

Abstract
Background. The evaluation of effective disease-modifying therapies for neurodegenerative disorders relies on objective and accurate measures of progression in at-risk individuals. Here we used a computational approach to identify a functional brain network associated with the progression of preclinical Huntington’s disease (HD).
Methods. Twelve premanifest HD mutation carriers were scanned with [18F]-fluorodeoxyglucose PET to measure cerebral metabolic activity at baseline and again at 1.5, 4, and 7 years. At each time point, the subjects were also scanned with [11C]-raclopride PET and structural MRI to measure concurrent declines in caudate/putamen D2 neuroreceptor binding and tissue volume. The rate of metabolic network progression in this cohort was compared with the corresponding estimate obtained in a separate group of 21 premanifest HD carriers who were scanned twice over a 2-year period.
Results. In the original premanifest cohort, network analysis disclosed a significant spatial covariance pattern characterized by progressive changes in striato-thalamic and cortical metabolic activity. In these subjects, network activity increased linearly over 7 years and was not influenced by intercurrent phenoconversion. The rate of network progression was nearly identical when measured in the validation sample. Network activity progressed at approximately twice the rate of single region measurements from the same subjects.
Conclusion. Metabolic network measurements provide a sensitive means of quantitatively evaluating disease progression in premanifest individuals. This approach may be incorporated into clinical trials to assess disease-modifying agents.
Trial registration. Registration is not required for observational studies.
Funding. NIH (National Institute of Neurological Disorders and Stroke, National Institute of Biomedical Imaging and Bioengineering) and CHDI Foundation Inc.
Introduction
The identification of objective and accurate biomarkers of disease progression will help expedite the evaluation of effective disease-modifying therapies for neurodegenerative disorders. Brain imaging provides one such opportunity through the characterization of large-scale networks associated with the specific disease process (1–4). To date, disease-related brain networks have been identified mainly by analyzing functional imaging data from clinically affected individuals who vary in the duration and severity of symptoms (1, 4, 5). Nonetheless, few approaches have been developed to isolate specifically those networks that relate to the progression of the underlying process. Such networks are particularly relevant to the study of “at-risk” individuals, in whom consistent clinical manifestations of the disease have yet to appear. Indeed, accurate determination of the preclinical progression rate is crucial for the evaluation of interventions designed to delay, or altogether prevent, the onset of symptoms.
Huntington’s disease (HD) is an inherited neurodegenerative disorder characterized by progressive impairments in motor, cognitive, and affective functions. Because it is an autosomal dominant disorder with near 100% penetrance, offspring of affected individuals can be tested for the presence of unstable CAG expansions in the gene encoding the huntingtin protein years before the onset of clinical symptoms. This presents an unusual opportunity to use brain imaging to identify a potential progression biomarker in premanifest carriers of this mutation, which would greatly facilitate the development of disease-modifying therapies for this illness.
Clinical rating scales, such as the Unified Huntington’s Disease Rating Scale (UHDRS), are currently the “gold standard” for assessing HD severity. However, such measures are insensitive to disease progression in the preclinical period as well as during symptom onset. Alternatively, imaging tools such as [11C]-raclopride with PET (to measure reductions in binding to caudate and putamen dopamine D2 neuroreceptors) and volumetric MRI (to assess tissue loss in these brain areas) have been used to determine the rate of disease progression in at-risk individuals (6). That said, in vivo measurements of neurodegeneration in single brain regions provide scant information concerning the broader functional topography of the underlying disease process (7, 8).
Network analysis has been increasingly applied to rest-state brain imaging data to identify and measure the expression of characteristic spatial covariance patterns in patients with neurodegenerative disorders (1, 4, 5, 9). In an earlier crosssectional analysis of metabolic images from premanifest HD mutation carriers and healthy control subjects, we reported a reproducible disease-related topography that discriminated between the 2 groups (10). Nonetheless, the expression of this pattern in gene carriers did not change consistently over time, thus limiting its utility as a network biomarker of preclinical disease progression. Recently, however, a spatial covariance algorithm has been developed for longitudinal imaging applications (11). By searching for functional brain networks with monotonically changing expression over time, this approach can be used to identify significant progression-related covariance topographies in disease populations. Once validated, such networks can be used to quantify the rate of progression of a neurodegenerative process at the systems level. This strategy can also be applied to assess rates of progression in at-risk individuals, such as presymptomatic carriers of disease mutations.
In the current study, we demonstrate the feasibility of this network-based approach by using longitudinal metabolic imaging data from premanifest HD carriers to identify and prospectively validate a distinct spatial covariance pattern associated with the underlying disease progression. Changes in pattern expression over time were used to quantify the rate of progression in the preclinical period, which was then confirmed in an independent cohort of at-risk individuals.
Results
The HD metabolic progression pattern
Pattern identification.
In this study, we posited that as a fully penetrant, dominantly inherited neurodegenerative disorder, preclinical HD is likely to exhibit consistent subject-by-subject changes in network activity over time. To examine this proposition, we analyzed longitudinal metabolic imaging data from a group of 12 premanifest mutation carriers (designated HD1; Figure 1 and Supplemental Table 1; supplemental material available online with this article; doi: 10.1172/JCI69411DS1) using a special computational algorithm (11) designed to detect patterns of regional functional connectivity (i.e., brain networks) with monotonically changing expression over time (the details of this procedure for pattern discovery and validation are provided in Methods). Analysis of [18F]-fluorodeoxyglucose (FDG) PET scans acquired at baseline, 1.5, and 4 years revealed a significant metabolic progression pattern (Figure 2A) that accounted for 9.7% of the overall variance. This network (Table 1) was characterized by declining metabolic activity in the caudate, putamen, thalamus, insula, and posterior cingulate gyrus, and in the prefrontal and occipital cortex, associated with covarying increases in the cerebellum, pons, hippocampus, and orbitofrontal cortex.
Figure 1. Flow diagram illustrating the design of the study.

109 participants (47 HD gene carriers and 62 age-matched healthy control [HC] subjects) were enrolled. A metabolic progression pattern was identified from the FDG PET scans obtained in a longitudinal cohort (HD1) comprising 12 premanifest HD carriers. Pattern expression was prospectively validated in a crosssectional validation cohort (HD2) comprising 9 premanifest carriers (for the evaluation of test-retest reliability) and 5 early-stage symptomatic HD subjects. The rate of network progression was validated in an independent longitudinal cohort (HD3) comprising 21 premanifest carriers. Metabolic network values were computed in a control group (HC1) comprising 12 healthy subjects and were used to standardize pattern expression in each subject. A second control group (HC2) comprising 20 healthy subjects was used to prospectively evaluate pattern expression in gene-negative individuals. A volume-loss progression pattern was identified in the structural MRI scans obtained in the longitudinal HD1 subjects. Progression rates for this pattern were assessed in both the HD1 and HD3 longitudinal cohorts. Individual scores of the volume-loss pattern were standardized with reference to values computed from a control group (HC4) comprising 18 healthy subjects. In the longitudinal HD1 cohort, the rates of network progression were compared with analogous regional measurements obtained in the caudate and putamen with [11C]-raclopride (RAC) PET (D2 receptor binding) and structural MRI (tissue volume). These regional values were standardized with reference to the corresponding HC3 (comprising 12 healthy subjects) and HC4 control groups.
Figure 2. HD metabolic progression pattern.

(A) This spatial covariance pattern was characterized by areas of declining (blue) and increasing (red) metabolic activity over time. The pattern is displayed as a reliability map of voxel weights thresholded at z = 2.33, P < 0.01 (1-tailed), using a bootstrap resampling procedure (ICV range = –6.02, 5.63, P < 0.0001; 1,000 iterations). (B) All premanifest HD1 subjects exhibited a monotonic increase in pattern expression (P < 0.001; permutation test) across the first 3 time points. (C) In the HD1 longitudinal cohort, pattern expression increased linearly with disease progression (P < 0.0001; IGM) at an estimated progression rate of 0.21/year (95% CI = 0.15, 0.27). Values from the 5 early symptomatic members of the HD2 testing cohort (yellow triangles) are provided for reference. (D) In the HD3 longitudinal testing cohort, pattern expression exhibited a similar linear increase with advancing disease (P < 0.0001; IGM) at a nearly identical rate of 0.19/year (95% CI = 0.11, 0.26). The longitudinal data from each subject are connected by lines. Red lines denote the initially premanifest HD1 subjects who subsequently phenoconverted (i.e., were clinically diagnosed as HD); blue lines denote their counterparts who did not phenoconvert during the study. Values before and after phenoconversion are represented by open and filled symbols. The horizontal broken line represents the mean (equal to 0) of the HC1 healthy control group; the dotted lines represent 2 SD above and below the normal mean. In C and D, the solid lines represent the best model-fitted lines with a 95% CI (broken curves).
Table 1.
Regions with significant loadings on the HD metabolic progression pattern
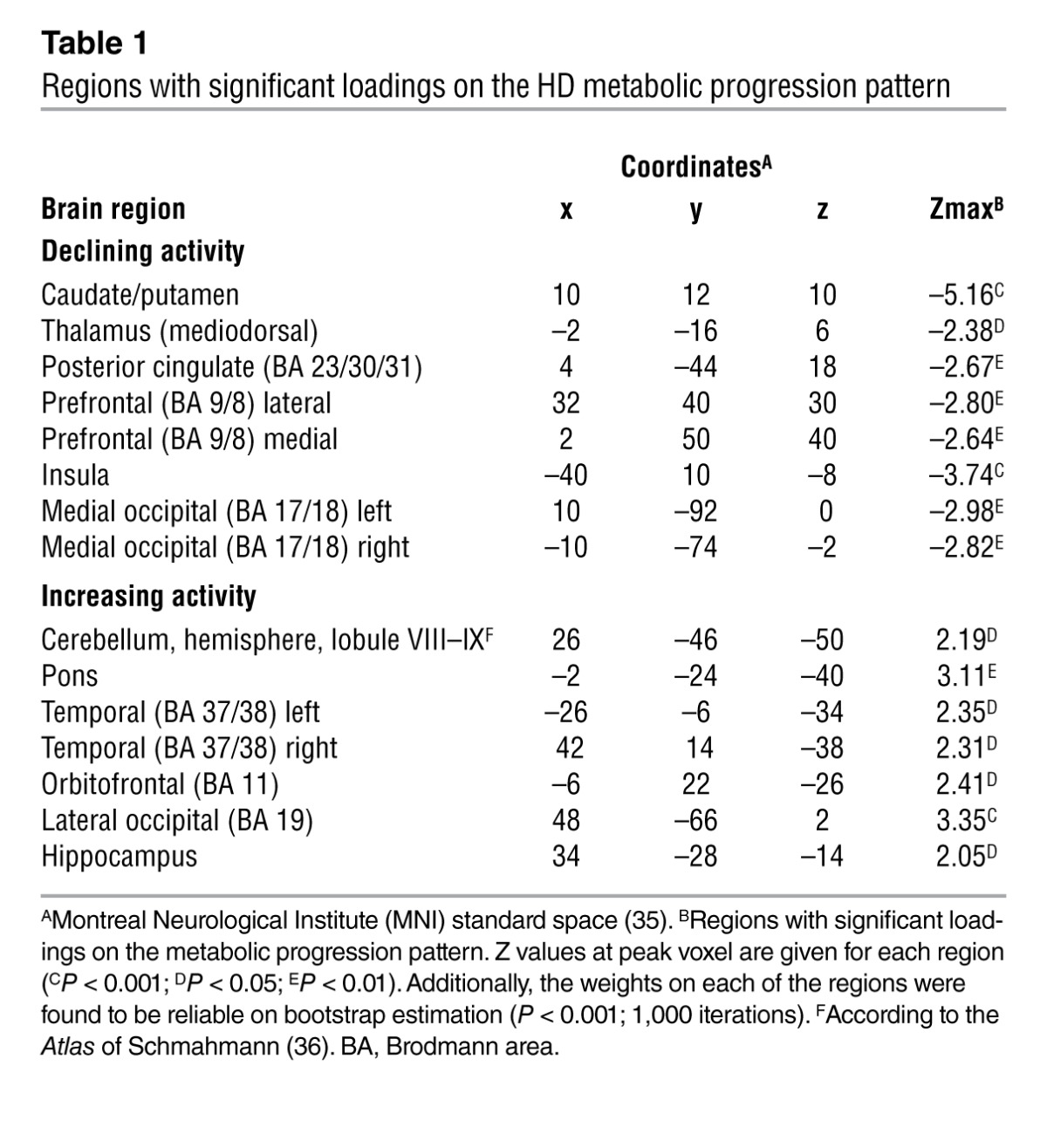
This metabolic network was quantified by computing subject scores (i.e., pattern expression) in the individual scans that the derivation set comprised (used to identify the network) and prospectively in the scans the various testing sets comprised (used to validate the network) (Figure 1). The resulting values for each subject and time point were standardized by z transformation to corresponding subject scores computed in the scans from the healthy control (HC1) reference cohort (Supplemental Table 1). In this way, these measures assayed the degree to which network activity deviated from normal in each subject and time point. Without exception, each of the 12 premanifest subjects in the HD1 cohort (Figure 2B) exhibited monotonic increases in pattern expression over time (P < 0.001, permutation test). Pattern expression at each time point was elevated in this group relative to the 12 neurologically normal subjects of the HC1 healthy control group (baseline: P < 0.05; 1.5 year: P = 0.001; 4 year: P < 0.001; Student’s t tests). Of note, this covariance pattern was specified by the significant ordinal trend in subject expression that was present over time in the individual HD1 gene carriers, without requiring or using descriptors such as age, CAG repeat length, or disease burden indices, such as the predicted years to onset (YTO) (an empirical estimate of the time remaining until phenoconversion based on a subject’s age and CAG repeat length; ref. 12). Likewise, demographic and imaging data from healthy subjects were not employed in the identification of the progression-related metabolic pattern. Although the network was derived entirely from the imaging data acquired in the gene carriers without regard to genotypic descriptors or age, post hoc analysis revealed significant correlations (r > 0.70, P < 0.05) between pattern expression values and the YTO at each time point (Supplemental Figure 1, A–C). This suggests an association between network activity and individual subject differences in measures of disease burden.
Pattern validation.
Nine HD1 subjects returned for final imaging assessment at 7 years, by which time 4 had phenoconverted, i.e., received a clinically definite diagnosis of HD by an experienced movement disorders specialist. Pattern expression was computed on a prospective single-case basis for these 9 subjects. Network activity values at 7 years were significantly higher than at baseline (P < 0.001, paired Student’s t test) and were also elevated (P = 0.001, Student’s t test) with respect to HC1 reference values. Of note, each of the phenoconverted subjects exhibited substantial elevations in network activity (>3 SD above the normal mean; range 3.6–6.3 SD) at 7 years. As with the first 3 longitudinal time points, pattern expression at the final time point correlated (r = 0.69, P < 0.05) with predicted YTO (Supplemental Figure 1D).
We next validated the metabolic progression pattern in HD2, a separate testing group comprising 14 gene carriers (9 premanifest and 5 early symptomatic) (Figure 1 and Supplemental Table 1). Each subject was scanned with FDG PET at 1 of 4 collaborative imaging sites (see Methods). We also quantified pattern expression in HC2, a second healthy control group comprising 20 age-matched normal subjects. Prospectively computed network values were elevated in the HD2 gene carriers (1.8 ± 2.2, mean ± SD) relative to HC2 (–0.1 ± 1.3) control subjects (P < 0.005, Student’s t test). Moreover, values in the 5 early symptomatic HD2 subjects (mean 3.0 years after diagnosis) were elevated (range: 3.4–4.8 SD above the normal mean) to levels similar to those observed in the 7-year scans of the HD1 phenoconverters (mean 4.5 years after diagnosis). Scans in the 9 premanifest HD2 subjects were repeated over a 3-week interval to evaluate the test-retest reproducibility of the pattern expression measure. Network values computed in these premanifest carriers were highly reproducible (intraclass correlation coefficient = 0.96, P < 0.001) on repeat measurements at multiple imaging sites (Figure 3). As in the HD1 cohort, the premanifest HD2 carriers exhibited correlations between network activity and predicted YTO in both the test (r = 0.65, P = 0.06) and retest (r = 0.72, P < 0.03) data (Supplemental Figure 1, E and F). Of note, pattern expression in the HC2 control subjects did not differ from HC1 values (P = 0.76), confirming that pattern expression is low or absent in subjects lacking the HD mutation.
Figure 3. Measurements of the HD metabolic progression pattern expression: test-retest reproducibility.

Test-retest reproducibility was excellent (intraclass correlation coefficient [ICC] = 0.96, P < 0.001) for metabolic pattern expression values computed prospectively in the 9 premanifest HD2 subjects who underwent repeat FDG PET studies at 4 imaging sites as part of the PREDICT-HD consortium. The line of identity (dotted line) falls within the 95% CI (broken curves) of the test-retest regression line (solid line). Data from the 4 imaging laboratories that participated in the test-retest study are signified by color. Site 1: North Shore University Hospital (n = 1); site 2: Indiana University (n = 2); site 3: University of Iowa (n = 3); site 4: University of Toronto (n = 3).
Nodal analysis.
The regional topography of the metabolic progression network was examined by post hoc analysis of the changes in local activity at the major network nodes. Over time, regional activity declined in the caudate/putamen (P < 0.0001, repeated measures ANOVA (RMANOVA); Figure 4A), mediodorsal thalamus (P < 0.005; Figure 4B), insula (P < 0.0001), posterior cingulate cortex (P < 0.005) as well as in the prefrontal (P < 0.0001; Figure 4C) and medial occipital (P < 0.005) regions. Progressive increases in regional activity were present concurrently in the pons (P < 0.001; Figure 4D), orbitofrontal cortex (P < 0.05), hippocampus (P < 0.01), and in the temporal (P < 0.01) and lateral occipital (P < 0.005) cortices.
Figure 4. HD metabolic progression pattern: regional changes.

In the premanifest HD1 cohort, progressive declines (black lines) in regional metabolic activity (P < 0.005, RMANOVA) were present in (A) the caudate nucleus and anterior putamen, (B) the mediodorsal thalamus, and (C) the prefrontal cortex. In these regions, metabolic activity at all time points was lower for the subjects who phenoconverted at a later point in the study (red) as compared with those who remained clinically premanifest at follow-up (blue). Regional metabolic activity concurrently increased in several regions (Table 1), including (D) the pons (P < 0.001), in which the subsequent phenoconverters exhibited higher values than their nonphenoconverting counterparts at each time point. For each region, MNI coordinates at the peak voxel are given in parentheses. The y axes denote regional metabolic activity ratio-normalized to the global mean. Error bars represent 1 SE above or below the mean of the HD subjects. The broken line represents mean metabolic activity for the HC1 healthy control group; the dotted lines represent 1 SE above and below the normal mean.
Measurement and confirmation of the network progression rate
After identifying a significant metabolic progression topography in the HD1 data, we used individual growth modeling (IGM), a statistical technique designed to assess longitudinal changes occurring in individual subjects (13), to evaluate the changes in pattern expression that were observed in the HD1 and HD3 longitudinal cohorts as a function of “disease time,” defined as the estimated YTO for each subject. (For the 4 HD1 premanifest carriers who subsequently phenoconverted during the study, we used the actual number of years until the clinical diagnosis of HD was made.) The details of the IGM analyses that were performed are provided in Methods.
A significant linear relationship was evident between increasing network activity and declining time to phenoconversion (Figure 2C), with an estimated progression rate of 0.21/year (P < 0.0001, 95% CI = 0.15, 0.27). To determine whether phenoconversion altered the rate of network progression, we reanalyzed the data using only the 8 HD1 premanifest carriers who did not phenoconvert (i.e., the “nonphenoconverters”) over the course of the follow-up period. The findings were very similar to those for the whole group, with a highly significant rate of increase in pattern expression (Figure 5A) of 0.20/years (P = 0.001, 95% CI = 0.09, 0.31; IGM). Importantly for the whole cohort as well as the nonphenoconverter subgroup, the growth models rejected the possibility that network progression was a nonlinear function of disease time (see Methods). Hence, the results suggest that pattern expression progresses at a constant rate in premanifest gene carriers, independently of the proximity of an individual premanifest mutation carrier to clinical diagnosis. Indeed, network values for the 5 early symptomatic HD2 subjects (Figure 2C) fell close to the best-fit line for the HD1 longitudinal data, compatible with a continuous linear trajectory spanning the period of phenoconversion. In this vein, we also note that the metabolic network increases observed in the individual HD1 subjects did not correlate with concurrent changes in UHDRS motor ratings (r = 0.25, P = 0.20, Bland-Altman within-subject correlation). This is perhaps not surprising, given that the nonphenoconverters had low UHDRS motor scores at baseline (2.5 ± 2.5, mean ± SD), which did not change over time (Supplemental Figure 2 and Supplemental Table 2). This contrasted with the pattern expression measured concurrently in the same subjects, which exhibited a significant increase over the same time period (Figure 5A).
Figure 5. Longitudinal changes in preclinical HD progression markers: analysis of nonphenoconverters.

(A) In the nonphenoconverting HD1 subjects, pattern expression increased linearly with disease progression (P = 0.001; IGM) at an estimated rate of 0.20/year (95% CI = 0.09, 0.31). (B) Caudate D2 receptor–binding values measured in the [11C]-raclopride PET scans of the nonphenoconverters exhibited a linear decline with advancing disease (–2.1% of normal mean per year, P < 0.0001; IGM). (C) A similar decline was noted for concurrent MRI measurements of caudate tissue volume in the same subjects (–1.5% of the normal mean per year, P = 0.001; IGM). The solid line depicts the best fitted line with the corresponding 95% CI (broken curves). The dotted lines represent 2 SD above and below the normal mean (dotted horizontal line) for each measure. In B and C, the individual data are represented as percentage of the mean value for age-matched healthy controls. (D) The rate of increase in the expression of the HD metabolic pattern (red) was greater (P ≤ 0.001, IGM) than the rates of decline measured for caudate D2 receptor binding (light blue) and tissue volume (dark blue). To compare network progression (increasing time course) with the corresponding changes in the region-based measures (declining time course), the latter values were flipped (i.e., reflected above the x axes) and plotted as absolute values (dotted lines). The y axes represent the standard z-scale. The horizontal dotted line represents the normal mean (equal to 0) for each parameter. The vertical dotted line represents the estimated time of phenoconversion (i.e., when YTO was 0).
To confirm the estimate of the progression rate, network activity values were computed on an individual basis in metabolic scans from a separate longitudinal cohort of gene carriers, designated HD3 (Figure 1 and Supplemental Table 1). This group consisted of 21 premanifest carriers scanned twice with FDG PET over a period of 2.3 ± 0.3 (mean ± SD) years. None of these individuals progressed clinically to receive a diagnosis of HD during this time interval. As observed in the HD1 and HD2 groups, network activity in the HD3 subjects correlated with predicted YTO (r > 0.48, P < 0.03) at both time points (Supplemental Figure 1, G and H). More importantly, a significant linear relationship (Figure 2D) was present between increases in network activity and concurrent changes in disease time in this testing group. The rate of network progression estimated in this sample was 0.19/year (P < 0.0001, 95% CI = 0.11, 0.26), which was nearly identical to that determined in the original HD1 cohort (0.21/year). Additionally, we note that in the 2 longitudinal data sets, variability in pattern expression declined as subjects neared phenoconversion. Thus, the reliability of the network measure is likely to improve with the emergence of clinical manifestations. In aggregate, these findings point to a stable and reproducible rate of metabolic network progression in premanifest HD subjects.
Network progression and volume loss
HD is associated with progressive volume loss in multiple brain regions, even prior to symptom onset (14–16). Thus, the measured rate of network progression may represent the effects of ongoing atrophy in key nodal regions rather than metabolic dysfunction per se. To determine whether this was indeed the case, we conducted a parallel spatial covariance analysis of longitudinal volumetric MRI scans from the HD1 subjects. These scans were obtained concurrently with FDG PET at each longitudinal time point. The analysis disclosed a significant pattern of progressive regional volume loss in the subjects (Figure 6A) that accounted for 5.2% of the overall variance. Similar to the metabolic pattern, a monotonic increase in the expression of the volume-loss pattern (Figure 6B) was evident in all subjects (P < 0.005, permutation test).
Figure 6. HD volume-loss progression pattern.

(A) This spatial covariance pattern was characterized by progressive loss of tissue volume (blue) in several brain regions (see text). The pattern is displayed as a reliability map thresholded at z = 2.33, P < 0.01 (1-tailed) using a bootstrap resampling procedure (ICV = –8.78, 6.81, P < 0.0001; 1,000 iterations). (B) All premanifest HD1 subjects exhibited a monotonic increase in pattern expression (P < 0.005; permutation test) across the first 3 time points. (C) In the HD1 longitudinal cohort, pattern expression increased linearly with disease progression (P < 0.0001; IGM) at a rate of 0.16/year (95% CI = 0.12, 0.21). (D) In the HD3 cohort, however, longitudinal changes in the expression of the volume-loss pattern were not significant (P = 0.40). Red lines denote the initially premanifest HD1 subjects who subsequently phenoconverted. Blue lines denote their counterparts who did not phenoconvert by the end of the study. Post-phenoconversion values are represented by filled symbols. The horizontal broken line represents the mean (equal to 0) for the healthy control group; the dotted lines represent 2 SD above and below the normal mean. In C and D, the solid line represents the best fit according to the model; the broken curves represent the 95% CI of the fitted line.
This pattern (Supplemental Table 3) was characterized by covarying volume loss in the caudate, putamen, insula, and in the prefrontal and occipital cortical regions — areas found consistently to exhibit atrophy in volumetric MRI studies of premanifest gene carriers (14, 15). These regions, notably, were also distinguished by declining functional activity as part of the HD metabolic progression pattern (Figure 2A). Nonetheless, despite qualitative similarities between the metabolic and volume-loss progression topographies, a quantitative comparison disclosed only a modest voxel-wise correlation (R2 = 0.14) between the regional loadings on the 2 patterns. Indeed, volume loss was present in many regions not involved in the metabolic progression topography, such as the premotor cortex, supplementary motor area, and precuneus. Importantly, the metabolic progression pattern was distinguished by areas of increasing activity (Figure 2A) that were not present as part of the volume-loss topography.
The differences between the metabolic and volume-loss progression patterns were more striking on analysis of the subject expression data. First, in contrast with the metabolic pattern, volume-loss pattern expression did not correlate (P > 0.12) with the predicted YTO at any of the HD1 time points. That said, longitudinal analysis of the volumetric MRI data from these subjects revealed a significant linear relationship between increasing volume-loss pattern expression and changes in disease time (Figure 6C). The estimated rate of progression for this pattern was 0.16/year (P < 0.0001, 95% CI = 0.12, 0.21), which was 30% slower (P < 0.05, IGM) than for measurements of metabolic pattern expression in the same subjects (estimated difference in slope = –0.063/year, 95% CI = –0.003, –0.123). Similarly, mean subject expression of the volume-loss pattern at phenoconversion (i.e., when predicted YTO equals zero) was estimated to be 2.63, which was lower (P = 0.005; IGM) than for its metabolic counterpart (estimated difference in Y-intercept = –1.47, 95% CI = –0.55, –2.39). Indeed, in the 5 early symptomatic HD2 subjects (Figure 6C), the mean expression of the volume-loss pattern was lower (less abnormal) than for the metabolic pattern (2.54 vs. 4.24, P < 0.004; Student’s t test).
Of note, the volume-loss pattern was found to be less generalizable when applied to prospective data from independent groups of gene carriers. Prospective computation of subject scores for the volume-loss pattern in the longitudinal HD3 MRI data did not reveal significant correlations with YTO values at the individual time points (P > 0.83) or between changes in these variables over time (P = 0.40, Figure 6D). These findings contrasted with the significant relationships noted above (Figure 2D and Supplemental Figure 1, G and H) for the metabolic pattern in the same subjects.
To directly evaluate the influence of volume loss on the metabolic network progression rate, we included volume-loss pattern expression as a covariate in the growth model. We found that the relationship between increasing metabolic network activity and declining YTO remained significant after adjusting for volume loss at the network level (slope = 0.07/year, P = 0.01). These results support the notion that progression in metabolic network activity is not driven by concurrent volume loss.
Striatal D2 receptor binding and tissue volume
Measurements of the rate of progression in the premanifest HD1 cohort using conventional region-based methods, such as caudate/putamen D2 binding measured in [11C]-raclopride PET scans and tissue volume measured in MRI scans (Supplemental Table 2), yielded results similar to those appearing in the published literature (15, 17–20). In this cohort, baseline caudate and putamen D2-binding values were lower (P < 0.005, Student’s t test) than for control subjects (Figure 7A), with reductions of 35.7% and 33.8% of the normal mean. Over time, D2 binding exhibited a linear decline, with disease progression in the caudate (Figure 7B) (P < 0.0001; IGM) and putamen (P < 0.005). The rate of decline was faster (P < 0.002) in the caudate (–2.1% of the normal mean/year, 95% CI = –2.7%, –1.5%) relative to the putamen (–1.8%/year, 95% CI = –2.9%, –0.8%).
Figure 7. Longitudinal changes in striatal D2 receptor binding and tissue volume.

(A) Composite [11C]-raclopride PET images from healthy control subjects (left) and premanifest HD carriers (right) scanned at baseline, 1.5, 4, and 7 years. In each image, dopamine D2 receptor binding was computed voxel-wise as (voxel/occipital-1) and displayed in standard space. The display was thresholded from 1.0 to 3.5. (B) Caudate (left) and putamen (right) D2 receptor–binding values measured using [11C]-raclopride PET exhibited a linear decrease with advancing disease (–2.1% and –1.8% of normal mean per year, P < 0.005; IGM); the decline in the caudate was faster than for the putamen (P < 0.002). (C) Gray matter tissue probability maps from healthy control subjects (left) and the premanifest HD carriers described above (right). Each map represents the average of the gray matter–segmented MRI scans from each group/time point. The display was thresholded from 0.0 to 1.0. (D) Caudate (left) and putamen (right) MRI-based tissue volume measurements also declined linearly with advancing disease (–2.3% and –1.7% of the normal mean per year, P < 0.0001; IGM). Progression rates did not differ for the 2 regions (P = 0.27). In B and D, individual values are represented as percentage of the mean (broken line) for an age-matched healthy control group; the dotted lines represent 2 SD above and below the normal mean. The data from the phenoconverters and nonphenoconverters in the longitudinal HD1 cohort are presented by red and blue lines, respectively. Caudate and putamen values for the symptomatic HD2 subjects (yellow triangles) are provided for reference.
At baseline, caudate volume (Figure 7C) was lower in the HD1 subjects than control values (P < 0.02, Student’s t test), with a reduction of 21.5% of the normal mean. Baseline putamen volume was reduced by only 12.4%, which did not differ from normal (P = 0.13). Over time, there was a linear decline in tissue volume (P < 0.0001) in the caudate (Figure 7D) (–2.3% of the normal mean/year, 95% CI = –2.9%, –1.6%) and putamen (–1.7%/year, 95% CI = –2.3%, –1.2%). In contrast to D2 binding, the rate of decline in tissue volume did not differ for these regions (P = 0.27).
Restricting the analysis to the nonphenoconverters (Figure 5, B and C) revealed significant declines in caudate D2 binding (–2.1% of the normal mean/year, 95% CI = –3.0%, –1.2%, P < 0.0001) and volume loss (–1.5%/year, 95% CI = –2.3%, –0.7%, P = 0.001) at rates similar to those for the whole HD1 cohort.
Comparison of the network and regional measures
Following z transformation of the imaging measures (see Methods), we directly compared the rates of progression that were estimated at the network level (from changes in the expression of the metabolic and volume-loss patterns) with the corresponding region-level estimates (from parallel measurements of caudate D2 binding and tissue volume) in the longitudinal HD1 data. Substantial differences (P < 0.0001; IGM) were evident in these estimates (Figure 8) in that the rate of increase in metabolic pattern expression was faster than for its volume-loss counterpart (P < 0.05) as well as for region-level estimates based on either caudate D2 binding (P < 0.0001) or tissue volume (P < 0.0005).
Figure 8. Time course of disease progression: network vs. regional biomarkers.

Linear trajectories (solid lines) of the network and regional imaging measures for the premanifest HD1 longitudinal cohort according to the best-fitting models. The rate of increase in the HD metabolic pattern expression (red) was greater than that for the volume-loss progression pattern (orange; P < 0.05, IGM) and the rates of decline measured for caudate D2 receptor binding (light blue; P < 0.0001) and tissue volume (dark blue; P < 0.0005) (Table 2). To allow for direct comparison of network progression (increasing time course) with corresponding changes in regional measures (decreasing time course), the values for caudate D2 receptor binding and tissue volume were flipped and analyzed as increasing mirror lines (dotted lines). The y axis represents the standard z scale. The horizontal dotted line represents the normal mean (equal to 0) for each parameter. The vertical dotted line represents the time of phenoconversion (i.e., YTO was zero). The estimated value for the metabolic progression pattern at phenoconversion (i.e., the y axis intercept) is signified by a red arrow. The estimated “start time” for the decline of caudate D2 receptor binding (i.e., the x axis intercept) is signified by a light blue arrow. Inset: Bubble plots depicting the estimated rates of disease progression and values at phenoconversion (see Methods) for the HD metabolic and volume-loss patterns (red and orange discs) and for caudate D2 receptor binding and tissue volume (light and blue discs).The diameter of each disc is proportional to the SE for each parameter estimate.
These measures also differed with regard to estimated values at phenoconversion (P = 0.001; IGM), with higher expression of the metabolic pattern (Figure 8) relative to the volume-loss pattern (P = 0.001) as well as the 2 regional measures (P < 0.001 for caudate D2 binding and tissue volume). Of note, volume-loss pattern expression progressed faster than concurrent declines in either caudate D2 binding (P < 0.005) or tissue volume (P < 0.02). That said, no difference was evident in the progression rates estimated for the 2 region-level measures (P = 0.62), although the mean value at phenoconversion was lower (more abnormal) for caudate D2 binding relative to tissue volume (P < 0.05). Progression rates and estimated values at phenoconversion are shown for the various imaging markers in Figure 8 (inset) and Table 2.
Table 2.
Time course of disease progression: network vs. regional biomarkers
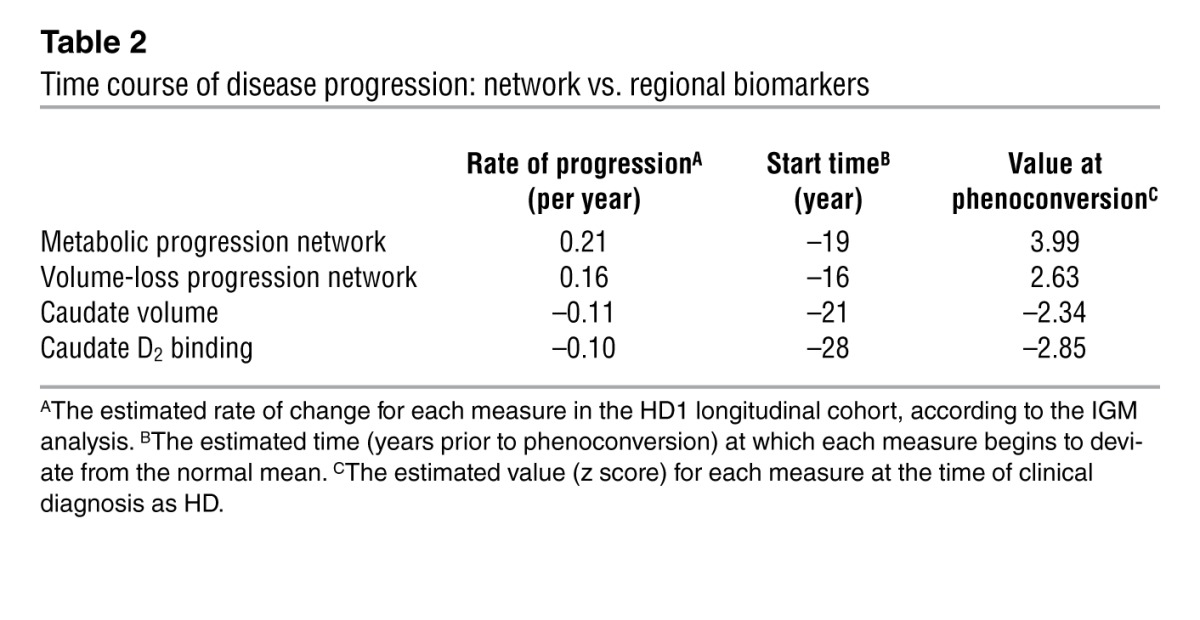
Of note, restricting the analysis to the nonphenoconverters (Figure 5D), we found that the rate of metabolic network progression remained faster (P ≤ 0.001, IGM) than for either caudate D2 binding or tissue volume. The estimated mean value for metabolic network expression at phenoconversion was also higher than for the 2 regional measures (P < 0.03). Thus, the findings of the restricted analysis are consistent with those of the whole cohort. It is therefore unlikely that the observed differences in the sensitivity of the various imaging measures to disease progression are attributable to the inclusion of the premanifest subjects who were near phenoconversion at the time of enrollment.
It is noteworthy that the findings do not change when a different descriptor of disease burden (21) is used as the predictor variable in the growth models. Indeed, significant linear relationships were present between changes in this index and increasing metabolic network activity (P < 0.0001) as well as declining caudate D2 binding (P < 0.01) and tissue volume (P < 0.0001) (Supplemental Figure 3, A–C). As above, metabolic network activity was comparatively more sensitive to progression (Supplemental Figure 3D), with a faster rate of change (P < 0.0001, IGM) and higher values at phenoconversion (P < 0.005) than the other imaging measures.
Discussion
In this study, we identified a specific metabolic brain network associated with disease progression in premanifest HD mutation carriers. In addition to exhibiting excellent test-retest replicability at multiple imaging sites, pattern expression measurements were found to increase linearly at nearly identical rates in 2 independent longitudinal cohorts of premanifest individuals. Moreover, of the imaging markers analyzed, metabolic network activity proved to be the most sensitive to disease progression, as demonstrated by its rapid rate of progression and high expression at the time of phenoconversion. This has practical implications for the assessment of disease-modifying therapies for HD and potentially also for other neurodegenerative disorders.
The longitudinal data also allow for the creation of a theoretical timeline for the various anatomical/functional changes that underlie disease progression in the preclinical period of HD. Growth modeling suggests that metabolic network progression begins 19–20 years before phenoconversion, anteceding the rise of the volume-loss pattern by 3 years and continuing at a constant rate in the remaining preclinical years and in the period following phenoconversion. Moreover, metabolic network activity reaches the 2 SD threshold for abnormality approximately 10 years before phenoconversion, roughly at the same time as caudate D2 receptor binding and before regional or network-level measures of volume loss. Metabolic network values progressively increase to approximately 4 SD above the normal mean at phenoconversion — a level significantly greater than corresponding measures of volume-loss pattern expression or of caudate D2 binding and tissue volume.
Of note, the progressive increase in metabolic network activity remained significant after adjustment for volume loss. This suggests that metabolic network activity is a reflection of structural as well as functional progression effects. Moreover, the data indicate that metabolic pattern expression, even without adjustment for volume loss, can be viewed as a sensitive measure of progression that can potentially be incorporated into disease modification trials. Indeed, a preliminary power/sample-size calculation was performed using this measure as a progression biomarker. We found that randomizing 120 premanifest HD carriers may be sufficient for detecting a 30% reduction in the progression rate at a power of 0.90 in a 2-year disease modification trial. This sample size is substantially smaller than that required based on clinical outcome measures or volumetric MRI measurements from single brain regions (22).
The IGMs also provide valuable insights into the natural history of preclinical HD. Analysis of the longitudinal data suggests that striatal D2 receptor binding is perhaps the earliest observable imaging change (Figure 8), beginning approximately 28 years before phenoconversion. Although [11C]-raclopride PET and volumetric MRI provided similar estimates of the rate of decline in striatal signal, D2 binding loss is likely to have begun at an earlier stage of the neurodegenerative process (23). Thus, despite its comparatively slow rate of deterioration, the early “start time” of the caudate D2-binding measure highlights its potential use as a progression biomarker in the period between 25 and 10 years before phenoconversion.
Additionally, the data suggest that the presence of abnormal elevations in metabolic network activity (i.e., subject scores >2 SD above the normal mean) at baseline is associated with a high likelihood of phenoconversion over the ensuing years. Indeed, each of the 4 premanifest gene carriers who were ultimately diagnosed with clinically definite HD had baseline expression above this threshold. In other words, a critical threshold of metabolic pattern expression may exist above which premanifest subjects have a substantially greater risk of phenoconversion in the ensuing 5-year period.
Several of the current observations warrant additional study. For example, it is important to determine whether metabolic network activity continues to increase beyond the first 5 years after phenoconversion and whether a plateau in pattern expression is reached at longer disease durations. It is important to know whether, at later disease stages, the metabolic pathology of HD becomes dominated by covariance topographies relating specifically to symptom severity (7). Of note, changes in regional glucose metabolism have been reported in a preclinical mouse model of HD (24). Network analysis may be applied to these data to identify a progression-related metabolic covariance pattern in the mouse analogous to that observed in the human cohort. Changes in pattern expression in the mouse can be monitored during experimental treatment with potential disease-modifying agents.
In aggregate, the metabolic progression network provides a sensitive and robust descriptor of the HD disease process in the decade prior to clinical diagnosis. Moreover, the linear increase in network activity observed before symptom onset appears to evolve further after diagnosis. Whether treatment can slow the progression of these changes in at-risk individuals is a topic for future study.
Methods
Subjects
Derivation.
Demographic data for the HD and healthy control samples are presented in Supplemental Table 1. For pattern identification and measurement of the progression rate, we studied a group of 12 premanifest HD mutation carriers, designated HD1 (male/female: 5/7; baseline age: 46.8 ± 11.0 [mean ± SD], range 25–62 years; CAG repeat length: 41.6 ± 1.7, range 39–45; predicted YTO: 10.3 ± 8.6, range 1–25 years) who underwent serial imaging with FDG and [11C]-raclopride PET, structural MRI, and UHDRS clinical ratings at baseline and again after 1.6 ± 0.1 (n = 12), 3.7 ± 0.3 (n = 10), and 7.2 ± 0.4 (n = 9) years. Limited metabolic and caudate/putamen D2 receptor–binding data from the first 3 time points have been reported previously (10). Mean total motor UHDRS ratings are presented in Supplemental Table 2 and Supplemental Figure 2. At baseline, all HD1 subjects were considered to be “premanifest” in that none was judged to have had a clinically definite diagnosis by a trained movement disorders specialist who was blind to the imaging data. The mean diagnostic confidence score at baseline was 1.3; the distribution of scores was as follows: 0 (n = 5), 1 (n = 2), 2 (n = 2), and 3 (n = 3). Two of the initially premanifest HD1 subjects were judged at 1.5 years to have “phenoconverted,” i.e., received a clinical diagnosis of definite HD; 2 others were found to have phenoconverted between then and the 4-year time point. We refer to the 4 premanifest subjects who subsequently developed sufficient clinical manifestations for diagnosis as “phenoconverters” and the remaining 8 premanifest subjects as “nonphenoconverters.”
Validation.
We analyzed FDG PET scans from 2 preclinical/early symptomatic cohorts of HD gene carriers. The first cohort, designated HD2, comprised 14 gene carriers: 9 subjects were premanifest (male/female: 3/6; age: 38.5 ± 12.3, range 20–55 years; CAG repeat length: 41.4 ± 1.4, range 40–44; predicted YTO: 13.8 ± 5.9, range 7–21 years), and 5 subjects were early symptomatic HD (male/female: 0/5; age: 53.8 ± 6.3, range 43–59 years; UHDRS motor ratings: 42.8 ± 4.4, range 38–50 years). The premanifest HD2 subjects were scanned twice with FDG PET (between-scan interval of 24.5 ± 10.5 days) at 1 of 4 imaging sites (see below). This was done to assess the test-retest variability of prospectively computed network expression values in individuals not utilized for pattern discovery. The symptomatic subjects were scanned at a single time point between 2 and 4 years (3.0 ± 0.7 years) after diagnosis. The second testing group, designated HD3, was studied longitudinally. This cohort comprised 21 premanifest HD carriers (male/female: 9/12; age: 40.3 ± 6.8, range 29–57 years; CAG repeat length: 42.9 ± 2.3, range 39–47; predicted YTO: 11.7 ± 6.5, range 1–25 years) who were scanned at baseline and again after 2.3 ± 0.3 years. Limited regional data from this cohort have been reported previously (25, 26).
Healthy control subjects.
Two groups of age-matched healthy volunteer subjects served as controls for the FDG PET study. The first healthy control group (HC1) consisted of 12 subjects (male/female: 6/6; age: 40.8 ± 14.7, range 27–66 years) who were age matched to the HD1 subjects at baseline. Pattern expression values measured in this group were used to standardize the network values computed in all subjects and time points of the HD1, HD2, and HD3 cohorts. The second healthy control group (HC2) consisted of 20 subsequent age-matched healthy subjects (male/female: 10/10; age: 47.7 ± 13.5, range 21–68 years). Pattern expression in this group was used to verify the absence of network activity in prospectively imaged gene negative subjects. Separate age-matched groups of healthy subjects, each imaged at a single time point, served as controls for the [11C]-raclopride PET (HC3: n = 12, male/female: 5/7, age: 42.5 ± 15.6, range 22–64 years) and the MRI (HC4: n = 18, male/female: 7/11, age: 39.8 ± 15.1, range 22–66 years) studies.
Imaging procedures
PET.
The HD1 subjects underwent FDG and [11C]-raclopride PET over a 2-day period at each of the 4 time points using the GE Advance tomograph (General Electric Medical Systems) at North Shore University Hospital (Manhasset, New York, USA) (27). In the HD2 group, the 5 symptomatic patients and 1 of the 9 premanifest subjects participating in the test-retest study were scanned with FDG PET on the GE Advance device at North Shore University Hospital. The remaining 8 premanifest HD2 subjects underwent FDG PET test-retest studies on the Siemens HR+ scanners at Indiana University (Indianapolis, Indiana, USA) (n = 2) and the University of Iowa (n = 3), and on the Siemens HRRT tomograph at the University of Toronto (n = 3). The HD3 subjects were scanned with FDG PET using the Siemens ECAT Exact HR+ scanner (Siemens) at University Medical Center Groningen.
For FDG PET, a 10-minute scan was acquired in 3D mode beginning 35 minutes after the intravenous injection of 5 mCi of radiotracer. The studies were performed after an overnight fast, with the subjects’ eyes open in a dimly lit room and with minimal auditory stimulation. Longitudinal scans from each premanifest HD1 subject were realigned and spatially normalized to a standard Talairach-based FDG PET template, and smoothed with an isotropic Gaussian kernel (10 mm) in all directions to improve the signal-to-noise ratio (10, 28). The scans from the prospective HD and healthy control groups were individually preprocessed using the same method.
For [11C]-raclopride PET, the subjects received 15 mCi of radiotracer by intravenous injection and dynamic images were acquired over 70 minutes (7 × 10 minutes), as described previously (10). The individual frames were spatially realigned to compensate for potential movement during scanning and were transformed into standard Talairach brain space. All normalized images involving the striatum were integrated into a single slice. Regions of interest (ROIs) were defined anatomically on each image with reference to a template in standard space using an automated procedure. Specifically, ROIs were placed bilaterally on the caudate nucleus, putamen, and occipital regions of each scan, blind to subject identity, mutation status (gene positive or negative), clinical status (premanifest or symptomatic), and time point. D2 receptor–binding affinity was separately estimated for the caudate and putamen by computing the striatal-occipital ratio (ROI/occipital-1) between 50 and 60 minutes after injection. The same set of standardized ROIs was used for the longitudinal scans from the premanifest subjects and for the prospective HD and healthy control scans. For each ROI, left and right values from the gene carriers were averaged and compared with the corresponding control values.
MRI.
Premanifest subjects in the HD1 longitudinal cohort and early symptomatic members of the HD2 cohort were scanned on the 1.5T GE Signa Echo Speed scanner at North Shore University Hospital. T1-weighted images were acquired with a 3D spoiled gradient recall sequence (SPGR) (echo time [TE] = 5 ms, reception time [TR] = 24 ms, flip angle = 20°), with matrix size 256′256′124, giving a resolution of 1–1.5 mm in the transverse and axial planes. These images were used to measure caudate and putamen volume at each time point and to identify and validate a volume-loss progression pattern in the MRI data from these subjects (see below).
To assess changes in striatal volume over time, manual segmentation was performed to measure caudate and putamen volume in the original MRI scans. This was performed with MRIcro software (available at http://www.cabiatl.com/mricro/mricro/index.html) utilizing the aligned MRI scans in native space. In each MRI scan, contours of caudate and putamen were separately outlined in the axial slices in which these structures were clearly visible. For each region, volumes on the left and right sides of the brain were separately calculated across all slices and then averaged across hemispheres. Caudate and putamen volumes were measured for the premanifest HD1 subjects at all 4 longitudinal time points and for the prospective HD and control groups at a single time point.
Similar procedures were applied to the 2 time-point longitudinal MRI data from the HD3 cohort. These subjects were scanned at each time point using a 1.5T Siemens scanner at the Groningen University Medical Center. T1-weighted images were acquired using a 3D magnetization-prepared 180° radio-frequency pulses and rapid gradient-echo (MPRAGE) sequence with 1.5-mm contiguous slices covering the whole brain. The images at baseline and follow-up were preprocessed and segmented into gray matter tissue probability maps using the same methods as for the HD1 cohort.
Network analysis
The metabolic progression pattern.
In the current study, we used Ordinal Trends Canonical Variance Analysis (OrT/CVA) (software available at http://www.nitrc.org/projects/gcva_pca), a within-group network modeling approach (11, 29), to search for a significant HD progression covariance pattern in the longitudinal FDG PET data of the premanifest HD1 mutation carriers. Specifically, we used OrT/CVA to identify patterns of regional connectivity in the premanifest HD1 mutation carriers that increase monotonically in their expression with disease progression. In general terms, this multivariate repeated measures technique interrogates the data for the presence of networks with consistent changes in activity across conditions on a subject-by-subject basis. This contrasts with mass-univariate methods that require significant change to be present at the group mean level.
More specifically, OrT/CVA is a special form of supervised principal component analysis. This mathematical-statistical algorithm searches for specific patterns of functional connectivity (i.e., large-scale brain networks) in serial imaging data acquired over multiple ordered experimental conditions. OrT/CVA uses a specially formulated transformation of the data matrix prior to single-value decomposition. In this way, the analysis seeks to detect a specific class of spatial covariance patterns characterized by monotonically increasing pattern expression over time on an individual case basis, while the functional relationships between the brain regions that make up the pattern topography remain constant. Thus, the algorithm identifies significant functional brain networks that exhibit an ordinal trend in subject activity, i.e., a consistent increase in pattern expression in all or most members of the derivation cohort, while rejecting other patterns that do not satisfy this requirement. Notably, OrT/CVA is guided solely by the design variables, which in this analysis encode the temporal ordering of the scans for each subject; other subject variables such as CAG repeat length, age, or predicted YTO are not utilized in the pattern identification procedure. The significance of candidate progression topographies is determined by nonparametric inferential tests (4). Permutation tests of the associated principal component scalars (subject scores) are performed to assess the possibility that the monotonic changes in pattern expression (i.e., the ordinal trend) observed across subjects/conditions in the derivation data set had occurred by chance. The voxel loadings (region weights) on the covariance pattern specify the spatial topography of the network, reflecting local contributions to its overall activity. The reliability of each voxel weight can be estimated and mapped using bootstrap procedures (30). In addition to the identification of relevant spatial covariance pattern(s) in the longitudinal imaging data, OrT/CVA quantifies the expression of the corresponding pattern(s) in each subject and condition in the derivation cohort and in prospective testing populations.
In this study, the HD progression covariance pattern was sought among the linearly independent (orthogonal) principal component (PC) patterns that resulted from the OrT/CVA of the metabolic imaging data from the first 3 time points of the HD1 longitudinal cohort, based upon the following criteria: (a) the search for appropriate patterns was limited to the PCs with the highest eigenvalues; and (b) subject scores for these PCs were entered singly and in all possible combinations to achieve the maximal separation between subject contrast scores. This analysis was conducted within a standard brain mask defined by thresholding at 0.5 the tissue probability map of gray matter derived from MRI scans of 421 healthy subjects as part of statistical parametric mapping software (available at: http://www.fil.ion.ucl.ac.uk/spm). The Akaike information criterion (AIC) was used to specify the optimal linear combination of subject contrast scores, i.e., the set of PCs with the best bias-variance trade-off (31). The resulting HD progression covariance pattern was considered significant if the associated subject scores exhibited a monotonically increasing trend over time that differed from chance (P < 0.05, permutation test). The reliability of the voxel weights on the pattern was tested using a bootstrap resampling procedure with 1,000 iterations (30). The significance level for voxel weight reliability was set at an inverse coefficient of variation (ICV) threshold of z = 1.64, corresponding to P < 0.05, 1-tailed.
Pattern validation and regional analysis.
Following pattern identification, we quantified pattern expression on a single subject/scan basis in several prospective testing data sets: (a) the fourth time point (7 years) of the HD1 cohort (n = 9); (b) the HD2 group (n = 14); (c) both time points of the HD3 cohort (n = 21); and (d) the HC1 (n = 12) and HC2 (n = 20) healthy control groups. Pattern expression values for all the scans were individually standardized by z transformation with respect to the HC1 control group, such that these normatives had a mean subject score of 0 and an SD of 1. All network quantification procedures were performed blind to time point, subject, YTO, clinical diagnosis, and UHDRS ratings.
Finally, a nodal analysis was performed in which we tracked the time course of local metabolic activity in the major network regions. Spherical volumes of interest (VOIs) (radius = 4 mm) were centered on the peak voxel of each region. The mean metabolic activity in each VOI was ratio-normalized by the global metabolic rate measured in the corresponding scan and plotted for each longitudinal time point.
The volume-loss progression pattern.
A segmentation algorithm (available at http://dbm.neuro.uni-jena.de/vbm/) was used in standard space to delineate gray matter voxels in each MRI scan (32, 33). Using the same OrT/CVA procedures described above, we analyzed the voxel-based morphometric scans from the first 3 time points (acquired on the same day as the FDG PET scans) of the longitudinal HD1 cohort. To facilitate spatial comparison with the metabolic network topography, covariance analysis of the volumetric MRI data from the HD1 cohort (to derive the volume-loss progression pattern) was conducted using the same gray matter mask and threshold as was used in the analysis of metabolic PET data from the same subjects. In addition, we determined the degree of similarity/difference between the metabolic and volume-loss topographies by computing the variance shared (R2) among all the corresponding nonzero voxel weights on the 2 pattern images utilizing the MATLAB statistical routine “corr” (34). Subject scores for the volume-loss covariance patterns were standardized with respect to values from an age-matched group of 18 healthy control subjects (HC4; Supplemental Table 1). For validation, pattern expression was quantified on a prospective case basis in the fourth time point MRI scans of the HD1 cohort and in the longitudinal scans of the HD3 cohort.
Statistics
Calculation of progression rates.
For each of the network or regional progression measures, the longitudinal HD1 data from all 4 time points were entered into IGMs (13), including the cases with incomplete data. Longitudinal trajectories were examined as a function of disease time, i.e., YTO, in linear and curvilinear (YTO2 or ln[YTO]) models. One of the 8 nonphenoconverters declined release of CAG information because of confidentiality concerns. The subject’s scan data were therefore not used in the IGM analysis. The model with the best fit to the data, i.e., that with the lowest AIC value, was selected (31). For validation, subject scores for the metabolic and volume-loss progression patterns were computed prospectively in the independent HD3 testing sample. The resulting progression rates were compared with the corresponding estimates from the original HD1 sample.
Comparison of progression indices.
Growth models were also used for the direct comparison of the progression rates of the different imaging measures in the HD1 cohort. To compare the rates of the regional measures with those determined at the network level, the striatal progression indices were z scored with respect to the corresponding healthy control values such that each normal reference group had a mean of 0 and an SD of 1. This allowed the network and regional imaging measures, and their corresponding longitudinal trajectories, to be rigorously compared and displayed on a common z scale. Moreover, because caudate/putamen D2 receptor binding and tissue volume measurements declined over time while pattern scores increased, the corresponding regression lines were flipped for the 2 region-based measures (Figure 8) so that the slopes of all the progression parameters were positive. This allowed for a direct comparison of the magnitude of the various imaging measures within a single growth model. In addition to the estimated rate of progression for each measure, the growth models provided several other key parameters: (a) the “start time,” at which each measure was estimated to have begun to deviate from the normal mean (represented by the x axis intercept when the value equaled zero); (b) the time at which each measure reached an abnormal expression level (defined as 2 SD above the normal mean value for that measure); and (c) the estimated mean value for each measure at phenoconversion (represented by the y axis intercept, when the YTO equaled zero).
All statistical analyses were performed using SAS 9.1 (SAS Institute Inc.), and the significance level was set at P < 0.05.
Study approval
Ethical permission for these studies was obtained from the Institutional Review Boards of the participating institutions. Written informed consent was obtained from each subject following detailed explanation of the procedures.
Supplementary Material
Acknowledgments
We thank the HD subjects and the healthy volunteer subjects who participated in this study. The authors greatly appreciate the assistance of Patricia J. Allen in data analysis, and Toni Fitzpatrick and Yoon Young Choi in manuscript preparation. Special thanks to the PREDICT-HD site coordinators at the University of Iowa, Indiana University, University of Toronto, and Columbia University for their help in organizing and executing the test-retest validation studies, and to James Brewer at the University of California San Diego for his invaluable guidance in the analysis of the volumetric MRI data.
Footnotes
Conflict of interest: David Eidelberg has received consultant fees from Pfizer Inc. Mark Guttman has equity in NeuroDerm and has received research funding from Merck for a clinical trial in Parkinson’s disease.
Citation for this article: J Clin Invest. 2013;123(9):4076–4088. doi:10.1172/JCI69411.
References
- 1.Eidelberg D. Metabolic brain networks in neurodegenerative disorders: a functional imaging approach. Trends Neurosci. 2009;32(10):548–557. doi: 10.1016/j.tins.2009.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Seeley WW, Crawford RK, Zhou J, Miller BL, Greicius MD. Neurodegenerative diseases target large-scale human brain networks. Neuron. 2009;62(1):42–52. doi: 10.1016/j.neuron.2009.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Raj A, Kuceyeski A, Weiner M. A network diffusion model of disease progression in dementia. Neuron. 2012;73(6):1204–1215. doi: 10.1016/j.neuron.2011.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Habeck C, Stern Y, Alzheimer’s Disease Neuroimaging Initiative. Multivariate data analysis for neuroimaging data: overview and application to Alzheimer’s disease. Cell Biochem Biophys. 2010;58(2):53–67. doi: 10.1007/s12013-010-9093-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Niethammer M, Eidelberg D. Metabolic brain networks in translational neurology: concepts and applications. Ann Neurol. 2012;72(5):635–647. doi: 10.1002/ana.23631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Paulsen JS. Functional imaging in Huntington’s disease. Exp Neurol. 2009;216(2):272–277. doi: 10.1016/j.expneurol.2008.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Eidelberg D, Surmeier DJ. Brain networks in Huntington’s disease. J Clin Invest. 2011;121(2):484–492. doi: 10.1172/JCI45646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Novak MJ, Warren JD, Henley SM, Draganski B, Frackowiak RS, Tabrizi SJ. Altered brain mechanisms of emotion processing in pre-manifest Huntington’s disease. Brain. 2012;135(pt 4):1165–1179. doi: 10.1093/brain/aws024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Spetsieris PG, Eidelberg D. Scaled subprofile modeling of resting state imaging data in Parkinson’s disease: methodological issues. Neuroimage. 2011;54(4):2899–2914. doi: 10.1016/j.neuroimage.2010.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Feigin A, et al. Thalamic metabolism and symptom onset in preclinical Huntington’s disease. Brain. 2007;130(pt 11):2858–2867. doi: 10.1093/brain/awm217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Habeck C, et al. A new approach to spatial covariance modeling of functional brain imaging data: ordinal trend analysis. Neural Comput. 2005;17(7):1602–1645. doi: 10.1162/0899766053723023. [DOI] [PubMed] [Google Scholar]
- 12.Langbehn DR, Brinkman RR, Falush D, Paulsen JS, Hayden MR, International Huntington’s Disease Collaborative Group. A new model for prediction of the age of onset and penetrance for Huntington’s disease based on CAG length. Clin Genet. 2004;65(4):267–277. doi: 10.1111/j.1399-0004.2004.00241.x. [DOI] [PubMed] [Google Scholar]
- 13.Singer JD. Applied Longitudinal Data Analysis: Modeling Change Event Occurrence. London, United Kingdom: Oxford University Press; 2003. [Google Scholar]
- 14.Rosas HD, Salat DH, Lee SY, Zaleta AK, Hevelone N, Hersch SM. Complexity and heterogeneity: what drives the ever-changing brain in Huntington’s disease? Ann N Y Acad Sci. 2008;1147:196–205. doi: 10.1196/annals.1427.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Aylward EH. Change in MRI striatal volumes as a biomarker in preclinical Huntington’s disease. Brain Res Bull. 2007;72(2–3):152–158. doi: 10.1016/j.brainresbull.2006.10.028. [DOI] [PubMed] [Google Scholar]
- 16.Aylward EH, et al. Onset and rate of striatal atrophy in preclinical Huntington disease. Neurology. 2004;63(1):66–72. doi: 10.1212/01.WNL.0000132965.14653.D1. [DOI] [PubMed] [Google Scholar]
- 17.Antonini A, et al. Striatal glucose metabolism and dopamine D2 receptor binding in asymptomatic gene carriers and patients with Huntington’s disease. Brain. 1996;119(pt 6):2085–2095. doi: 10.1093/brain/119.6.2085. [DOI] [PubMed] [Google Scholar]
- 18.Antonini A, Leenders KL, Eidelberg D. [11C]raclopride-PET studies of the Huntington’s disease rate of progression: relevance of the trinucleotide repeat length. Ann Neurol. 1998;43(2):253–255. doi: 10.1002/ana.410430216. [DOI] [PubMed] [Google Scholar]
- 19.Tabrizi SJ, et al. Potential endpoints for clinical trials in premanifest and early Huntington’s disease in the TRACK-HD study: analysis of 24 month observational data. Lancet Neurol. 2012;11(1):42–53. doi: 10.1016/S1474-4422(11)70263-0. [DOI] [PubMed] [Google Scholar]
- 20.Majid DS, et al. Automated structural imaging analysis detects premanifest Huntington’s disease neurodegeneration within 1 year. Mov Disord. 2011;26(8):1481–1488. doi: 10.1002/mds.23656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tabrizi SJ, et al. Biological and clinical manifestations of Huntington’s disease in the longitudinal TRACK-HD study: cross-sectional analysis of baseline data. Lancet Neurol. 2009;8(9):791–801. doi: 10.1016/S1474-4422(09)70170-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Aylward EH, et al. Longitudinal change in regional brain volumes in prodromal Huntington disease. J Neurol Neurosurg Psychiatry. 2011;82(4):405–410. doi: 10.1136/jnnp.2010.208264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cha JH. Transcriptional signatures in Huntington’s disease. Prog Neurobiol. 2007;83(4):228–248. doi: 10.1016/j.pneurobio.2007.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cepeda-Prado E, et al. R6/2 Huntington’s disease mice develop early and progressive abnormal brain metabolism and seizures. J Neurosci. 2012;32(19):6456–6467. doi: 10.1523/JNEUROSCI.0388-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.van Oostrom JC, et al. Striatal dopamine D2 receptors, metabolism, and volume in preclinical Huntington disease. Neurology. 2005;65(6):941–943. doi: 10.1212/01.wnl.0000176071.08694.cc. [DOI] [PubMed] [Google Scholar]
- 26.van Oostrom JC, Dekker M, Willemsen AT, de Jong BM, Roos RA, Leenders KL. Changes in striatal dopamine D2 receptor binding in pre-clinical Huntington’s disease. Eur J Neurol. 2009;16(2):226–231. doi: 10.1111/j.1468-1331.2008.02390.x. [DOI] [PubMed] [Google Scholar]
- 27.Ma Y, Tang C, Spetsieris PG, Dhawan V, Eidelberg D. Abnormal metabolic network activity in Parkinson’s disease: test-retest reproducibility. J Cereb Blood Flow Metab. 2007;27(3):597–605. doi: 10.1038/sj.jcbfm.9600358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huang C, et al. Changes in network activity with the progression of Parkinson’s disease. Brain. 2007;130(pt 7):1834–1846. doi: 10.1093/brain/awm086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Moeller JR, Habeck CG. Reciprocal benefits of mass-univariate and multivariate modeling in brain mapping: applications to event-related functional MRI, H215O- and FDG-PET. . Int J Biomed Imaging. 2006;2006:79862. doi: 10.1155/IJBI/2006/79862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Efron B, Tibshirani R. An Introduction To The Bootstrap. New York, New York, USA: CRC Press LLC; 1994. [Google Scholar]
- 31.Burnham KP, Anderson DR. Model Selection And Multimodel Inference. New York, New York, USA: Springer Verlag; 2002. [Google Scholar]
- 32.Keller SS, Wilke M, Wieshmann UC, Sluming VA, Roberts N. Comparison of standard and optimized voxel-based morphometry for analysis of brain changes associated with temporal lobe epilepsy. Neuroimage. 2004;23(3):860–868. doi: 10.1016/j.neuroimage.2004.07.030. [DOI] [PubMed] [Google Scholar]
- 33.Ruocco HH, Bonilha L, Li LM, Lopes-Cendes I, Cendes F. Longitudinal analysis of regional grey matter loss in Huntington disease: effects of the length of the expanded CAG repeat. J Neurol Neurosurg Psychiatry. 2008;79(2):130–135. doi: 10.1136/jnnp.2007.116244. [DOI] [PubMed] [Google Scholar]
- 34.Mure H, et al. Parkinson’s disease tremor-related metabolic network: characterization, progression, and treatment effects. Neuroimage. 2011;54(2):1244–1253. doi: 10.1016/j.neuroimage.2010.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Collins DL, Neelin P, Peters TM, Evans AC. Automatic 3D intersubject registration of MR volumetric data in standardized Talairach space. J Comput Assist Tomogr. 1994;18(2):192–205. doi: 10.1097/00004728-199403000-00005. [DOI] [PubMed] [Google Scholar]
- 36.Schmahmann JD, Doyon J, Toga AW, Petrides M, Evans AC. MRI atlas of the human cerebellum. San Diego, California, USA: Academic Press; 2000. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.