

Abstract
In a report reading like a fascinating detective story, Vincent and colleagues crack the mysterious case of east Texas bleeding disorder. They show that affected individuals have a mutation in exon 13 of the coagulation F5 gene that causes increased expression of an alternatively spliced transcript, which encodes a previously unrecognized factor V (FV) isoform they call FV-short. This FV isoform lacks a large portion of the B domain of FV, which is normally released upon the proteolytic activation of FV by thrombin and binds tightly to the coagulation regulator tissue factor pathway inhibitor-α (TFPIα). This interaction leads to an approximately 10-fold increase in the level of TFPIα circulating in plasma and a resultant anticoagulant effect that produces a hemorrhagic diathesis.
The players: TFPIα and FV
Full-length tissue factor pathway inhibitor-α (TFPIα) contains an acidic N terminus followed by three tandem Kunitz-type protease inhibitory domains and a basic C terminus. It regulates coagulation by producing factor Xa–dependent (FXa-dependent) feedback inhibition of the factor VIIa/tissue factor complex (FVIIa/TF) (1), which is responsible for the initiation of coagulation, and by directly inhibiting FXa in a process that is enhanced by protein S (PS) (2, 3). Full-length TFPIα circulates at a low concentration (∼0.16 nM) in plasma, but platelets carry approximately 50% of total blood TFPIα and release it at sites of injury where they aggregate.
Plasma levels of TFPIα are reduced in patients with factor V (FV) or PS deficiency (4, 5). Full-length TFPIα (MW 42 kDa) circulating in plasma is bound in two high molecular complexes (MW > 700 kDa) that require the presence of FV and the basic C terminus of TFPIα, which is needed for its interaction with FV (6). These high molecular weight complexes may contain additional constituents (e.g., PS).
The activated form of FV (FVa) is a coagulation cofactor that dramatically accelerates FXa activation of prothrombin to thrombin. FV is a 330-kDa single-chain protein that circulates in plasma at a concentration of approximately 20 nM. About 20% of the total FV in blood is carried by platelets as fragmented, partially activated forms (7). FV is activated by FXa or thrombin through the proteolytic release of its large, intervening B domain with the ultimate production of the heavy (105 kDa) and light (74 kDa) chains of FVa that associate in a Ca2+-dependent fashion (Figure 1A).
Figure 1. Forms of factor V and alignment of the sequences of the B domain BR and TFPIα.
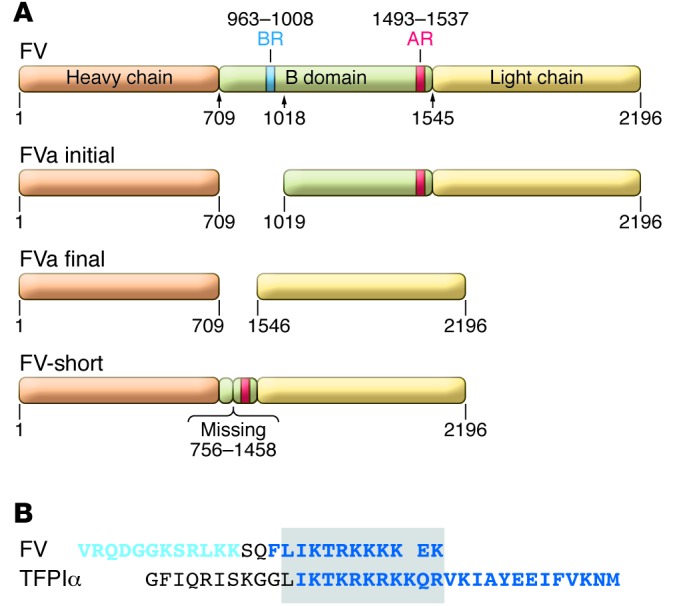
(A) Structure of FV. FV: the heavy chain (orange), light chain (yellow) and B domain (green) are shown. The BR (blue) and AR (red) of the B domain are depicted as well as the sites of cleavage produced by FXa and thrombin during the activation of FV. FVa initial: initial cleavage of FV by FXa and thrombin occurs at R709 and R1018. FVa final: cleavage at R1545 is produced by thrombin, but slowly or not at all by FXa, producing the heavy chain and light chains of FVa that associate in a calcium ion–dependent fashion. FV-short: lacks residues 756–1458 of the FV B domain, but contains the AR. (B) Alignment of B domain and TFPIα sequences. Two sequences within the BR domain of the FV B domain found to be involved in its presumed interaction with AR are shown: peptide 983–994 (light blue lettering) and peptide 997–1008 (dark blue lettering), with the latter appearing to be much more important (8). The sequence of TFPIα required for its interaction with FV is shown in dark blue lettering (6). Boxed is a region of substantial homology.
Elegant studies by Bos and Camire demonstrated that the B domain (aa 710–1545) serves to maintain FV in an inactive, procofactor state and that an interaction between a basic region (BR) (aa 963–1008) and an acidic region (AR) (aa 1493–1537) within the B domain are required for this effect (Figure 1A and ref. 8). Two peptides within the BR (aa 983–994 and 997–1008) were shown to be most important for its presumed binding to the AR (Figure 1B). Deletion of either the basic or acidic portions of the B domain produces derivatives of FV with cofactor activity.
The plot
In 2001, Kuang et al. described a large Texas kindred with a moderately severe bleeding disorder that was characterized by bruising, epistaxis, menorrhagia, and hemorrhage following trauma or surgery that frequently required blood transfusion (9). The prothrombin time (PT) and the activated partial thromboplastin time (aPTT) were both prolonged, suggesting an abnormality in the “common” coagulation pathway that consists of FX, FV, prothrombin, and fibrinogen. Assays of all the coagulation factors, however, produced normal results. Autosomal dominant inheritance and a mild inhibitor pattern in coagulation studies in which patient plasma is mixed with normal plasma suggested a gain-of-function mutation that limited coagulation.
One possible explanation of these results was enhanced inactivation of FXa and thrombin by a hyperactive form of the coagulation inhibitor antithrombin. Initial linkage analysis using an intragenic microsatellite marker showed that the disorder indeed mapped to a locus near the antithrombin gene. Sequencing of the antithrombin gene, however, failed to identify a mutation. More defined linkage studies narrowed the involved locus to a 1.5-Mb region (1q24) that was centromeric to the antithrombin gene and contained the gene for FV (F5) (9). Subsequent sequencing of the F5 gene in an affected individual identified a novel A2440G nucleotide alteration in exon 13 that segregated with the disease and produced a S756G substitution in the B domain of FV. Since the B domain of FV is not required for FV activity and FV clotting activity in affected family members was normal, the alteration was felt to represent a private polymorphism within the family and unlikely to be associated with the bleeding disorder. Thus, east Texas bleeding disorder remained unexplained.
More detective work
To determine whether the A2440G alteration in the FV genome detected by Kuang et al. produced an effect on the level or size of the FV protein, Vincent and colleagues (10) analyzed the plasma of family members by Western blotting. They identified a unique 250-kDa band that was prominent in the plasma of affected and barely detectable in the plasma of unaffected family members and confirmed by mass spectrometry that it represented a form of FV. A subsequent search for potential mRNA splicing abnormalities using RT-PCR and mRNA from family members’ white blood cells detected the expected F5 transcript (∼2950 bp) and a shorter transcript (∼840 bp, encoding FV-short) that predominated in affected individuals and was a minor constituent in unaffected individuals. Thus, the A2440G mutation appeared to dramatically increase (∼20-fold) the expression of an alternatively spliced transcript that is normally in low abundance. Sequencing showed that FV-short lacked aa 756–1458 of the B domain of FV (Figure 1A).
The authors then turned their attention to the coagulation abnormalities evident in people with east Texas bleeding disorder. Using a sensitive thrombin generation assay in which coagulation was induced with low levels of TF, they showed that the inhibitory activity in patient plasma was associated with FV-short, but that recombinant FV-short itself did not possess intrinsic anticoagulant activity. As TFPIα had been shown to interact with FV in normal plasma (4, 6), TFPIα levels were measured and found to be approximately 10-fold higher in the affected family members. Immunoprecipitation experiments confirmed that TFPIα was bound to FV-short in patient plasma. Further, in normal plasma, TFPIα appeared to be equally distributed between FV and FV-short even though FV-short is at a greater than 20-fold lower concentration. Therefore the affinity of FV-short for TFPIα is considerably greater than that of FV.
Remaining questions
This report has solved the case of east Texas bleeding disorder, and in doing so, it has identified a previously unrecognized, alternatively spliced isoform of FV, demonstrated a gain-of-function mutation in the F5 gene, and shown that increased levels of TFPIα cause bleeding. As is common with a scientific advance, the findings raise additional questions:
What mechanism underlies the tight binding of TFPIα to FV-short?
The basic C-terminal peptide of full-length TFPIα (FL-TFPIα) is required for the binding of TFPIα to both FV-short and to FV. FV is maintained in an inactive, procofactor state through an interaction between a BR and an AR within its B domain (8). The BR is not present in FV-short, leaving its AR as a potential site for TFPIα binding (Figure 1A). Consistent with this notion, alignment of the peptides in the BR that affect its interaction with the AR and the C-terminal peptide of TFPIα reveals an area of remarkable homology (Figure 1B). The much lower affinity of TFPIα for FV might be explained by its need to compete with the endogenous BR in FV for AR binding.
Does the TFPIα:FV-short complex have FV cofactor activity?
The studies of Bos and Camire suggest that FV-short should display FV cofactor activity (8). It is conceivable that, by interacting with the AR, TFPIα functions as a surrogate for the BR, thereby keeping FV-short in a procofactor state by interfering with its binding to FXa.
Is east Texas bleeding disorder simply caused by increased circulating TFPIα, or does the TFPIα:FV-short complex possess intrinsic anticoagulant activity?
The presence of FVa in the prothrombinase complex increases TFPIα inhibition of FXa by producing a 20-fold decrease in the Ki of the TFPIα-FXa encounter complex (11). This effect is completely abrogated by the presence of the substrate prothrombin (12). It is possible that the TFPIα:FV-short complex has enhanced anti-FXa activity that is less sensitive to the presence of prothrombin. The inhibitory activity of such a complex would presumably be circumvented by thrombin cleavage of FV-short at R1545, releasing the AR and TFPIα (Figure 1A).
What is the effect of FL-TFPIα on forms of FV that are produced normally during coagulation and that, like FV-short, lack the BR of FV?
Proteolysis of FV by FXa and thrombin involves cleavage at R709, R1018, and R1545 (Figure 1A). Initial cleavages by both FXa and thrombin occur at R709 and R1018 and produce a form of FV that lacks the BR and would exhibit cofactor activity. This form of FV, and perhaps the partially proteolyzed forms of FV released by platelets, would be potential targets of TFPIα until cleavage at R1545 releases the AR from the activated FV molecule. An important difference in the activation of FV by FXa and by thrombin, however, is that cleavage at R1545 occurs much more slowly or not at all in the case of FXa (13). This implies that FXa-activated FV would be considerably more susceptible to an effect of TFPIα. Whether FXa generated by FVIIa/TF or thrombin produced weakly by FXa in the absence of FVa is responsible for the initial activation of FV during hemostasis is not known. TFPIα released at relatively high local concentrations by platelets that adhere and aggregate at a site of injury could limit thrombin generation that was dependent on FXa-activated FV. Once generated, thrombin would counter this effect of TFPIα by producing forms of activated FV that have been cleaved at R1545 and lack the potential TFPIα-binding site AR.
Is there a role for PS?
PS is known to interact with FXa, FV(a), and TFPIα and is also released by stimulated platelets. Thus, its potential role in the reactions discussed above warrants investigation.
Most important to the affected members of the east Texas family, however, is that the elucidation of the underlying pathophysiology suggests that TFPI inhibitors currently in development may provide a means of treatment (14, 15).
Acknowledgments
George Broze is funded by NIH grant R37 077193.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Citation for this article: J Clin Invest. 2013;123(9):3710–3712. doi:10.1172/JCI71220.
See the related article beginning on page 3777.
References
- 1.Broze GJ, Jr, Warren LA, Novotny WF, Higuchi DA, Girard JJ, Miletich JP. The lipoprotein-associated coagulation inhibitor which inhibits the factor VII-tissue factor complex also inhibits factor Xa: Insight into its possible mechanism of action. Blood. 1988;71(2):335–343. [PubMed] [Google Scholar]
- 2.Hackeng TM, Sere KM, Tans G, Rosing J. Protein S stimulates inhibition of the tissue factor pathway by tissue factor pathway inhibitor. Proc Natl Acad Sci U S A. 2006;103(9):3106–3111. doi: 10.1073/pnas.0504240103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ndonwi M, Tuley EA, Broze GJ., Jr The Kunitz-3 domain of TFPI-alpha is required for protein S-dependent enhancement of factor Xa inhibition. Blood. 2010;116(8):1344–1351. doi: 10.1182/blood-2009-10-246686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Duckers C, et al. Low plasma levels of tissue factor pathway inhibitor in patients with congenital factor V deficiency. Blood. 2008;112(9):3615–3623. doi: 10.1182/blood-2008-06-162453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Castoldi E, Simioni P, Tormene D, Rosing J, Hackeng TM. Hereditary and acquired protein S deficiencies are associated with low TFPI levels in plasma. J Thromb Haemost. 2010;8(2):294–300. doi: 10.1111/j.1538-7836.2009.03712.x. [DOI] [PubMed] [Google Scholar]
- 6.Ndonwi M, Girard TJ, Broze GJ., Jr The C-terminus of tissue factor pathway inhibitor α is required for its interaction with factors V and Va. J Thromb Haemost. 2012;10(9):1944–1946. doi: 10.1111/j.1538-7836.2012.04834.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Monkovic DD, Tracy PB. Functional characterization of human platelet-released factor V and its activation by factor Xa and thrombin. J Biol Chem. 1990;265(28):17132–17140. [PubMed] [Google Scholar]
- 8.Bos MHA, Camire RM. A bipartite autoinhibitory region within the B-domain suppresses function in factor V. J Biol Chem. 2012;287(31):26342–26351. doi: 10.1074/jbc.M112.377168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kuang SQ, et al. Characterization of a novel autosomal dominant bleeding disorder in a large kindred from east Texas. Blood. 2001;97(6):1549–1554. doi: 10.1182/blood.V97.6.1549. [DOI] [PubMed] [Google Scholar]
- 10.Vincent LM, et al. Coagulation factor VA2440G causes east Texas bleeding disorder via TFPIα. . J Clin Invest. 2013;123(9):3777–3787. doi: 10.1172/JCI69091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huang Z-F, Wun T-C, Broze GJ., Jr Kinetics of factor Xa inhibition by tissue factor pathway inhibitor. J Biol Chem. 1993;268(36):26950–26955. [PubMed] [Google Scholar]
- 12.Mast AE, Broze GJ., Jr Physiological concentrations of tissue factor pathway inhibitor do not inhibit prothrombinase. Blood. 1996;87(5):1845–1850. [PubMed] [Google Scholar]
- 13.Monkovic DD, Tracy PB. Activation of human factor V by factor Xa and thrombin. Biochemistry. 1990;29:1118–1128. doi: 10.1021/bi00457a004. [DOI] [PubMed] [Google Scholar]
- 14.Hilden I, et al. Hemostatic effect of a monoclonal antibody mAb 2021 blocking the interaction between FXa and TFPI in a rabbit hemophilia model. Blood. 2012;119(24):5871–5878. doi: 10.1182/blood-2012-01-401620. [DOI] [PubMed] [Google Scholar]
- 15.Gorczyca ME, et al. Inhibition of tissue factor pathway inhibitor by the aptamer BAX499 improves clotting of hemophilic blood and plasma. J Thromb Haemost. 2012;10(8):1581–1590. doi: 10.1111/j.1538-7836.2012.04790.x. [DOI] [PubMed] [Google Scholar]