was not observed for combined NPM1 and IDH1 mutations (57). Whether the differences in the prognostic relevance between IDH1 and IDH2 mutations are due to level of expression, differential cellular localizations, or relative production of 2HG remains to be delineated. In addition, the studies showing a favorable outcome for patients with FLT3-ITD–negative, NPM1/IDH-mutant AML were conducted in younger adults, and whether elderly patients with FLT3-ITD–negative, NPM1/IDH-mutant AML have similarly favorable outcomes with dose-intensive therapy or with less aggressive regimens needs to be delineated.
In other hematological disorders, prognosis in IDH mutation has not been explored as extensively. In one study of 88 ALL patients, all three patients with R132H IDH1 mutations relapsed or died within six months (54). In MDS, mutant IDH1 has been associated with shorter leukemia-free survival (65). IDH mutations are a common event in the progression from MPN to AML, and the incidence of IDH mutation seems to be independent of V617F JAK2 mutation (42). However, concurrent IDH and V617F JAK2 mutations have been shown to correlate with reduced leukemia-free survival in MPN patients (66). These data suggest that IDH mutations can serve as a marker of incipient transformation, which may predate clinical evidence of overt transformation in patients with chronic myeloid malignancies.
Detecting IDH mutation in clinical specimens
Several methods for detecting IDH mutations have been explored in a clinical context, including Sanger sequencing and multiplex assays, and these approaches have been used to track IDH mutation status over the course of disease and treatment. For instance, the SNaPshot Multiplex assay employs fluorophore-labeled allele-specfic primers to identify hotspot mutations (67, 68). When a SNaPshot assay was used to track IDH mutation status of three patients, the only patient with detectable R132H IDH1 mutations in the bone marrow after induction chemotherapy was also the only one of these patients to relapse (69). One study used a high-sensitivity multiplex PCR method to screen for R132 IDH1 mutations by creating a pool of mutation-specific primers that generate PCR products of different lengths in mutant versus wild-type samples (70). Another approach is to utilize PCR-denaturing high performance liquid chromatography (PCR-DHPLC) to detect heterozygous mutation before further examination with Sanger sequencing (58). Similarly, high-resolution melting is a rapid method that capitalizes on differential DNA melting curves to detect mutations in specific genes. This method has been used to detect different mutations and SNPs in IDH genes in patients with AML (71). This technique may serve as a valuable screening technique to make screening of clinical samples more efficient in comparison with Sanger sequencing (72).
Since the neomorphic function of IDH was identified, 2HG has been considered as a potential biomarker of IDH-related oncogenesis. Several studies have employed liquid-chromatography mass spectroscopy to explore this. A prospective study of patients with newly diagnosed AML serially examined 2HG levels in serum and urine, with parallel evaluation of IDH1/2 mutant allele burden in bone marrow. Both of these parameters correlated with treatment response, suggesting that 2HG might serve as an appropriate proxy for IDH mutation and as a marker of residual disease (69). Recently, DiNardo and colleagues measured serum 2HG levels in patients from the E1900 AML patient cohort and found that 2HG correlated with the presence of IDH1/2 mutations, regardless of the specific mutant allele (73). Consistent with the mutational data, high 2HG levels with cooccuring NPM1 mutations or without cooccuring DNMT3a mutations were associated with improved survival (73). Most importantly, residual 2HG detected in samples taken when patients were in clinical remission were associated with impaired survival, suggesting that 2HG may serve as a sensitive measure of minimal residual disease in IDH-mutant AML.
As 2HG emerges as a biomarker, alternative assays have been developed for more convenient measurement. An enzymatic assay for 2HG has been developed that assesses levels of NADH, which is produced stoichiometrically as 2HG is metabolized by recombinant 2HG dehydrogenase (74). Alternatively, given the well-explored relationship between 2HG production and TET2 function, hydroxymethylcytosine (5hmC) levels have been found to correlate with IDH mutational status and response to treatment in leukemia patients, indicating that hydroxymethylation of genomic DNA may serve as a biomarker of IDH mutation activity in AML (75).
IDH inhibitors
The development and preclinical validation of IDH inhibitors has emerged as an important goal in order to test the viability of mutant IDH1/2 as a therapeutic target and as a tool to help dissect the IDH/2HG pathway in different malignant contexts. One compound under investigation is compound 35, which is active against R132H IDH1 mutants, and preclinical tests have indicated that its application reduces 2HG production in cell lines and mouse xenograft models (35). A similar selective compound was more recently used in combination with an exogenous 2HG system to show that the cellular phenotype induced by IDH mutation is reversed by this drug, whereas the cellular phenotype induced by exogenous 2HG production is not (34). Recently, compounds targeting mutant IDH2 have been explored in leukemia cell lines (36) and glioma cell models (37). In both cases, IDH inhibitors were found to reduce 2HG production and inhibit the growth of leukemia or glioma cells in a mutant-specific manner. In addition, IDH inhibition led to global changes in DNA methylation/histone state and to induction of hematopoietic/neural differentiation, suggesting that these agents might induce differentiation in IDH-mutant cells through alterations in the epigenetic state. However, extensive in vivo studies in IDH-mutant transformation models remain to be reported, and the role of IDH in malignant cells after oncogenic transformation requires additional, extensive investigation.
IDH mutations in leukemia
The discovery of mutation in metabolic genes has given long-awaited credence to concept that altered metabolism is a hallmark of human cancers. However, the implications of mutations in genes such as IDH to oncogenesis are complex, and enormous potential lies in understanding the ramifications of this mutation, 2HG production, and downstream effectors such as TET2 in leukemia. Understanding this system will require the production of better models of IDH-mutant leukemias that incorporate the genetic complexity observed in human leukemia, particularly incorporation of many combinations of mutations. Clinically, the significance of IDH mutation must be examined in the context of other mechanistically important factors in larger cohorts with more comprehensive mutational and epigenetic analysis — and new technologies in screening, sequencing, and otherwise evaluating patient samples will help allow this to happen. As IDH inhibitors continue to be developed and to move closer to clinical testing, it will be come paramount to identify patient populations that stand to benefit the most from IDH-targeted therapy and to determine whether combination therapies should be investigated. Finally, while the role of IDH mutations in prognosis and therapeutic response requires further evaluation, there is clearly significant evidence that understanding its function in this disease will unlock significant mechanistic features of leukemogenesis. Particularly given its differential prognostic impact in glioma, a deeper understanding of these mutations and their neomorphic, oncogenic function will improve our understanding of carcinogenesis.
Acknowledgments
A.S. McKenney was supported by NIH MSTP grant GM07739.
Footnotes
Conflict of interest: Ross L. Levine reports receiving research funding from Agios Pharmaceuticals.
Citation for this article: J Clin Invest. 2013;123(9):3672–3677. doi:10.1172/JCI67266.
References
- 1.Sjöblom T, et al. The consensus coding sequences of human breast and colorectal cancers. Science. 2006;314(5797):268–274. doi: 10.1126/science.1133427. [DOI] [PubMed] [Google Scholar]
- 2.Parsons DW, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321(5897):1807–1812. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mardis ER, et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med. 2009;361(11):1058–1066. doi: 10.1056/NEJMoa0903840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gross S, et al. Cancer-associated metabolite 2-hydroxyglutarate accumulates in acute myelogenous leukemia with isocitrate dehydrogenase 1 and 2 mutations. J Exp Med. 2010;207(2):339–344. doi: 10.1084/jem.20092506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Marcucci G, et al. IDH1 and IDH2 gene mutations identify novel molecular subsets within de novo cytogenetically normal acute myeloid leukemia: a Cancer and Leukemia Group B study. J Clin Oncol. 2010;28(14):2348–2355. doi: 10.1200/JCO.2009.27.3730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ward PS, et al. Identification of additional IDH mutations associated with oncometabolite R(–)-2-hydroxyglutarate production. Oncogene. 2012;31(19):2491–2498. doi: 10.1038/onc.2011.416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dang L, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2009;462(7274):739–744. doi: 10.1038/nature08617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ward PS, et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell. 2010;17(3):225–234. doi: 10.1016/j.ccr.2010.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Figueroa ME, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell. 2010;18(6):553–567. doi: 10.1016/j.ccr.2010.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xu W, et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer Cell. 2011;19(1):17–30. doi: 10.1016/j.ccr.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Reitman ZJ, Yan H. Isocitrate dehydrogenase 1 and 2 mutations in cancer: alterations at a crossroads of cellular metabolism. J Natl Cancer Inst. 2010;102(13):932–941. doi: 10.1093/jnci/djq187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yen KE, Bittinger MA, Su SM, Fantin VR. Cancer-associated IDH mutations: biomarker therapeutic opportunities. Oncogene. 2010;29(49):6409–6417. doi: 10.1038/onc.2010.444. [DOI] [PubMed] [Google Scholar]
- 13.Yan H, et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med. 2009;360(8):765–773. doi: 10.1056/NEJMoa0808710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhao S, et al. Glioma-derived mutations in IDH1 dominantly inhibit IDH1 catalytic activity and induce HIF-1α. Science. 2009;324(5924):261–265. doi: 10.1126/science.1170944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lu C, et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature. 2012;483(7390):474–478. doi: 10.1038/nature10860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ono R, Taki T, Taketani T, Taniwaki M, Kobayashi H, Hayashi Y. LCX, leukemia-associated protein with a CXXC domain, is fused to MLL in acute myeloid leukemia with trilineage dysplasia having t(10;11)(q22;q23). Cancer Res. 2002;62(14):4075–4080. [PubMed] [Google Scholar]
- 17.Lorsbach RB, Moore J, Mathew S, Raimondi SC, Mukatira ST, Downing JR. TET1, a member of a novel protein family, is fused to MLL in acute myeloid leukemia containing the t(10;11)(q22;q23). Leukemia. 2003;17(3):637–641. doi: 10.1038/sj.leu.2402834. [DOI] [PubMed] [Google Scholar]
- 18.Tahiliani M, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324(5929):930–935. doi: 10.1126/science.1170116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ito S, D’Alessio AC, Taranova OV, Hong K, Sowers LC, Zhang Y. Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature. 2010;466(7310):1129–1133. doi: 10.1038/nature09303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cimmino L, Abdel-Wahab O, Levine RL, Aifantis I. TET family proteins and their role in stem cell differentiation and transformation. Cell Stem Cell. 2011;9(3):193–204. doi: 10.1016/j.stem.2011.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guo JU, Su Y, Zhong C, Ming GL, Song H. Hydroxylation of 5-methylcytosine by TET1 promotes active DNA demethylation in the adult brain. Cell. 2011;145(3):423–434. doi: 10.1016/j.cell.2011.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Williams K, et al. TET1 and hydroxymethylcytosine in transcription and DNA methylation fidelity. Nature. 2011;473(7347):343–348. doi: 10.1038/nature10066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ficz G, et al. Dynamic regulation of 5-hydroxymethylcytosine in mouse ES cells and during differentiation. Nature. 2011;473(7347):398–402. doi: 10.1038/nature10008. [DOI] [PubMed] [Google Scholar]
- 24.Wu H, et al. Dual functions of Tet1 in transcriptional regulation in mouse embryonic stem cells. Nature. 2011;473(7347):389–393. doi: 10.1038/nature09934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ito S, et al. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science. 2011;333(6047):1300–1303. doi: 10.1126/science.1210597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.He YF, et al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science. 2011;333(6047):1303–1307. doi: 10.1126/science.1210944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Delhommeau F, et al. Mutation in TET2 in myeloid cancers. N Engl J Med. 2009;360(22):2289–2301. doi: 10.1056/NEJMoa0810069. [DOI] [PubMed] [Google Scholar]
- 28.Langemeijer SM, et al. Acquired mutations in TET2 are common in myelodysplastic syndromes. Nat Genet. 2009;41(7):838–842. doi: 10.1038/ng.391. [DOI] [PubMed] [Google Scholar]
- 29.Jankowska AM, et al. Loss of heterozygosity 4q24 and TET2 mutations associated with myelodysplastic/myeloproliferative neoplasms. Blood. 2009;113(25):6403–6410. doi: 10.1182/blood-2009-02-205690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Abdel-Wahab O, et al. Genetic characterization of TET1, TET2, and TET3 alterations in myeloid malignancies. Blood. 2009;114(1):144–147. doi: 10.1182/blood-2009-03-210039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ko M, et al. Impaired hydroxylation of 5-methylcytosine in myeloid cancers with mutant TET2. Nature. 2010;468(7325):839–843. doi: 10.1038/nature09586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Moran-Crusio K, et al. Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer Cell. 2011;20(1):11–24. doi: 10.1016/j.ccr.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sasaki M, et al. IDH1(R132H) mutation increases murine haematopoietic progenitors and alters epigenetics. Nature. 2012;488(7413):656–659. doi: 10.1038/nature11323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Losman JA, et al. (R)-2-hydroxyglutarate is sufficient to promote leukemogenesis its effects are reversible. Science. 2013;339(6127):1621–1625. doi: 10.1126/science.1231677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Popovici-Muller J, et al. Discovery of the first potent inhibitors of mutant IDH1 that lower tumor 2-HG in vivo. ACS Med Chem Lett. 2012;3(10):850–855. doi: 10.1021/ml300225h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang F, et al. Targeted inhibition of mutant IDH2 in leukemia cells induces cellular differentiation. Science. 2013;340(6132):622–626. doi: 10.1126/science.1234769. [DOI] [PubMed] [Google Scholar]
- 37.Rohle D, et al. An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science. 2013;340(6132):626–630. doi: 10.1126/science.1236062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zou Y, Zeng Y, Zhang DF, Zou SH, Cheng YF, Yao YG. IDH1 and IDH2 mutations are frequent in Chinese patients with acute myeloid leukemia but rare in other types of hematological disorders. Biochem Biophys Res Commun. 2010;402(2):378–383. doi: 10.1016/j.bbrc.2010.10.038. [DOI] [PubMed] [Google Scholar]
- 39.Patel KP, et al. Acute myeloid leukemia with IDH1 or IDH2 mutation: frequency clinicopathologic features. Am J Clin Pathol. 2011;135(1):35–45. doi: 10.1309/AJCPD7NR2RMNQDVF. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ibáñez M, et al. Screening for IDH mutations in chronic myelomonocytic leukemia. Leuk Lymphoma. 2013;54(2):406–407. doi: 10.3109/10428194.2012.701295. [DOI] [PubMed] [Google Scholar]
- 41.Caramazza D, et al. IDH mutations and trisomy 8 in myelodysplastic syndromes and acute myeloid leukemia. Leukemia. 2010;24(12):2120–2122. doi: 10.1038/leu.2010.213. [DOI] [PubMed] [Google Scholar]
- 42.Pardanani A, Lasho TL, Finke CM, Mai M, McClure RF, Tefferi A. IDH1 and IDH2 mutation analysis in chronic- and blast-phase myeloproliferative neoplasms. Leukemia. 2010;24(6):1146–1151. doi: 10.1038/leu.2010.77. [DOI] [PubMed] [Google Scholar]
- 43.Abdel-Wahab O, et al. Genetic analysis of transforming events that convert chronic myeloproliferative neoplasms to leukemias. Cancer Res. 2010;70(2):447–452. doi: 10.1158/0008-5472.CAN-09-3783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang SJ, et al. Genetic analysis of patients with leukemic transformation of myeloproliferative neoplasms shows recurrent SRSF2 mutations that are associated with adverse outcome. Blood. 2012;119(19):4480–4485. doi: 10.1182/blood-2011-11-390252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tefferi A, et al. IDH mutations in primary myelofibrosis predict leukemic transformation and shortened survival: clinical evidence for leukemogenic collaboration with JAK2V617F. Leukemia. 2012;26(3):475–480. doi: 10.1038/leu.2011.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ho PA, et al. Molecular alterations of the IDH1 gene in AML: a Children’s Oncology Group and Southwest Oncology Group study. Leukemia. 2010;24(5):909–913. doi: 10.1038/leu.2010.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ho PA, et al. Leukemic mutations in the methylation-associated genes DNMT3A and IDH2 are rare events in pediatric AML: a report from the Children’s Oncology Group. Pediatr Blood Cancer. 2011;57(2):204–209. doi: 10.1002/pbc.23179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Damm F, et al. Prevalence and prognostic value of IDH1 and IDH2 mutations in childhood AML: a study of the AML-BFM and DCOG study groups. Leukemia. 2011;25(11):1704–1710. doi: 10.1038/leu.2011.142. [DOI] [PubMed] [Google Scholar]
- 49.Oki K, et al. IDH1 and IDH2 mutations are rare in pediatric myeloid malignancies. Leukemia. 2011;25(2):382–384. doi: 10.1038/leu.2010.307. [DOI] [PubMed] [Google Scholar]
- 50.Liang DC, et al. Cooperating gene mutations in childhood acute myeloid leukemia with special reference on mutations of ASXL1, TET2, IDH1, IDH2, and DNMT3A. Blood. 2013;121(15):2988–2995. doi: 10.1182/blood-2012-06-436782. [DOI] [PubMed] [Google Scholar]
- 51.Schnittger S, Haferlach C, Ulke M, Alpermann T, Kern W, Haferlach T. IDH1 mutations are detected in 6.6% of 1414 AML patients and are associated with intermediate risk karyotype and unfavorable prognosis in adults younger than 60 years and unmutated NPM1 status. Blood. 2010;116(25):5486–5496. doi: 10.1182/blood-2010-02-267955. [DOI] [PubMed] [Google Scholar]
- 52.Makishima H, et al. CBL, CBLB, TET2, ASXL1, and IDH1/2 mutations and additional chromosomal aberrations constitute molecular events in chronic myelogenous leukemia. Blood. 2011;117(21):e198–e206. doi: 10.1182/blood-2010-06-292433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tang JY, Chang CC, Lin PC, Chang JG. Isocitrate dehydrogenase mutation hot spots in acute lymphoblastic leukemia and oral cancer. Kaohsiung J Med Sci. 2012;28(3):138–144. doi: 10.1016/j.kjms.2011.10.023. [DOI] [PubMed] [Google Scholar]
- 54.Zhang Y, et al. Mutation analysis of isocitrate dehydrogenase in acute lymphoblastic leukemia. Genet Test Mol Biomarkers. 2012;16(8):991–995. doi: 10.1089/gtmb.2011.0323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Koszarska M, et al. Type and location of isocitrate dehydrogenase mutations influence clinical characteristics and disease outcome of acute myeloid leukemia. Leuk Lymphoma. 2013;54(5):1028–1035. doi: 10.3109/10428194.2012.736981. [DOI] [PubMed] [Google Scholar]
- 56.Boissel N, et al. Prognostic impact of isocitrate dehydrogenase enzyme isoforms 1 and 2 mutations in acute myeloid leukemia: a study by the Acute Leukemia French Association group. J Clin Oncol. 2010;28(23):3717–3723. doi: 10.1200/JCO.2010.28.2285. [DOI] [PubMed] [Google Scholar]
- 57.Green CL, et al. The prognostic significance of IDH2 mutations in AML depends on the location of the mutation. Blood. 2011;118(2):409–412. doi: 10.1182/blood-2010-12-322479. [DOI] [PubMed] [Google Scholar]
- 58.Chotirat S, Thongnoppakhun W, Promsuwicha O, Boonthimat C, Auewarakul CU. Molecular alterations of isocitrate dehydrogenase 1 and 2 (IDH1 and IDH2) metabolic genes and additional genetic mutations in newly diagnosed acute myeloid leukemia patients. J Hematol Oncol. 2012;5:5. doi: 10.1186/1756-8722-5-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Thol F, et al. Prognostic impact of IDH2 mutations in cytogenetically normal acute myeloid leukemia. Blood. 2010;116(4):614–616. doi: 10.1182/blood-2010-03-272146. [DOI] [PubMed] [Google Scholar]
- 60.Chou WC, et al. Distinct clinical and biologic characteristics in adult acute myeloid leukemia bearing the isocitrate dehydrogenase 1 mutation. Blood. 2010;115(14):2749–2754. doi: 10.1182/blood-2009-11-253070. [DOI] [PubMed] [Google Scholar]
- 61.Patel JP, et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med. 2012;366(12):1079–1089. doi: 10.1056/NEJMoa1112304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Abbas S, et al. Acquired mutations in the genes encoding IDH1 and IDH2 both are recurrent aberrations in acute myeloid leukemia: prevalence and prognostic value. Blood. 2010;116(12):2122–2126. doi: 10.1182/blood-2009-11-250878. [DOI] [PubMed] [Google Scholar]
- 63.Patel JP, et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med. 2012;366(12):1079–1089. doi: 10.1056/NEJMoa1112304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Slovak ML, et al. Karyotypic analysis predicts outcome of preremission and postremission therapy in adult acute myeloid leukemia: a Southwest Oncology Group/Eastern Cooperative Oncology Group Study. Blood. 2000;96(13):4075–4083. [PubMed] [Google Scholar]
- 65.Patnaik MM, et al. Differential prognostic effect of IDH1 versus IDH2 mutations in myelodysplastic syndromes: a Mayo Clinic study of 277 patients. Leukemia. 2012;26(1):101–105. doi: 10.1038/leu.2011.298. [DOI] [PubMed] [Google Scholar]
- 66.Tefferi A, et al. IDH mutations in primary myelofibrosis predict leukemic transformation and shortened survival: clinical evidence for leukemogenic collaboration with JAK2V617F. Leukemia. 2012;26(3):475–480. doi: 10.1038/leu.2011.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Borger DR, et al. Frequent mutation of isocitrate dehydrogenase (IDH)1 and IDH2 in cholangiocarcinoma identified through broad-based tumor genotyping. Oncologist. 2012;17(1):72–79. doi: 10.1634/theoncologist.2011-0386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dias-Santagata D, et al. Rapid targeted mutational analysis of human tumours: a clinical platform to guide personalized cancer medicine. EMBO Mol Med. 2010;2(5):146–158. doi: 10.1002/emmm.201000070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Fathi AT, et al. Prospective serial evaluation of 2-hydroxyglutarate, during treatment of newly diagnosed acute myeloid leukemia, to assess disease activity and therapeutic response. Blood. 2012;120(23):4649–4652. doi: 10.1182/blood-2012-06-438267. [DOI] [PubMed] [Google Scholar]
- 70.Chou WC, Huang YN, Huang CF, Tseng MH, Tien HF. A single-tube, sensitive multiplex method for screening of isocitrate dehydrogenase 1 (IDH1) mutations. Blood. 2010;116(3):495–496. doi: 10.1182/blood-2010-04-280636. [DOI] [PubMed] [Google Scholar]
- 71.Ibáñez M, et al. Rapid screening of ASXL1, IDH1, IDH2, and c-CBL mutations in de novo acute myeloid leukemia by high-resolution melting. J Mol Diagn. 2012;14(6):594–601. doi: 10.1016/j.jmoldx.2012.06.006. [DOI] [PubMed] [Google Scholar]
- 72.Patel KP, et al. Diagnostic testing for IDH1 and IDH2 variants in acute myeloid leukemia an algorithmic approach using high-resolution melting curve analysis. J Mol Diagn. 2011;13(6):678–686. doi: 10.1016/j.jmoldx.2011.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.DiNardo CD, et al. Serum 2-hydroxyglutarate levels predict isocitrate dehydrogenase mutations and clinical outcome in acute myeloid leukemia. Blood. 2013;121(24):4917–4924. doi: 10.1182/blood-2013-03-493197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Balss J, et al. Enzymatic assay for quantitative analysis of (D)-2-hydroxyglutarate. Acta Neuropathol. 2012;124(6):883–891. doi: 10.1007/s00401-012-1060-y. [DOI] [PubMed] [Google Scholar]
- 75.Pollyea DA, et al. 2-Hydroxyglutarate in IDH mutant acute myeloid leukemia: predicting patient responses, minimal residual disease and correlations with methylcytosine and hydroxymethylcytosine levels. Leuk Lymphoma. 2013;54(2):408–410. doi: 10.3109/10428194.2012.701009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tefferi A, et al. IDH1 and IDH2 mutation studies in 1473 patients with chronic-, fibrotic- or blast-phase essential thrombocythemia, polycythemia vera or myelofibrosis. Leukemia. 2010;24(7):1302–1309. doi: 10.1038/leu.2010.113. [DOI] [PMC free article] [PubMed] [Google Scholar]