

Abstract
Molecularly targeted agents promise to revolutionize therapeutics by reducing morbidity and mortality in patients with cancer. However, despite an urgent need for more effective anticancer compounds, current preclinical drug evaluations largely fail to satisfy the demand. New preclinical strategies, including the improvement of sophisticated mouse models and co-clinical study designs, are being used to augment the predictive value of animal-based translational cancer research. Here, we review the development of successful preclinical antineoplastic agents, their associated limitations, and alternative methods to predict clinical outcomes.
Introduction
The laboratory mouse Mus musculus is frequently used for preclinical studies to evaluate anticancer drugs, because approximately 99% of its genes overlap with those of Homo sapiens and the mouse genome can be readily manipulated. However, the genomic identity between H. sapiens and M. musculus cannot compensate for significant species differences in physiology, anatomy, metabolism, biochemistry, pharmacokinetics, and toxicokinetics. Several studies have demonstrated that the efficacy of drug regimens established in mouse models is infrequently translated to the clinic (1–3), indicating that conventional mouse cancer models are historically poor predictors of clinical efficacy. Although recent studies have suggested that alternative, more sophisticated murine models may be more predictive of drug responses and resistance observed in the clinic (1, 4–10), comprehensive comparative studies are still missing today.
The inconsistencies in translating results from mouse models to predict clinical treatment outcomes may be technical and thus may be overcome with models that better recapitulate disease biology and response criteria that are more closely aligned between mouse and human. Recently, the concept of “co-clinical trials” has been introduced, in which drug efficacy is tested in parallel in humans and mice (8, 11). These trials require that the animal model mirrors the human counterpart as closely as possible and that the study designs for both species are strictly aligned.
Here, we focus on emerging murine models of cancer — genetically engineered mouse (GEM) and patient-derived xenograft (PDX) models — and illustrate aspects of an enhanced preclinical study design that may improve the predictive value of preclinical trials.
Current challenges
Although genome-sequencing technologies have elucidated molecular mechanisms and complexities that underlie neoplastic disease, the in vivo validation of targeted compounds is still a major hurdle for accurate prediction and reliable translation to humans. In fact, many compounds yield encouraging preclinical efficacy, but only 9% of candidates demonstrate robust clinical performance and are eventually approved by the FDA (12). This enormous attrition rate can be attributed to several phenomena. First, conventional models rely on cell lines that are selected for growth under nonphysiological conditions. Second, disease complexity and limited genome-engineering technologies make it impossible to develop GEM models that identically mimic the full complexity of human malignancies. Third, xenograft models require immunodeficient hosts that do not replicate the normal tumor-host microenvironment. Fourth, the measures of success in preclinical studies (e.g., slowing tumor growth) are not congruent with clinical criteria for success (e.g., tumor regression). Finally, the emergence of novel molecularly targeted compounds requires assessment methods that differ from those used to validate cytotoxic agents. Hence, the question remains: what elements are required for the successful development of novel anticancer therapeutics?
Finding the perfect cast
No performance can go on without the cast of characters; ideally, the actors will be perfectly selected as to make the show succeed. Although three mouse models are currently used for cancer research (reviewed in refs. 13–15), two vie for best proxy for patients with cancer: PDX models and GEM models (compared in Figure 1). The third model, conventional xenografts, is still widely used for drug response assessment in complex biological systems, briefly introduced here.
Figure 1. Comparative analysis of GEM and PDX models to mimic human malignancies.
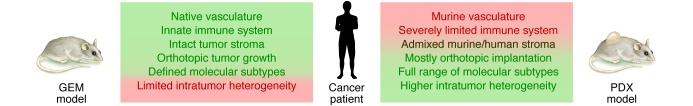
Green indicates similar features, red indicates divergent attributes.
Conventional xenograft models use subcutaneous implants of cultured human cells in immunocompromised host mice. However, these cell lines themselves can be a source of artifact, due to artificial genetic and epigenetic changes induced by in vitro propagation under nonphysiological growth conditions (16). Subsequently, these cells become less differentiated, more homogeneous, and have accelerated doubling times when cultured (17–19). Although subcutaneous engraftment allows for easy assessment of tumor size, this is generally an ectopic location, which does not fully replicate the natural tumor microenvironment, as tissue- or organ-specific properties significantly contribute to tumor progression and modulate therapeutic response (20). Finally, xenografts exclude the important interactions between immune and cancer cells during tumor initiation, maintenance, and response to treatment (21). Two emerging trends in mouse modeling have sought to address some of these known shortcomings of conventional xenograft models.
PDX models.
Over the past decades, PDX models have gained popularity as an alternative to conventional cell line–based xenografts. These models consist of explanted fragments of tumor tissue (usually 20–50 mm3) that are directly transferred into immunosuppressed recipient animals and can be propagated over several generations. The key advantages of this model for examining therapeutic responses include the preservation of molecular diversity among tumor cells, recapitulation of the human tumor microenvironment from cotransplantation of supporting human stroma, and the availability of molecular characterization of tumor material. Several groups have published data that demonstrate the potential of PDX models to anticipate personalized anticancer treatment (4, 22, 23). In a recent pilot study, Hidalgo et al. used this approach to identify effective personalized treatments for patients with various advanced refractory cancers (22). After propagation of tumor samples from 14 patients, the resulting PDX-bearing mice, or xenopatients, were treated with 63 drugs in 232 treatment regimens. Based on the response data, clinical treatments were effective for 11 out of 14 patients. After the successful translation from PDX models into the clinical setting, these personalized models were dubbed mouse avatars (24).
Despite the success of PDX, the model has some weaknesses. Genomic and stromal stability of PDX upon serial transplantation is a common concern, as genetic drift and/or replacement of human stroma by murine fibroblasts would significantly alter the original composition of the tumor. Comparative genome hybridization (25, 26) or comprehensive genome-wide gene-expression analysis (27, 28) performed on early-passage tumor grafts demonstrated genomic stability between the original tumor and its progeny, which can even be extended over multiple tumor graft generations (29–32). Direct analysis addressing the fate of stromal components of the original graft or stromal changes upon propagation are still warranted today (28). However, recent studies by Hylander et al. demonstrated that the human microvasculature of a colon PDX is partially replaced by host vessels as early as three weeks (33). Additionally, Lin et al. reported that more than 40% of analyzed pancreatic PDXs harbored less human than mouse DNA, supporting the presence of infiltrating murine cells (34).
A critical limitation of the PDX model is the lack of a functional immune system. Although cancer treatments increasingly incorporate immune-based therapies, it remains to be determined whether mouse models faithfully mimic human immunological cancer defense. Grafting of PDXs into “humanized’ rodent models seems a logical strategy to overcome this ambiguity. Although restoration of the human immune system in mice is largely successful, functionality was reported incomplete due to missing HLA class I and class II elements for T cell differentiation in the murine host (35).
Two emerging concepts potentially circumvent this species-specific immune barrier. Kalscheuer et al. demonstrated that injection of human HSCs from bone marrow of adults, along with implantation of HLA-matched human fetal thymic tissue into immunodeficient mice, resulted in functional T cell, B cell, and myeloid cell reconstitution (36). Specifically, newly generated T cells were self tolerant and had a diverse repertoire. In contrast, Wege et al. simultaneously transplanted human breast cancer cells and human fetal HSCs into neonatal NOD-scid Il2rg–/– mice, which led to specific T cell maturation and tumor cell–specific T cell activation (37). Further, despite MHC incompatibility of injected tumor cells and human HSCs, no signs of rejection were detected. However, the presence of human tumor cells triggered activation of CD4+ T cells and expansion of natural killer cells (37); notably, this immune response did not result in immunological elimination of the engrafted tumor cells, thus mirroring the human response.
Another obstacle is failed engraftment of transplanted material, which can be greater than 90%, depending on the type of cancer (30). These engraftment failures may be due to the absence of a tumor-supporting niche enriched with human stromal tissue or due to ectopic implantation. DeRose et al. provoked increased tumor growth and graft stability, without improving engraftment rate, by coinjecting primary human mesenchymal stem cells with breast tumor grafts into the mammary fat pads of NOD-scid mice (38). This enhanced tumor bed, likely associated with enhanced vascularization, suggests that stabilizing the tumor-supporting niche can improve engraftment. In support of the importance of implantation site, Dong et al. showed that implantation of resected non–small cell lung cancers under the renal capsules of NOD-scid mice not only significantly increased the engraftment rate (90% vs. 30%–40%; refs. 27, 39), but also allowed rapid drug testing (within 6–8 weeks) to prevent unnecessary treatment delays (40). Although only 16 out of 32 resected tumors provided sufficient tissue to assess the response to three different chemotherapy regimens, good concordance of treatment response was observed in 11 patients and corresponding PDX models (40). A particularly high correlation of resistance to conventional therapy was observed between mice and humans.
Although several reports of highly correlated therapeutic outcomes using ectopic engraftment may encourage use of this methodology (4, 22, 23), the majority of PDX is currently implanted at the original anatomical site to model a more relevant microenvironment providing a complex mixture of proneoplastic and antineoplastic factors (41–43). Subsequently, the propensity for metastasis was found greatly enhanced after orthotopical engraftment (44–47). Although the techniques of orthotopic implantation are time consuming, technically challenging, and require sufficient methodology for labeling and imaging these tumors (48), a more accurate tumor model is generated.
Understandably, the advent of PDX models has fueled hope of patients and clinicians that these models can identify patient-tailored treatment options — being either optimized first-line therapy or alternative options bypassing treatment-associated resistance. Nevertheless, a noticeable caveat of this approach is the long latency, as generation and “screening” may take several months depending on the engraftment rate and therapeutic regimen, which patients with advanced disease might not survive. Instead, PDXs are currently much more beneficial for establishing tumor type–specific panels, which harbor a representative spectrum of molecularly alterations found in human malignancies. These platforms are valuable to test multiple therapeutic strategies and to identify mechanisms of drug sensitivity or resistance in genetically defined models. The continuous expansion of PDX repositories might further allow for significant, genetically stratified study cohorts to parallel human trials in a co-clinical trial.
The demonstrated predictive value of the PDX model is a cornerstone of modern biomedical research; however, further efforts are warranted to transform the PDX model into a true avatar of patients with cancer.
GEM models.
To mimic alterations found in human malignancies, the genetic profiles of GEM models are manipulated such that one or more genes is mutated, deleted, or overexpressed. In comparison with PDX models, altering cancer-associated genes allows orthotopic tumor initiation and progression supported by autochthonous tumor stroma and vasculature. Additionally, the presence of a functional innate immune system influences tumorigenic development (49).
Each GEM model should meet several general criteria: (a) accurate representation of precise molecular events that occur in human tumors; (b) expression of manipulated genes within endogenous loci, rather than as transgenes; (c) temporal activation of the mutated gene(s) to recapitulate human tumorigenesis; (d) tissue-specific mutations; (e) occurrence of disease-defining lesions in a limited number of cells; (f) concordance of tumor type and histopathological features with the human malignancy; and (g) similar initiation-to-progression sequence with the human malignancy (50).
Although GEM models can dissect the effects of specific genomic alterations, they are frequently criticized for incomplete representation of human tumor genetic complexity. Indeed, the relatively benign cytogenetic profile in murine tumors contrasts the genomic instability of human cancers (51, 52). Experimental destabilization of murine cancer genomes can be modeled through telomere dysfunction, impaired DNA damage checkpoints, and defective DNA repair to allow emergence of resistance mechanisms in response to antineoplastic therapies (53–57).
The functional significance of tumor-promoting genetic lesions can be divided into driver mutations, which are essential for malignant growth, and passenger mutations, which appear to be less significant (58). Whereas murine tumors usually form upon activation of single-dominant driver mutations, human tumor initiation depends on multiple alterations in key signaling pathways. Similarly, close investigation is warranted to determine whether the temporal accumulation of mutations in human malignancies needs equal consideration in GEM models. Attempts to address the relevance of these model deficiencies included comparative analyses of regulatory interaction networks, namely interactomes, of murine and human prostate cancers to distinguish causally responsible driver mutations and facilitate identification of new targets within dysregulated signaling pathways in prostate cancer (Cory Abate-Shen, personal communication).
GEM models notoriously fail to recapitulate the metastatic features of human malignancies. Whether syngeneic implantation of GEM-derived allografts allows similar enhanced metastasis, as in orthotopic xenograft models, is currently under intense investigation. However, induction of multifocal cancer lesions in syngeneic recipients is well established for GEM models of hematologic diseases (59, 60). Nonetheless, GEM models have gained an invaluable role in molecularly targeted drug development and will be valuable tools to dissect drug resistance mechanisms.
The leading role.
Direct comparison of PDX and GEM models might equally predict a patient’s response to antineoplastic therapy. Applying the criteria of an “ideal” preclinical model for novel antineoplastic compound development, as described by Ocana et al. (validation of the target, information about the mechanism of action, identification of pharmacodynamics [PD] activity markers, characterization of the toxicity profile, and identification of resistance mechanisms and how to bypass them) (61), might lead to the assumption that GEM models better fit these criteria over PDX. Although the former provides a powerful experimental platform for mining disease progression or drug response biomarkers and can be further used for drug resistance characterization, the strengths of the latter comprise genetic complexity of human cancers and potential for cross-species drug evaluation (e.g., biologics).
Along these lines, the debate on the rationale of using GEM models to assess immune-modulating cancer therapeutics often centers on the dissimilarity of the human and murine immune system. However, in the absence of any alternative model harboring a functional innate immune system, only GEM models can be tested for their predictability of clinical outcome. Ideally, PDX and GEM models are ultimately used in a complementary manner to improve the predictions for clinical trials.
Writing the perfect script
Even if an actor’s performance is flawless, a promising story will not become a blockbuster without a compelling script; the same holds true for preclinical trial design. Close alignment and synchronization of murine and human trials is integral to predicting treatment responses, although detailed comparison of preclinical and clinical drug intervention studies reveals unintentional design disparities. For example, preclinical trials rarely meet external and internal validation criteria, such as the use of several different mouse models, hypothesis-driven design and statistics, and discrimination against proof-of-principle experiments, that are required for clinical trials (62).
How well do mice play the role?
An ideal murine preclinical study should not only be randomized and blindly evaluated, but also should consist of a sufficient sample size with gender and genetic background stratification to ensure internal validity. Beyond these parameters, a number of aspects potentially impact the translation of murine results to humans.
Appropriate age-related translation remains difficult because the life expectancy of mice (approximately 18–24 months) and humans (approximately 70 years) varies significantly. While the majority of patients with cancer are older than 55 years, murine tumor induction is initiated shortly after sexual maturity (6–12 weeks). Tumor latency is also prolonged in humans compared with that in mice. Finally, a significant number of patients with cancer face preexisting age-related comorbidities at diagnosis, which are difficult to model in mice. This discrepancy is usually balanced by requiring good performance status and adequate organ function in study-eligible patients with cancer to minimize clinical deterioration.
Additionally, the cancer-killing potential of novel drugs in preclinical studies is mostly assessed in treatment-naive tumors. Conversely, based on ethical considerations, only human patients with tumors refractory to several lines of treatment are enrolled in phase I and phase II clinical trials. These differences can lead to misinterpretation and prediction errors for drug responses. Further, the majority of enrolled patients suffer from advanced metastatic disease in multiple organ sites, which is insufficiently modeled in PDX and GEM. This inconsistency additionally biases novel cancer compound evaluation across species.
Finally, whereas human tolerance of antineoplastics is improved by several kinds of supportive drugs, these additives are rarely administered in mice. Whether absence of modeled drug-drug interactions is of prime relevance remains enigmatic even today. However, recent in vivo studies suggest that these concerns may be minor, because therapeutic response rates in mouse models closely mirror those in the clinic (8, 9).
Evaluation of therapeutic response.
Treatment efficacy evaluation is at the core of every drug intervention study. Despite the importance of parallel features of preclinical and clinical trials, identical “success criteria” are frequently disregarded in murine experiments. Whereas complete arrest of tumor growth upon antineoplastic treatment translates into the minimal clinical “success” (with tumor regression being more desired), simple diminution of tumor growth in preclinical studies is regarded efficacious as long as statistical significance is achieved.
Classically, tumor shrinkage reflects efficacy after cytotoxic therapies, because these compounds inhibit cell division. These observations led to the development of internationally standardized clinical Response Evaluation Criteria In Solid Tumors (RECIST), which are based on size changes of the target lesion (63). Accordingly, the most widely used techniques for tumor assessment after cytotoxic therapies in patients are noninvasive imaging modalities like PET, MRI, and/or CT. Although the parameters and biomarkers used to quantify treatment responses in mice and humans should prove testable and relevant in both settings, sophisticated imaging technologies for small animal use are not always available or affordable.
Molecularly targeted anticancer compounds, not directed toward the cell division machinery, frequently exert cytostatic effects, which result in tumor stabilization without induction of tumor shrinkage (64). Functional metabolomic changes rather than tumor mass dynamics are therefore preferred response parameters for these drug types. Although PET scans offer sufficient accuracy to assess treatment responses, their cost and availability is prohibitive for murine studies. Alternatively, fluorescence (65, 66) and bioluminescence (67, 68) optical imaging can be used to evaluate functional drug effects. Genetic modification of the target cells to express either luciferase or a fluorescent protein is inevitable and applicable for PDX cultured ex vivo for a short time (e.g., leukemia, glioblastoma multiforme) (69, 70). Although fluorescence-based imaging offers the possibility to simultaneously monitor several different fluorescent proteins, the poor tissue penetration of a few millimeters limits its use to GEM models with superficially growing tumors (e.g., lymphoma) (71, 72). In comparison, luciferase-mediated bioluminescence has superior tissue penetration and is used in GEM models with deep tumor localization (73, 74).
Adequate drug efficacy assessment is not only a matter of measuring response, but also depends on the optimal measurement time and suitable end points. Novel targeted agents differ significantly from traditional cytotoxic agents in terms of administration schedules and routes and toxicity profiles. Among the essential differences are daily oral application versus cyclic intravenous application and nonhematologic toxicity (e.g., cardiovascular, refs. 75, 76; gastrointestinal, ref. 77; cutaneous, ref. 78; or renal, ref. 79) versus hematologic toxicity. Therefore, the methodology of early clinical trials developed for agents targeting the cell division machinery might not be suitable to evaluate different targeted compounds.
Clinical trials often evaluate the overall survival of participating patients; the translation of this efficacy criterion is lethality in mice. This assumption might be misleading for several reasons. In humans, metastatic spread of the primary tumor is considered the major cause of death from cancer (80); however, only a few PDX and GEM models recapitulate this feature of human neoplasia. Further, detailed assessment of potential toxicities caused by the compound under study is usually missing in preclinical trials, which could be another mechanism of mortality in mice rather than advanced tumor burden. Thus, the underlying causes leading to overall survival data differ significantly between preclinical and clinical studies. Recent reports from an in-depth series of preclinical case studies performed in two highly validated Kras-mutant GEM models also suggest that progression-free survival may be better qualified for outcome prediction (9).
The act
Even if the cast and script are optimal, the act itself can fall short. Another major roadblock to expedient cancer drug development remains — identifying the most accurate treatment regimen during preclinical trials. Presently, there is no satisfactory solution to address this problem. Although we identified the GEM model as a close model of human malignancies, species-specific differences remain and therefore hinder assumptions on effective dosing schedules. Validated PD biomarkers have increasing roles in phase I clinical trials (81), suggesting that they may also help inform preclinical study design and define the biologically active dose for phase II clinical trials. This potential paradigm shift has an even more pronounced impact on resulting toxicities.
The popularity of PD biomarkers is associated with the emergence of additional molecularly targeted agents, which do not require the maximum tolerated dose to be most effective. Daily oral administration of these agents, as opposed to intermittent intravenous administration of cytotoxic agents, may induce chronic toxicities that are difficult to estimate. While dose-limiting toxicity is currently evaluated after one treatment cycle in phase I trials, this time period might be insufficient to evaluate tolerance of novel targeted compounds.
To model systemic effects of chronic molecularly targeted modulation, Barbacid and colleagues genetically inactivated target genes throughout the whole body of GEM lung adenocarcinoma models (Mariano Barbacid, personal communication, and ref. 82). Despite significant breeding efforts, this experimental setup allows analysis of potential combination therapies of several targeted drugs. However, the time frame required to reveal deleterious side effects in these models is still unknown, illustrating why preclinical trials sometimes offer compelling data that fail to hold up in the clinic.
An example of unexpected translation was recently observed in patients diagnosed with pancreatic ductal adenocarcinoma and experimentally treated with gemcitabine and saridegib (IPI-926). Saridegib inhibits Smoothened, a component of the Hedgehog signaling pathway, and results in depletion of tumor-associated stromal tissues (83). Preclinical studies showed that this drug combination significantly extends survival in tumor-bearing mice (84); however, Infinity Pharmaceuticals halted the phase II clinical trial (NCT01130142) because interim analyses revealed inferior survival in the experimental study arm. Detailed reanalysis yielded several key differences between the preclinical and clinical trial structures and led to a hypothesis that the agent may produce different effects in a chronic versus acute treatment regimen. Subsequent postclinical experiments in mice with early-stage disease found that long-term, continuous targeting of Smoothened induces a more aggressive phenotype and reduced overall survival (Kenneth P. Olive, personal communication).
Current clinical investigation
As rapid translation of novel targeted therapies represents a crucial bottleneck in modern antineoplastic development, innovative concepts are urgently needed. The parallel implementation of murine-based preclinical and human phase I/II clinical trials offers such an opportunity (8, 11). The power of co-clinical trial design in addition to utilization of GEM models was initially demonstrated by turning formerly fatal acute promyelocytic leukemia into a curable disease (11).
In our experience, a co-clinical study evaluating treatment response in murine Kras-driven lung adenocarcinoma revealed predictive response data in just nine months (8). Valuable real-time findings from the much quicker progressing mouse component allow for immediate adjustment of the human counterpart. Early evaluation of distinct outcome-related questions is facilitated through murine studies and detailed in Figure 2 (11).
Figure 2. Co-clinical trials in patients with lung cancer and in GEM models of human lung cancer.

Depicted is a schematic of our recent co-clinical trial assessing selumetinib and docetaxel combination therapy in KRAS-driven lung adenocarcinomas. Murine tumor response evaluation revealed that loss of the tumor suppressor LKB1 serves as a predictive biomarker of treatment outcome (8). The colors of the different GEM models correspond to the color of the data in the bar graph.
Our analyses identified that the loss of the tumor suppressor Lkb1 significantly influences overall response and drug resistance to docetaxel and selumetinib combination therapy in lung cancer–bearing mice. As these genetic lesions were not prospectively evaluated in the enrolled patients (85), the preclinical studies spurred a retrospective analysis and influenced the design of the subsequent registration trial.
Co-clinical trials can bridge some of the gaps in clinical drug development evolving from insufficient patient numbers. Particularly, less common cancer types or genetically distinct cancer subsets can readily be modeled in GEM models and allow numerous drug evaluations for single agents and combination therapies. Eventually, this tailored preclinical assessment facilitates prioritization of the most promising treatment option for evaluation in humans.
Future directions
Despite initial advances in antineoplastic drug development based on improved murine models, enhanced preclinical study design, and optimal drug response criteria, our advances in understanding cancer biology are not keeping up with the increasing knowledge of molecular cancer genetics. The potential improvements in efficient drug development outlined above illustrate the importance of multi-institutional efforts to conduct successful co-clinical trials. Ideally, efficiently running clinical test centers would install preclinical units to provide complementary infrastructure for comparative pathology and bioinformatics (11). Ultimately, these efforts will culminate in rapid identification of powerful weapons to convert fatal advanced malignancies into manageable chronic disease.
Acknowledgments
We apologize to our colleagues for the omission of references to many important contributions due to space limitations. We thank members of the Wong and Kung laboratories for helpful discussions. This work was supported by the Deutsche Forschungsgemeinschaft (HE 6897/1-1 to G.S. Herter-Sprie), the National Cancer Institute (CA122794, CA166480, CA163896, P01CA154303, R01CA140594, U01CA141576 to K.-K. Wong), the Susan Spooner Research Fund (to K.-K. Wong), and the National Institutes of Health (to A.L. Kung).
Footnotes
Conflict of interest: Andrew L. Kung serves as a consultant to Novartis, Molecular Imaging, and Stemergie and has sponsored research agreements with Novartis, Agios, and Karyopharm. Kwok-Kin Wong owns equity in and receives compensation from G1 Therapeutics, serves as a consultant to MolecularMD, and has sponsored research agreements with AstraZeneca, Infinity Pharmaceuticals, and Millennium Pharmaceuticals.
Citation for this article: J Clin Invest. 2013;123(9):3639–3645. doi:10.1172/JCI68340.
References
- 1.Johnson JI, et al. Relationships between drug activity in NCI preclinical in vitro and in vivo models and early clinical trials. Br J Cancer. 2001;84(10):1424–1431. doi: 10.1054/bjoc.2001.1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Talmadge JE, Singh RK, Fidler IJ, Raz A. Murine models to evaluate novel and conventional therapeutic strategies for cancer. Am J Pathol. 2007;170(3):793–804. doi: 10.2353/ajpath.2007.060929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Takimoto CH. Why drugs fail: of mice and men revisited. Clin Cancer Res. 2001;7(2):229–230. [PubMed] [Google Scholar]
- 4.Fiebig HH, Maier A, Burger AM. Clonogenic assay with established human tumour xenografts: correlation of in vitro to in vivo activity as a basis for anticancer drug discovery. Eur J Cancer. 2004;40(6):802–820. doi: 10.1016/j.ejca.2004.01.009. [DOI] [PubMed] [Google Scholar]
- 5.Fidler IJ. Orthotopic implantation of human colon carcinomas into nude mice provides a valuable model for the biology and therapy of metastasis. Cancer Metastasis Rev. 1991;10(3):229–243. doi: 10.1007/BF00050794. [DOI] [PubMed] [Google Scholar]
- 6.Hoffman RM. Orthotopic metastatic mouse models for anticancer drug discovery and evaluation: a bridge to the clinic. Invest New Drugs. 1999;17(4):343–359. doi: 10.1023/A:1006326203858. [DOI] [PubMed] [Google Scholar]
- 7.Killion JJ, Radinsky R, Fidler IJ. Orthotopic models are necessary to predict therapy of transplantable tumors in mice. Cancer Metastasis Rev. 1998;17(3):279–284. doi: 10.1023/A:1006140513233. [DOI] [PubMed] [Google Scholar]
- 8.Chen Z, et al. A murine lung cancer co-clinical trial identifies genetic modifiers of therapeutic response. Nature. 2012;483(7391):613–617. doi: 10.1038/nature10937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Singh M, et al. Assessing therapeutic responses in Kras mutant cancers using genetically engineered mouse models. Nat Biotechnol. 2010;28(6):585–593. doi: 10.1038/nbt.1640. [DOI] [PubMed] [Google Scholar]
- 10.Zhou Y, et al. Chimeric mouse tumor models reveal differences in pathway activation between ERBB family- and KRAS-dependent lung adenocarcinomas. Nat Biotechnol. 2010;28(1):71–78. doi: 10.1038/nbt.1595. [DOI] [PubMed] [Google Scholar]
- 11.Nardella C, Lunardi A, Patnaik A, Cantley LC, Pandolfi PP. The APL paradigm and the “co-clinical trial” project. Cancer Discov. 2011;1(2):108–116. doi: 10.1158/2159-8290.CD-11-0061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Thomas D. Release of BIO/Biomedtracker Drug Approval Rates Study [press release]. Washington DC, USA: BIOtechNow; February 15, 2011. [Google Scholar]
- 13.Herter-Sprie GS, Wong KK. Can mouse models of cancer reliably improve clinical trial outcome? Clinical Investigation. 2013;3(2):119–130. doi: 10.4155/cli.12.149. [DOI] [Google Scholar]
- 14.Singh M, Murriel CL, Johnson L. Genetically engineered mouse models: closing the gap between preclinical data and trial outcomes. Cancer Res. 2012;72(11):2695–2700. doi: 10.1158/0008-5472.CAN-11-2786. [DOI] [PubMed] [Google Scholar]
- 15.Dranoff G. Experimental mouse tumour models: what can be learnt about human cancer immunology? Nat Rev Immunol. 2012;12(1):61–66. doi: 10.1038/nri3129. [DOI] [PubMed] [Google Scholar]
- 16.Beroukhim R, et al. Patterns of gene expression and copy-number alterations in von-hippel lindau disease-associated and sporadic clear cell carcinoma of the kidney. Cancer Res. 2009;69(11):4674–4681. doi: 10.1158/0008-5472.CAN-09-0146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Engelholm SA, et al. Genetic instability of cell lines derived from a single human small cell carcinoma of the lung. Eur J Cancer Clin Oncol. 1985;21(7):815–824. doi: 10.1016/0277-5379(85)90221-4. [DOI] [PubMed] [Google Scholar]
- 18.Hausser HJ, Brenner RE. Phenotypic instability of Saos-2 cells in long-term culture. Biochem Biophys Res Commun. 2005;333(1):216–222. doi: 10.1016/j.bbrc.2005.05.097. [DOI] [PubMed] [Google Scholar]
- 19.Nielsen KV, Madsen MW, Briand P. In vitro karyotype evolution and cytogenetic instability in the non-tumorigenic human breast epithelial cell line HMT-3522. Cancer Genet Cytogenet. 1994;78(2):189–199. doi: 10.1016/0165-4608(94)90089-2. [DOI] [PubMed] [Google Scholar]
- 20.Wilmanns C, Fan D, O’Brian CA, Bucana CD, Fidler IJ. Orthotopic and ectopic organ environments differentially influence the sensitivity of murine colon carcinoma cells to doxorubicin and 5-fluorouracil. Int J Cancer. 1992;52(1):98–104. doi: 10.1002/ijc.2910520118. [DOI] [PubMed] [Google Scholar]
- 21.Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science. 2011;331(6024):1565–1570. doi: 10.1126/science.1203486. [DOI] [PubMed] [Google Scholar]
- 22.Hidalgo M, et al. A pilot clinical study of treatment guided by personalized tumorgrafts in patients with advanced cancer. Mol Cancer Ther. 2011;10(8):1311–1316. doi: 10.1158/1535-7163.MCT-11-0233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Julien S, et al. Characterization of a large panel of patient-derived tumor xenografts representing the clinical heterogeneity of human colorectal cancer. Clin Cancer Res. 2012;18(19):5314–5328. doi: 10.1158/1078-0432.CCR-12-0372. [DOI] [PubMed] [Google Scholar]
- 24. New evidence for the usefulness of “avatar” mice for cancer patients [press release]. Madrid, Spain: CNIO, Centro Nacional de Investigaciones Oncologicas; January 2012. [Google Scholar]
- 25.Cutz JC, et al. Establishment in severe combined immunodeficiency mice of subrenal capsule xenografts and transplantable tumor lines from a variety of primary human lung cancers: potential models for studying tumor progression-related changes. Clin Cancer Res. 2006;12(13):4043–4054. doi: 10.1158/1078-0432.CCR-06-0252. [DOI] [PubMed] [Google Scholar]
- 26.de Plater L, et al. Establishment and characterisation of a new breast cancer xenograft obtained from a woman carrying a germline BRCA2 mutation. Br J Cancer. 2010;103(8):1192–1200. doi: 10.1038/sj.bjc.6605900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fichtner I, et al. Establishment of patient-derived non-small cell lung cancer xenografts as models for the identification of predictive biomarkers. Clin Cancer Res. 2008;14(20):6456–6468. doi: 10.1158/1078-0432.CCR-08-0138. [DOI] [PubMed] [Google Scholar]
- 28.Tentler JJ, et al. Patient-derived tumour xenografts as models for oncology drug development. Nat Rev Clin Oncol. 2012;9(6):338–350. doi: 10.1038/nrclinonc.2012.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Monsma DJ, et al. Genomic characterization of explant tumorgraft models derived from fresh patient tumor tissue. J Transl Med. 2012;10:125. doi: 10.1186/1479-5876-10-125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Garber K. From human to mouse and back: ‘tumorgraft’ models surge in popularity. J Natl Cancer Inst. 2009;101(1):6–8. doi: 10.1093/jnci/djn481. [DOI] [PubMed] [Google Scholar]
- 31.Rubio-Viqueira B, et al. An in vivo platform for translational drug development in pancreatic cancer. Clin Cancer Res. 2006;12(15):4652–4661. doi: 10.1158/1078-0432.CCR-06-0113. [DOI] [PubMed] [Google Scholar]
- 32.Garber K. Personal mouse colonies give hope for pancreatic cancer patients. J Natl Cancer Inst. 2007;99(2):105–107. doi: 10.1093/jnci/djk046. [DOI] [PubMed] [Google Scholar]
- 33.Hylander BL, et al. Origin of the vasculature supporting growth of primary patient tumor xenografts. J Transl Med. 2013;11:110. doi: 10.1186/1479-5876-11-110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lin MT, et al. Quantifying the relative amount of mouse and human DNA in cancer xenografts using species-specific variation in gene length. Biotechniques. 2010;48(3):211–218. doi: 10.2144/000113363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bernard D, Peakman M, Hayday AC. Establishing humanized mice using stem cells: maximizing the potential. Clin Exp Immunol. 2008;152(3):406–414. doi: 10.1111/j.1365-2249.2008.03659.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kalscheuer H, et al. A model for personalized in vivo analysis of human immune responsiveness. Sci Transl Med. 2012;4(125):125ra130. doi: 10.1126/scitranslmed.3003481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wege AK, et al. Humanized tumor mice — a new model to study and manipulate the immune response in advanced cancer therapy. Int J Cancer. 2011;129(9):2194–2206. doi: 10.1002/ijc.26159. [DOI] [PubMed] [Google Scholar]
- 38.DeRose YS, et al. Tumor grafts derived from women with breast cancer authentically reflect tumor pathology, growth, metastasis and disease outcomes. Nat Med. 2011;17(11):1514–1520. doi: 10.1038/nm.2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Merk J, Rolff J, Becker M, Leschber G, Fichtner I. Patient-derived xenografts of non-small-cell lung cancer: a pre-clinical model to evaluate adjuvant chemotherapy? Eur J Cardiothorac Surg. 2009;36(3):454–459. doi: 10.1016/j.ejcts.2009.03.054. [DOI] [PubMed] [Google Scholar]
- 40.Dong X, et al. Patient-derived first generation xenografts of non-small cell lung cancers: promising tools for predicting drug responses for personalized chemotherapy. Clin Cancer Res. 2010;16(5):1442–1451. doi: 10.1158/1078-0432.CCR-09-2878. [DOI] [PubMed] [Google Scholar]
- 41.Condon MS. The role of the stromal microenvironment in prostate cancer. Semin Cancer Biol. 2005;15(2):132–137. doi: 10.1016/j.semcancer.2004.08.002. [DOI] [PubMed] [Google Scholar]
- 42.De Wever O, Mareel M. Role of tissue stroma in cancer cell invasion. J Pathol. 2003;200(4):429–447. doi: 10.1002/path.1398. [DOI] [PubMed] [Google Scholar]
- 43.Hideshima T, Mitsiades C, Tonon G, Richardson PG, Anderson KC. Understanding multiple myeloma pathogenesis in the bone marrow to identify new therapeutic targets. Nat Rev Cancer. 2007;7(8):585–598. doi: 10.1038/nrc2189. [DOI] [PubMed] [Google Scholar]
- 44.Fidler IJ, Naito S, Pathak S. Orthotopic implantation is essential for the selection, growth and metastasis of human renal cell cancer in nude mice [corrected]. Cancer Metastasis Rev. 1990;9(2):149–165. doi: 10.1007/BF00046341. [DOI] [PubMed] [Google Scholar]
- 45.Manzotti C, Audisio RA, Pratesi G. Importance of orthotopic implantation for human tumors as model systems: relevance to metastasis and invasion. Clin Exp Metastasis. 1993;11(1):5–14. doi: 10.1007/BF00880061. [DOI] [PubMed] [Google Scholar]
- 46.Stephenson RA, Dinney CP, Gohji K, Ordonez NG, Killion JJ, Fidler IJ. Metastatic model for human prostate cancer using orthotopic implantation in nude mice. J Natl Cancer Inst. 1992;84(12):951–957. doi: 10.1093/jnci/84.12.951. [DOI] [PubMed] [Google Scholar]
- 47.Waters DJ, Janovitz EB, Chan TC. Spontaneous metastasis of PC-3 cells in athymic mice after implantation in orthotopic or ectopic microenvironments. Prostate. 1995;26(5):227–234. doi: 10.1002/pros.2990260502. [DOI] [PubMed] [Google Scholar]
- 48.Suetsugu A, et al. Imageable fluorescent metastasis resulting in transgenic GFP mice orthotopically implanted with human-patient primary pancreatic cancer specimens. Anticancer Res. 2012;32(4):1175–1180. [PubMed] [Google Scholar]
- 49.Francia G, Cruz-Munoz W, Man S, Xu P, Kerbel RS. Mouse models of advanced spontaneous metastasis for experimental therapeutics. Nat Rev Cancer. 2011;11(2):135–141. doi: 10.1038/nrc3001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Richmond A, Su Y. Mouse xenograft models vs GEM models for human cancer therapeutics. Dis Model Mech. 2008;1(2–3):78–82. doi: 10.1242/dmm.000976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zender L, et al. Identification and validation of oncogenes in liver cancer using an integrative oncogenomic approach. Cell. 2006;125(7):1253–1267. doi: 10.1016/j.cell.2006.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sweet-Cordero A, et al. Comparison of gene expression and DNA copy number changes in a murine model of lung cancer. Genes Chromosomes Cancer. 2006;45(4):338–348. doi: 10.1002/gcc.20296. [DOI] [PubMed] [Google Scholar]
- 53.Artandi SE, et al. Telomere dysfunction promotes non-reciprocal translocations and epithelial cancers in mice. Nature. 2000;406(6796):641–645. doi: 10.1038/35020592. [DOI] [PubMed] [Google Scholar]
- 54.Zhu C, et al. Unrepaired DNA breaks in p53-deficient cells lead to oncogenic gene amplification subsequent to translocations. Cell. 2002;109(7):811–821. doi: 10.1016/S0092-8674(02)00770-5. [DOI] [PubMed] [Google Scholar]
- 55.Olive KP, et al. Mutant p53 gain of function in two mouse models of Li-Fraumeni syndrome. Cell. 2004;119(6):847–860. doi: 10.1016/j.cell.2004.11.004. [DOI] [PubMed] [Google Scholar]
- 56.Hingorani SR, et al. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell. 2005;7(5):469–483. doi: 10.1016/j.ccr.2005.04.023. [DOI] [PubMed] [Google Scholar]
- 57.Wong KK, et al. Telomere dysfunction and Atm deficiency compromises organ homeostasis and accelerates ageing. Nature. 2003;421(6923):643–648. doi: 10.1038/nature01385. [DOI] [PubMed] [Google Scholar]
- 58.Greenman C, Wooster R, Futreal PA, Stratton MR, Easton DF. Statistical analysis of pathogenicity of somatic mutations in cancer. Genetics. 2006;173(4):2187–2198. doi: 10.1534/genetics.105.044677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Harris AW, Pinkert CA, Crawford M, Langdon WY, Brinster RL, Adams JM. The E mu-myc transgenic mouse. A model for high-incidence spontaneous lymphoma and leukemia of early B cells. J Exp Med. 1988;167(2):353–371. doi: 10.1084/jem.167.2.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bric A, et al. Functional identification of tumor-suppressor genes through an in vivo RNA interference screen in a mouse lymphoma model. Cancer Cell. 2009;16(4):324–335. doi: 10.1016/j.ccr.2009.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ocana A, Pandiella A, Siu LL, Tannock IF. Preclinical development of molecular-targeted agents for cancer. Nat Rev Clin Oncol. 2011;8(4):200–209. doi: 10.1038/nrclinonc.2010.194. [DOI] [PubMed] [Google Scholar]
- 62.Jucker M. The benefits and limitations of animal models for translational research in neurodegenerative diseases. Nat Med. 2010;16(11):1210–1214. doi: 10.1038/nm.2224. [DOI] [PubMed] [Google Scholar]
- 63.Eisenhauer EA, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer. 2009;45(2):228–247. doi: 10.1016/j.ejca.2008.10.026. [DOI] [PubMed] [Google Scholar]
- 64.Llovet JM, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359(4):378–390. doi: 10.1056/NEJMoa0708857. [DOI] [PubMed] [Google Scholar]
- 65.Liu T, et al. An imageable metastatic treatment model of nasopharyngeal carcinoma. Clin Cancer Res. 2007;13(13):3960–3967. doi: 10.1158/1078-0432.CCR-07-0089. [DOI] [PubMed] [Google Scholar]
- 66.Hoffman RM. Cellular and subcellular imaging in live mice using fluorescent proteins. Curr Pharm Biotechnol. 2012;13(4):537–544. doi: 10.2174/138920112799436311. [DOI] [PubMed] [Google Scholar]
- 67.Zeng Q, et al. Treating triple-negative breast cancer by a combination of rapamycin and cyclophosphamide: an in vivo bioluminescence imaging study. Eur J Cancer. 2010;46(6):1132–1143. doi: 10.1016/j.ejca.2010.01.014. [DOI] [PubMed] [Google Scholar]
- 68. Ozawa T, James CD. Establishing intracranial brain tumor xenografts with subsequent analysis of tumor growth and response to therapy using bioluminescence imaging. J Vis Exp. 2010. (41).pii: 1986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jarzabek MA, et al. In vivo bioluminescence imaging validation of a human biopsy-derived orthotopic mouse model of glioblastoma multiforme. Mol Imaging. 2013;12(3):161–172. [PubMed] [Google Scholar]
- 70.Terziyska N, et al. In vivo imaging enables high resolution preclinical trials on patients’ leukemia cells growing in mice. PLoS One. 2012;7(12):e52798. doi: 10.1371/journal.pone.0052798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tung CH. Fluorescent peptide probes for in vivo diagnostic imaging. Biopolymers. 2004;76(5):391–403. doi: 10.1002/bip.20139. [DOI] [PubMed] [Google Scholar]
- 72.Gilbert LA, Hemann MT. DNA damage-mediated induction of a chemoresistant niche. Cell. 2010;143(3):355–366. doi: 10.1016/j.cell.2010.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hawes JJ, Reilly KM. Bioluminescent approaches for measuring tumor growth in a mouse model of neurofibromatosis. Toxicol Pathol. 2010;38(1):123–130. doi: 10.1177/0192623309357075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wu R, Hu TC, Rehemtulla A, Fearon ER, Cho KR. Preclinical testing of PI3K/AKT/mTOR signaling inhibitors in a mouse model of ovarian endometrioid adenocarcinoma. Clin Cancer Res. 2011;17(23):7359–7372. doi: 10.1158/1078-0432.CCR-11-1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Strevel EL, Ing DJ, Siu LL. Molecularly targeted oncology therapeutics and prolongation of the QT interval. J Clin Oncol. 2007;25(22):3362–3371. doi: 10.1200/JCO.2006.09.6925. [DOI] [PubMed] [Google Scholar]
- 76.Strevel EL, Siu LL. Cardiovascular toxicity of molecularly targeted agents. Eur J Cancer. 2009;45(suppl 1):318–331. doi: 10.1016/S0959-8049(09)70046-2. [DOI] [PubMed] [Google Scholar]
- 77.Asnacios A, Naveau S, Perlemuter G. Gastrointestinal toxicities of novel agents in cancer therapy. Eur J Cancer. 2009;45(suppl 1):332–342. doi: 10.1016/S0959-8049(09)70047-4. [DOI] [PubMed] [Google Scholar]
- 78.Segaert S, Chiritescu G, Lemmens L, Dumon K, Van Cutsem E, Tejpar S. Skin toxicities of targeted therapies. Eur J Cancer. 2009;45(suppl 1):295–308. doi: 10.1016/S0959-8049(09)70044-9. [DOI] [PubMed] [Google Scholar]
- 79.Carles J, Morales R, Perez JM, Suarez C, Rodon J, Valverde C. Management and interpretation of novel toxicities of molecular targeted therapies: renal toxicities. Eur J Cancer. 2009;45(suppl 1):309–317. doi: 10.1016/S0959-8049(09)70045-0. [DOI] [PubMed] [Google Scholar]
- 80. Revised 2013. Cancer. World Health Organization Web Site: World Health. http://www.who.int/mediacentre/factsheets/fs297/en/index.html .
- 81.Goulart BH, Clark JW, Pien HH, Roberts TG, Finkelstein SN, Chabner BA. Trends in the use and role of biomarkers in phase I oncology trials. Clin Cancer Res. 2007;13(22 pt 1):6719–6726. doi: 10.1158/1078-0432.CCR-06-2860. [DOI] [PubMed] [Google Scholar]
- 82.Choi YJ, et al. The requirement for cyclin D function in tumor maintenance. Cancer Cell. 2012;22(4):438–451. doi: 10.1016/j.ccr.2012.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Infinity Pharmaceuticals, Inc. Infinity Reports Update from Phase 2 study of saridegib plus gemcitabine in patients with metastatic pancreatic cancer [press release]. Cambridge, Massachusetts, USA: Business Wire; January 27, 2012. [Google Scholar]
- 84.Olive KP, et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science. 2009;324(5933):1457–1461. doi: 10.1126/science.1171362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Janne PA, et al. Selumetinib plus docetaxel for KRAS-mutant advanced non-small-cell lung cancer: a randomised, multicentre, placebo-controlled, phase 2 study. Lancet Oncol. 2013;14(1):38–47. doi: 10.1016/S1470-2045(12)70489-8. [DOI] [PubMed] [Google Scholar]