

Abstract
All currently approved antiviral drugs for the treatment of chronic hepatitis B virus (HBV) infection are nucleos(t)ide reverse transcriptase inhibitors (NRTI), which inhibit the DNA synthesis activity of the HBV polymerase. The polymerase is a unique reverse transcriptase (RT) that has a novel protein priming activity in which HP initiates viral DNA synthesis using itself as a protein primer. We have determined the ability of NRTI-triphosphates (TP) to inhibit HBV protein priming and their mechanisms of action. While entecavir-TP (a dGTP analog) inhibited protein priming initiated specifically with dGTP, clevudine-TP (a TTP analog) was able to inhibit protein priming independently of the deoxynucleoside triphosphate (dNTP) substrate and without being incorporated into DNA. We next investigated the effect of NRTIs on the second stage of protein priming, wherein two dAMP nucleotides are added to the initial deoxyguanosine nucleotide. The obtained results indicated that clevudine-TP as well as tenofovir DF-DP strongly inhibited the second stage of protein priming. Tenofovir DF-DP was incorporated into the viral DNA primer, whereas clevudine-TP inhibited the second stage of priming without being incorporated. Finally, kinetic analyses using the HBV endogenous polymerase assay revealed that clevudine-TP inhibited DNA chain elongation by HP in a noncompetitive manner. Thus, clevudine-TP appears to have the unique ability to inhibit HBV RT via binding to and distorting the HP active site, sharing properties with both NRTIs and nonnucleoside RT inhibitors.
INTRODUCTION
Chronic hepatitis B virus (HBV) infection remains a worldwide health problem, afflicting over 350 million patients and resulting in one million deaths per year (1). HBV has a ca. 3.2-kb relaxed circular (RC) DNA genome that is repaired to form a covalently closed circular (CCC) DNA in the host cell. CCC DNA is then transcribed by the host RNA polymerase II to produce a pregenomic RNA (pgRNA), the precursor to RC DNA (2, 3). Reverse transcription of pgRNA by the viral polymerase (HP), a multifunctional reverse transcriptase (RT), generates first the minus strand and then the plus strand of RC DNA using its RNA- and DNA-dependent DNA polymerization activities, respectively (2, 4).
Initiation of viral minus-strand DNA synthesis occurs via a novel protein priming mechanism in which HP, specifically a tyrosine residue (Y63) in its N-terminal domain, serves as a protein primer, while its central RT domain serves as the catalyst (5–9). Furthermore, protein-primed initiation of reverse transcription requires a short RNA stem-loop structure, termed epsilon (Hε), on the 5′ end of pgRNA, which is recognized specifically by HP in a host chaperone-dependent reaction (7, 10–15). Hε, specifically the last three nucleotides of its internal bulge, serves as the specific template for protein priming (7, 10, 16). Furthermore, it serves as an allosteric activator of the HP enzymatic activity (17, 18). The product of protein priming is a short, 3-nucleotide (nt)-long (dGAA) viral minus-strand DNA covalently attached to HP. Based on differential sequence and structural requirements defined using the duck hepatitis B virus (DHBV) polymerase, protein priming can be divided into two distinct stages, the initial attachment of the first dGMP residue to the polymerase (initiation or polymerase deoxyguanidinylation) followed by the addition of two (dAA; for HP) or three (dTAA; for the DHBV polymerase) nucleotide residues to the initiating dGMP (DNA polymerization) (7, 19, 20). After protein priming, the nascent minus-strand-HP complex is transferred from the Hε template to a complementary sequence at the 3′ end of pgRNA called DR1, where HP continues minus-strand DNA elongation (7, 10, 21).
Current treatments for chronic HBV infection include pegylated alpha interferon and nucleos(t)ide RT inhibitors (NRTIs) (22, 23). Interferon therapy is effective only in a minority of patients and is associated with side effects and toxicities. Five NRTIs have been approved by the FDA for HBV treatment, including lamivudine (2′,3′-dideoxy-3′-thiacytidine; 3TC), adefovir dipivoxil {9-[2-[[bis[(pivaloyloxy)methoxy]-phosphinyl]-methoxy]ethyl]adenine}, entecavir {2-amino-9-[(1S,3R,4S)-4-hydroxy-3-(hydroxymethyl)-2-methylidenecyclopentyl]-6,9-dihydro-3H-purin-6-one}, telbivudine [1-(2-deoxy-β-l-erythro-pentofuranosyl)-5-methylpyrimidine-2,4(1H,3H)-dione], and tenofovir disoproxil fumarate (DF) {9-[(R)-2-[[bis[[(isopropoxycarbonyl)oxy]methoxy]phosphinyl]methoxy]propyl]adenine fumarate}. NRTIs suppress HBV replication in most patients but are not curative, requiring potentially life-long treatment that may be associated with drug resistance and toxicity (22, 24).
Entecavir has been shown to potently inhibit HBV DNA synthesis and displays a high barrier for HBV drug resistance, which occurs at a very low frequency even after 5 years of treatment (25–27). In common with most other NRTIs, entecavir inhibits HBV DNA synthesis in a substrate (dGTP)-competitive manner. As dGTP is the initiating nucleotide for HBV protein priming, entecavir is thought to have the unique ability, among all approved NRTIs, to inhibit HBV protein priming by competing with dGTP. However, as the protein priming assay employed to measure entecavir effects relied on a recombinant HP protein isolated from insect cells to carry out Hε-independent DNA synthesis in vitro (5, 27), it remains uncertain if entecavir indeed inhibits authentic, Hε-dependent protein priming as proposed.
Tenofovir DF, an oral prodrug of tenofovir, is a potent inhibitor of both HIV and HBV replication and is approved for the treatment of both infections. With respect to HBV, tenofovir DF has a very high barrier to resistance; no HBV resistance mutations have been identified after 5 years of treatment in phase 3 extension studies (28). Tenofovir DF is an acyclic nucleotide analog lacking a 3′ hydroxyl group and has been shown to be a competitive inhibitor of recombinant HP (purified from insect cells) (29). While it is clear that tenofovir DF inhibits DNA elongation by HP in biochemical and cell-based assays, it may also inhibit the second stage of HBV protein priming in which two dAMP residues are added to form the complete dGAA primer; however, this has not been formally demonstrated. A thymidine analog, fialuridine [1-(2-deoxy-2-fluoro-1-d-arabinofuranosyl)-5-iodouracil; FIAU], was shown to inhibit the DNA polymerization stage of protein priming by the DHBV polymerase via a competitive mechanism (30). Due to the lack of TMP incorporation during protein priming by HP, FIAU is not predicted to inhibit HP priming.
Clevudine [1-(2-deoxy-2-fluoro-5-methyl-β-l-arabinofuranosyl) uracil; l-FMAU] is a recently introduced NRTI approved for treatment of chronic HBV infections in South Korea (31–34). Clevudine has the unnatural l-configuration and, compared to other NRTIs, has the unique ability to maintain suppression of HBV replication for an extended period of time (for at least 24 weeks) following drug withdrawal (35, 36). As with other NRTIs, clevudine-triphosphate (TP) directly inhibits HBV DNA synthesis as measured in the endogenous polymerase assay (EPA) (32), in which isolated HBV nucleocapsids carry out viral DNA synthesis (mostly plus-strand DNA elongation) using the packaged (endogenous) HP as the catalyst and the viral minus-strand DNA as the template (2, 37). In contrast to many other NRTIs, clevudine is not considered an obligate chain terminator because of the presence of the 3′-OH group (34). Furthermore, clevudine is not incorporated into DNA by the Epstein-Bar virus or cellular DNA polymerases (38), indicating it does not serve as a substrate for DNA synthesis by the polymerases. This is in contrast to some other arabinosides, such as FIAU, which can be used by mammalian cellular DNA polymerases as substrates (39). Thus, in contrast to all other known nucleoside analog therapies, clevudine appears to inhibit viral polymerase activity without being incorporated into viral DNA (34, 38, 40).
As protein priming represents a novel biochemical reaction that requires an HP conformation(s) distinct from that for generic DNA strand elongation (9, 19, 20, 30, 41), specific targeting of the priming conformation of HP with antivirals is expected to complement well those that target the HP elongation mode (true for most currently approved NRTIs), as measured by the commonly used EPA. Taking advantage of an authentic, Hε-dependent in vitro HBV protein priming assay that we have recently developed (7), we have analyzed the effects of several NRTIs on HBV protein priming. Furthermore, in our efforts to understand the novel mechanism of action of clevudine, we conducted a kinetic study with this compound using the classical EPA.
MATERIALS AND METHODS
Plasmids and compounds.
pcDNA-3FHP, for expressing the 3× FLAG-tagged full-length HP in human cells, and pCMV-HE, for expressing the Hε RNA derived from the 5′ end of HBV pgRNA, have been described previously (7). pCMV-HE-B6G or pCMV-HE-B6A produces a mutant Hε in which the last (6th) Hε internal bulge residue (B6; nt 1863), which serves as the template for the first nucleotide of viral minus-strand DNA, is changed from an rC to an rG (B6G) or rA (B6A) (7). Clevudine-monophosphate (MP), clevudine-diphosphate (DP), clevudine-TP, 3TC-TP, emtricitabine {4-amino-5-fluoro-1-[(2S,5R)-2-(hydroxymethyl)-1,3-oxathiolan-5-yl]-1,2-dihydropyrimidin-2-one; FTC}-TP, and tenofovir DF-DP were made at Pharmasset and Gilead, respectively. Entecavir-TP was purchased from Moravek Biochemicals. Hemin was purchased from Sigma and prepared as described previously (42). The structures of the nucleos(t)ide analogs used in this study are depicted in Fig. 1.
Fig 1.

Chemical structures of NRTIs used in this study. See the text for details.
Protein expression.
HP with bound Hε (wild type [WT] or mutant) was expressed and purified using HEK293T cells as previously described (7). Briefly, HEK293T cells were transfected with pcDNA-3FHP together with pCMV-HE, pCMV-HE-B6G, or pCMV-HE-B6A. Two days after transfection, cells were washed and then lysed at 4°C in FLAG lysis buffer (50 mM Tris, pH 7, 100 mM NaCl, 50 mM KCl, 10% glycerol, 1% NP-40, 1 mM EDTA) plus 1× Complete protease inhibitor (Roche), 1 mM phenylmethylsulfonyl fluoride (PMSF), 10 mM β-mercaptoethanol (β-ME), 2 mM dithiothreitol (DTT), and 250 U RNasin plus RNase inhibitor (Promega) per ml lysis buffer and scraped off the plate. HP was purified from the clarified lysate using protein A/G beads (Pierce) bound to the M2 anti-FLAG antibody (Sigma). Unbound materials were removed by washing 5 times with FLAG lysis buffer at 4°C with individual protease inhibitors (28 μM E-64, 1 mM PMSF, 5 μg/μl leupeptin), 2 mM DTT, 10 mM β-ME, and 10 U/ml RNasin plus RNase inhibitor. Purified HP was stored at −80°C until use in the same washing buffer while remaining bound to the M2 beads. Glutathione S-transferase (GST)-tagged HP fusion protein (HTPRT-Spe) for the gel mobility shift assay was expressed in Escherichia coli and purified by using glutathione affinity resin as described previously (11, 12). Chaperone proteins were expressed and purified as described previously (12).
In vitro RNA binding assay by co-IP.
The immunoprecipitation (IP) in vitro RNA binding assay was performed as previously described, with minor modifications (7). Briefly, FLAG lysis buffer from HP purification was removed from aliquots of the HP-bound M2 beads. Binding of HP to Hε or mutant Hε-dB RNA, with the Hε internal bulge deleted and being defective in HP binding (7, 12), was tested by incubating each aliquot of HP-bound beads with ca. 0.4 μg in vitro-transcribed and 32P-labeled ε RNAs in radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris [pH 7.5], 150 mM NaCl, 1 mM EDTA, 0.05% NP-40) with 1× Complete protease inhibitor cocktail, 2 mM DTT, 1 mM PMSF, 1 U per μl RNasin plus RNase inhibitor. Distilled water (dH2O; mock) or clevudine-TP (100 μM) was added before the addition of RNA. After 3 h of binding at room temperature with shaking, unbound materials were removed and the beads were washed in RIPA buffer with 2 mM DTT, individual protease inhibitors, and 10 U RNasin plus RNase inhibitor per ml buffer. Bound materials were eluted by boiling and resolved on a sodium dodecyl sulfate (SDS)-15% polyacrylamide gel. The gel containing the 32P-labeled ε RNA was dried and exposed to film to detect the labeled RNA bound to HP.
Gel mobility shift RNA binding assay.
The gel mobility shift assay was performed as previously described (12, 43). Briefly, 20 ng in vitro-transcribed 32P-labeled Hε RNA was incubated with 20 ng GST-HP fusion protein along with Hsp90 (360 ng), Hsp70 (3 μg), Hdj1 (200 ng), Hop (370 ng), p23 (100 ng), and an ATP regenerating system in a 10-μl reaction mix in the presence of dH2O, FTC-TP, clevudine-MP, clevudine-DP, clevudine-TP, or hemin at the indicated concentrations. Samples were incubated for 2 h at 30°C, after which the samples were resolved on a 5% native polyacrylamide gel and detected by autoradiography.
In vitro HP priming assay.
Priming assays were performed as recently published (7). Briefly, TMgNK buffer (20 mM Tris-HCl, pH 7.0, 15 mM NaCl, 10 mM KCl, 4 mM MgCl2) along with 1× EDTA-free protease inhibitor cocktail (Roche), 4 mM DTT, 1 mM PMSF, and 1 U RNasin plus RNase inhibitor (Promega) per μl buffer were added to the beads. Compounds dissolved in dH2O or dH2O alone (mock) was added individually to each priming assay. One microliter of the indicated radiolabeled nucleotide ([α-32P]dNTP; 10 mCi/ml; 3,000 Ci/mmol) was then added, and the reaction mixtures were incubated at 25°C for 4 h with shaking. After the priming reactions, the beads were washed in TNK buffer plus individual protease inhibitors and 10 mM β-ME. The washed beads were then boiled in 2× SDS sample buffer for 10 min. Radiolabeled HP resulting from protein priming was resolved by running the eluate on an SDS-12.5% polyacrylamide gel and detected by autoradiography. Priming signals were quantified by phosphorimaging.
To test if the nucleoside analogs could be used as substrates to initiate priming (becoming covalently attached to Y63 of HP) (see Fig. 4), HP copurified with Hε was first incubated in TMgNK buffer, as previously outlined, with dH2O (mock) or with 100 μM dGTP, clevudine-TP, tenofovir-DP, or 3TC-TP at 25°C for 2 h with shaking. Priming reactions were then washed twice with TNK buffer plus individual protease inhibitors and 10 mM β-ME to remove unincorporated nucleotides. After these washes, fresh TMgNK buffer was added along with 1 μl [α-32P]dATP and incubated for 2 more hours at 25°C with shaking. After priming, samples were washed extensively to remove unincorporated nucleotides, and labeled HP was resolved on an SDS-polyacrylamide gel as previously outlined.
Fig 4.
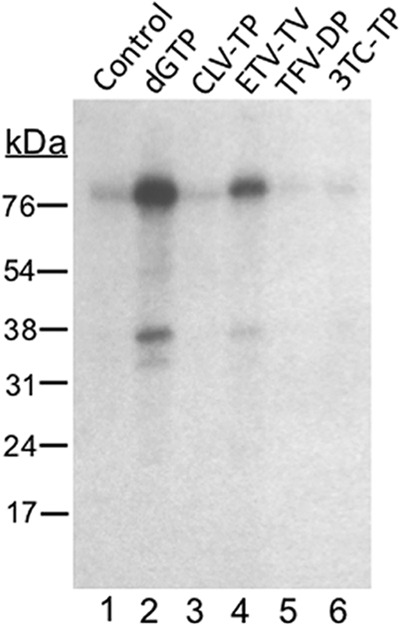
Entecavir, but not clevudine, could be used as a substrate to initiate protein priming. HP copurified with Hε was first incubated in TMgNK with 100 μM dGTP (lane 2), clevudine-TP (CLV-TP; lane 3), entecavir-TP (ETV-TP; lane 4), tenofovir DF-DP (TFV-DP; lane 5), or 3TC-TP (lane 6). To monitor dGTP (or analog)-independent (background) dATP incorporation, which could represent low-level misincorporation of dATP as the first nucleotide of priming (7), no dGTP was added to the reaction shown in lane 1 (control). Unincorporated nucleotides or analogs were then washed out, and fresh TMgNK buffer was added along with [α-32P]dATP to allow DNA polymerization. After extensive washes to remove unincorporated nucleotides, samples were resolved by SDS-PAGE and visualized by autoradiography. The positions of the protein molecular mass markers (in kDa) are indicated.
For assaying effects of the NRTIs on the DNA polymerization step of priming (see Fig. 5), HP was first primed (for initiation) with 100 μM dGTP or water (labeled −dG) for 2 h with shaking. Priming reaction mixes were then washed twice with TNK buffer plus individual protease inhibitors and 10 mM β-ME to remove unincorporated nucleotides, and fresh TMgNK buffer was added to the samples. Subsequently, compounds dissolved in dH2O or dH2O alone (mock) was individually added to the reactions, after which 1 μl [α-32P]dATP was added to allow for polymerization for an additional 2 h with shaking. To release the covalently linked nucleotide and DNA from HP, priming reactions were washed extensively and then treated with tyrosyl-DNA phosphodiesterase 2 (Tdp2), and the released DNA products in the supernatant were resolved by urea-polyacrylamide gel electrophoresis (PAGE), while bound HP was resolved by SDS-PAGE as previously outlined (7).
Fig 5.

Clevudine and tenofovir inhibited the DNA polymerization stage of HP protein priming in vitro. HP copurified with Hε in TMgNK buffer was primed with unlabeled dGTP (lanes 3 to 10). To monitor dGTP-independent (background) dATP incorporation, as described for Fig. 4, lane 1, no dGTP was added to the reactions shown in lanes 1 and 2 (−dG). Subsequently, samples were washed twice to remove unincorporated dGTP, and fresh TMgNK buffer was added along with dH2O (lanes 1 to 4) or 100 μM clevudine-TP (CLV-TP; lanes 5 and 6), tenofovir DF-DP (TFV-DP; lanes 7 and 8), or 3TC-TP (lanes 9 and 10). [α-32P]dATP was then added to all reactions to allow for polymerization in the presence or absence of potential drug inhibition. After priming, samples were washed extensively to remove unincorporated nucleotides and then were mock treated (odd-numbered lanes) or treated with Tdp2 (even-numbered lanes) to release DNA attached to HP. (a) The beads, which contained the primed HP, were processed for and resolved by SDS-PAGE and visualized by autoradiography. The positions of the protein molecular mass markers (in kDa) are indicated. (b) Priming signals (from samples not treated with Tdp2, i.e., the odd-numbered lanes in panel a) from three independent experiments were quantified by phosphorimaging, and after subtracting the background signal from lane 1 in panel a (−dG), they were expressed as percentages of the mock-treated control (lane 3). Error bars denote the standard errors of the means (SEM). (c) The supernatant, which contained the released DNA, was collected after Tdp2 digestion, resolved on an 8 M urea-20% polyacrylamide gel, and visualized by autoradiography. The positions of the dGAn (where n denotes the number of dA residues following the initial dG residue) oligonucleotide markers are indicated.
EPA.
Intracellular HBV nucleocapsids were harvested from induced (tetracycline-removed) HepAD38 cells (44) by polyethylene glycol precipitation as described previously (45). EPA was performed in a reaction volume of 60 μl containing the nucleocapsids (50%, vol/vol), 50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 10 mM MgCl2, 1 mM DTT, 0.1% NP-40, 100 μM dATP and dGTP, 1 μM unlabeled dCTP, 0.3 μCi/μl [α-32P]dCTP. Various concentrations of TTP (0.02 to ∼2 μM, as indicated) and clevudine-TP (0 to ∼5 μM) were included. The reaction mixtures were incubated at 37°C. After 20, 40, 60, 80, 100, and 120 min of incubation, 9 μl of the reaction mix was taken and mixed with 1 μl of quench solution (20 mM EDTA, 0.4% SDS [final concentration]). Five μl of each reaction mix was spotted on DE81 paper, washed 3× with 125 mM Na2HPO4 followed by dH2O and ethanol (EtOH), dried, and exposed to a phosphorscreen. The radioactive spots were cut out, and β-emissions were counted using a scintillation counter. The rate of product formation at each concentration of substrate and drug was calculated and plotted. The mode of inhibition was evaluated by fitting the data to the competitive equation, v = Vmax × [S]/{Km × (1 + [I]/Ki) + [S]}, or the noncompetitive equation, v = Vmax × [S]/{(Km+ [S]) × (1 + [I]/Ki)}, where v is the observed rate, [S] is substrate concentration, [I] is inhibitor concentration, Vmax is the maximum rate of metabolism, Km is the Michaelis constant, and Ki is the inhibition constant.
Statistical analysis.
Results, reported as 50% inhibitory concentrations (IC50; means ± SEM), were determined by plotting product formation as a function of drug concentrations on a scatterplot and creating a logarithmic trend line using Microsoft Excel. For the kinetic analysis, a nonlinear fit was performed using the GraFit program, version 5 (Erithacus Software, Horley, Surrey, United Kingdom).
RESULTS
Clevudine-triphosphate, as well as entecavir-triphosphate, strongly inhibited the initiation stage of Hε-dependent HBV protein priming in vitro.
To determine the effects of NRTIs on HBV protein priming, we tested their triphosphate (-TP) forms for their ability to inhibit Hε-dependent protein priming initiation in vitro carried out by HP-Hε complex purified from human cells (7). Among all NRTIs tested, clevudine-TP and entecavir-TP inhibited in vitro priming strongly, eliminating the priming signal at 100 μM concentration (Fig. 2a, lanes 7, 12, and 15, and b), with an IC50 for inhibiting protein priming estimated at 4.89 μM for clevudine-TP and 1.78 μM for entecavir-TP (see Fig. S1 in the supplemental material). Although the inhibitory effect of entecavir, a deoxyguanosine analog, was consistent with the suggestion in the literature that entecavir could inhibit initiation of protein priming via competition with the initiating dGTP priming substrate (27), the strong inhibitory effect of clevudine, a thymidine analog, was unexpected. To help understand how clevudine inhibited protein priming, we also tested the clevudine mono- and diphosphate (clevudine-MP and -DP, respectively) in the priming reaction and found they had no inhibitory activity (Fig. 2a, lanes 13 and 14, and b). These results suggest that clevudine-TP interacts with the HP dNTP-binding site, as other NRTIs do (see Discussion).
Fig 2.
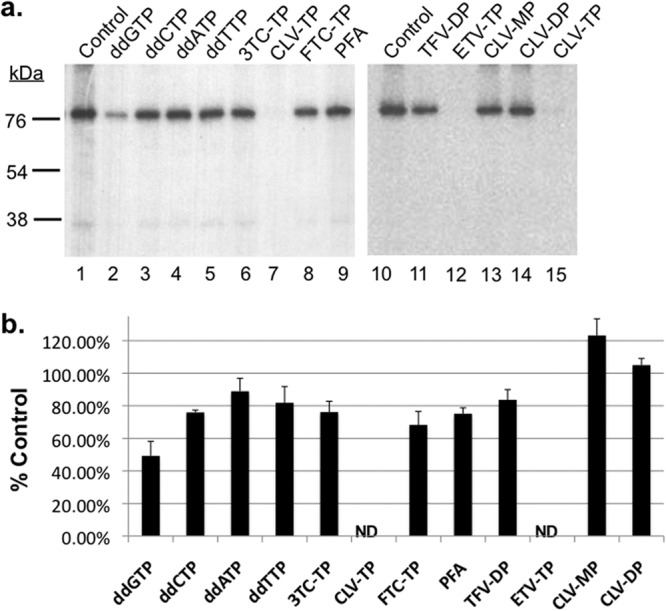
Clevudine and entecavir inhibited Hε-dependent initiation of protein priming. In vitro priming reactions were performed by incubating HP copurified with Hε in TMgNK buffer with [α-32P]dGTP in the presence of dH2O (control; lanes 1 and 10), 100 μM dideoxyguanosine (ddGTP; lane 2), dideoxycytosine (ddCTP; lane 3), dideoxyadenosine (ddATP; lane 4), dideoxythymidine (ddTTP; lane 5), 3TC-TP (lane 6), clevudine-TP (CLV-TP; lanes 7 and 15), emtricitabine-TP (FTC-TP; lane 8), tenofovir DF-DP (TFV-DP; lane 11), entecavir-TP (ETV-TP; lane 12), clevudine-MP (CLV-MP; lane 13), clevudine-DP (CLV-DP; lane 14), or 1 mM PFA (lane 9). (a) Priming products were resolved by SDS-PAGE and visualized by autoradiography. The positions of the protein molecular mass markers (in kDa) are indicated. (b) Priming signals from three independent experiments were quantified by phosphorimaging, and signals from the treated samples are expressed as percentages of controls. Error bars denote the standard errors of the means (SEM). ND, not detectable.
FTC and 3TC, two deoxycytidine analogs, and tenofovir DF, a dAMP analog, are all classical NRTIs whose triphosphates inhibit HP by competing for HP binding with the corresponding natural nucleotides (dCTP and dATP, respectively). Following binding to HP, these nucleoside analog triphosphates are incorporated into viral DNA and prevent chain elongation due to their lack of the 3′-OH groups (22, 23). None of these three NRTI-TPs showed any significant inhibition of this first step of protein priming (Fig. 2a, lanes 6, 8, and 11, and b) as expected, as these NRTI-TPs should not compete with the initiating dGTP priming substrate. Not surprisingly, ddGTP moderately inhibited protein priming initiation, whereas the other ddNTPs had little effect (Fig. 2a, lanes 2 to 5, and b). In addition, the pyrophosphate analog phosphonoformic acid (PFA), known to inhibit HP in EPA, showed little effect on priming, in support of the notion that HP adopts different conformations during protein priming and DNA elongation (Fig. 2a, lane 9, and b) (9, 41).
Clevudine-TP did not affect HP-Hε binding.
To further explore the mechanism whereby clevudine-TP inhibited protein priming, we tested its potential effect on HP-Hε binding, a prerequisite for protein priming. Using two different in vitro RNA binding assays (7, 43), we failed to detect any inhibitory effect of clevudine, in the -MP, -DP, or -TP form, or FTC-TP (used as a conventional NRTI control) on HP-Hε binding, while hemin strongly inhibited HP-Hε binding, as previously reported (see Fig. S2 in the supplemental material) (42). These results indicated that none of the phosphorylated forms of clevudine inhibited protein priming by blocking HP-Hε binding.
Inhibition of protein priming initiation by clevudine-TP independent of the template nucleotide sequence.
As clevudine-TP unexpectedly inhibited protein priming initiated with dGTP, we decided to determine if clevudine-TP could also inhibit protein priming initiated with other dNTP substrates. The dNTP used to initiate protein priming can be changed in a predictable manner by changing the nucleotide at the last template position (the 6th position of the internal bulge, or B6) of the Hε RNA; for example, Hε-B6G directs initiation of protein priming with dCTP and Hε-B6A with TTP instead of dGTP (7). As anticipated, 3TC-TP, a dCTP analog, inhibited priming with dCTP but not TTP (Fig. 3, lanes 4, 8, 12, and 16) or dGTP, as shown above. Also, entecavir-TP showed no inhibitory effect on priming initiated with dCTP or dTTP (Fig. 3, lanes 10 and 14), as expected. In sharp contrast, clevudine-TP inhibited priming initiated with either TTP or dCTP (better than ddTTP or ddCTP, respectively, which were used as controls) (Fig. 3, lanes 2, 3, 6, and 7), as well as with dGTP (in the case of the WT Hε template), as discussed above. Thus, these results clearly showed that clevudine-TP did not function as a competitive inhibitor of TTP incorporation; rather, it inhibited priming through an entirely novel mechanism (see Discussion).
Fig 3.

Clevudine-TP inhibited protein priming initiated by dCTP and TTP. HP was copurified with the mutant Hε-B6A (coding for priming initiation with TTP; lanes 1 to 4 and 9 to 12) or Hε-B6G (coding for initiation with dCTP; lanes 5 to 8 and 13 to 16). HP bound to Hε-B6A or Hε-B6G was assayed for protein priming as described in the legend to Fig. 2, except [α-32P]TTP (Hε-B6A) or [α-32P]dCTP (Hε-B6G) was used instead of [α-32P]dGTP, in the presence of dH2O (control; lanes 1, 5, 9, and 13), 100 μM clevudine-TP (CLV-TP; lanes 2 and 6), ddTTP (lanes 3 and 11), ddCTP (lanes 7 and 15), 3TC-TP (lanes 4, 8, 12, and 16), or entecavir-TP (ETV-TP; lanes 10 and 14). Samples were resolved by SDS-PAGE and visualized by autoradiography.
Entecavir, but not clevudine, could be used by HP as a substrate to initiate protein priming.
To further understand the mechanisms of inhibition of HBV protein priming by the NRTIs, we tested their capacity to be used by HP as a substrate in the priming reaction. To this end, we first incubated the purified HP-Hε complex with (unlabeled) dGTP, clevudine-TP, entecavir-TP, tenofovir DF-DP, or 3TC-TP in the priming reaction to allow the initiation of protein priming, i.e., the covalent attachment of dGMP or, potentially, the various analogs (in their MP forms) to HP. Unincorporated nucleotides/analogs were then removed before the addition of 32P-labeled dATP, which would be incorporated as the 2nd and 3rd nucleotide during DNA synthesis using an Hε template (7). Other than the natural dGTP, we found that entecavir-TP, but not clevudine-TP, 3TC-TP, or tenofovir DF-DP, could be used as a substrate for priming initiation, allowing subsequent dATP incorporation (Fig. 4). Compared to dGMP, incorporation of entecavir-MP apparently allowed less efficient subsequent addition of dAMP (Fig. 4, lane 4 versus 2). Alternatively, entecavir-MP might not have served as the initiating substrate as efficiently as the natural dGMP.
Clevudine-TP and tenofovir DF-DP inhibited the DNA polymerization stage of HP priming.
As we found, unexpectedly, that clevudine-TP could inhibit the initiation stage of HBV protein priming independent of the nucleotide substrate used, we decided to test if it could also inhibit the second stage of protein priming, which includes the incorporation of two dAMP residues (using UU of the Hε bulge as the template) after the initiating dGMP. As tenofovir DF is a dAMP analog, we also tested its potential effect on this stage of protein priming. We initiated protein priming with unlabeled dGTP; subsequently, clevudine-TP, tenofovir DF-DP, or 3TC-TP was added prior to addition of [α-32P]dATP for DNA polymerization. We found that DNA polymerization was inhibited most strongly by clevudine-TP (Fig. 5a, lane 5, and b; also see Fig. S3 in the supplemental material), followed by tenofovir DF-DP (Fig. 5a, lane 7, and b). 3TC-TP showed little effect on DNA polymerization (Fig. 5a, lane 9, and b). To analyze the specific DNA products that were covalently linked to HP during DNA polymerization in the presence of the nucleotide analog triphosphates, the labeled polymerization products were treated with tyrosyl-DNA phosphodiesterase 2 (Tdp2), an enzyme that specifically breaks the phosphotyrosyl-5′ DNA linkage, such as that between Y63 of HP and the 5′ end of the HBV minus-strand DNA (7, 41, 46). In the in vitro polymerization assay, we have previously observed that some of the covalent HP-dGAA (dGA2) complex, i.e., the complete priming product, is transferred to a complementary sequence at the 3′ DR1 on pgRNA, where three more dAMP residues can then be added as directed by the template pgRNA (7, 21). Visualization of the released DNA from HP by urea-PAGE revealed that clevudine-TP and tenofovir DF-DP inhibited polymerization both at Hε (formation of HP-dGA2) and at the 3′ DR1 following primer transfer (species above dGA2) (Fig. 5c, lanes 6 and 8). Furthermore, whereas tenofovir DF-DP induced the production of a distinct species (just below dGA2, most likely representing dG-tenofovir-labeled dA) (Fig. 5c, lane 8), indicative of its incorporation into the DNA followed by chain termination, clevudine showed no signs of incorporation into the DNA (Fig. 5c, lane 6).
Kinetic analysis revealed that clevudine inhibited HBV DNA chain elongation by HP in a noncompetitive manner.
To determine the mechanism of clevudine-mediated inhibition of HP activity during general DNA elongation (subsequent to protein priming), we performed a kinetic analysis of drug inhibition using the classical EPA, which measures HBV DNA elongation by HP within the nucleocapsids. Since clevudine is a thymidine analog, inhibition of HBV DNA polymerase by clevudine-TP was examined by varying both clevudine-TP and TTP concentrations. The data were fit to both competitive and noncompetitive equations (Fig. 6a and b, respectively). The plots clearly fit better to the noncompetitive equation with an inhibition constant (Ki) of 0.68 μM. The same inhibition experiment was also performed in the presence of a fixed (1 μM) concentration of TTP and various concentrations of dCTP, which should not compete with clevudine-TP. As expected, clevudine-TP did not compete with dCTP, and a similar noncompetitive inhibition profile was observed when the concentration of dCTP was varied and an apparent Ki of 1.15 μM was calculated (see Fig. S4a in the supplemental material). A control experiment was performed with 3TC-TP, and as expected, it competitively inhibited HBV DNA polymerase with respect to dCTP, giving an apparent Ki of 0.0075 μM (see Fig. S4b in the supplemental material), which was comparable to the published data (47, 48).
Fig 6.

Inhibition of the endogenous HBV DNA polymerase by clevudine in a noncompetitive manner. EPA was conducted using HBV nucleocapsids harvested from HepAD38 cells as described in Materials and Methods. Dependence of the rate of radioactive product formation on dTTP concentration was examined in the presence of 0 (○), 0.05 (●), 0.2 (□), and 1 μM (■) clevudine-TP (CLV-TP). The rate of product formation at each concentration was determined by measuring the radioactivity at 6 different time points and conducting a linear regression analysis. All of the rates were globally fit to either a competitive (a) or noncompetitive (b) inhibition equation. The goodness of the fit (R2) for competitive and noncompetitive fits was 0.80 and 0.95, respectively. Two independent experiments were performed, and consistent results with noncompetitive inhibition were observed. A representative result is shown.
DISCUSSION
By taking advantage of our newly developed in vitro HBV protein priming system (7), we have discovered that an NRTI, clevudine, inhibited HBV protein-primed DNA synthesis independent of the nucleotide substrates used (Fig. 7), rather than acting as a competitive inhibitor relative to TTP. Also, clevudine did not serve as a substrate for HP during protein priming to exert its inhibitory effect. Kinetic analysis using the classical EPA also revealed that clevudine inhibited HBV DNA elongation by a noncompetitive mechanism. To the best of our knowledge, clevudine represents the first nucleoside analog that can inhibit DNA synthesis via a noncompetitive mechanism and without being incorporated into DNA.
Fig 7.
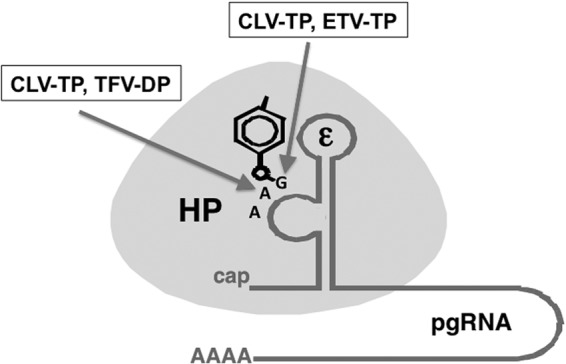
Summary of the inhibitory effects on HBV protein priming by NRTIs. HP is depicted as an oval and the viral RNA template (ε) for protein priming as a stem-loop structure with an internal bulge. As drawn, ε is part of the viral pgRNA with a 5′ cap and 3′ poly(A) tail. The phenol ring from Y63 in the RT protein, used to prime reverse transcription, is highlighted, as are the first three nucleotides of the viral minus-strand DNA that is covalently attached to HP as a result of protein priming. Both clevudine-TP (CLV-TP) and entecavir-TP (ETV-TP) are able to inhibit the initiation stage of protein priming (the covalent attachment of the first nucleotide, dGMP, to HP), and clevudine-TP (CLV-TP) and tenofovir DF-DP (TFV-DP) are able to inhibit the DNA polymerization stage of protein priming (the addition of two dAMPs following dGMP). See the text for details.
Virtually all nucleoside analog polymerase inhibitors are thought to be incorporated into the DNA or RNA product in place of the corresponding natural nucleotide in a competitive manner and subsequently terminate further DNA synthesis due to their lack of the 3′-OH group, or, when the 3′-OH group is present, induce DNA chain termination following the addition of a few more nucleotides (delayed chain termination; described below for entecavir) (23, 49, 50). One possible exception is ribavirin {1-[(2R,3R,4S,5R)-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]-1H-1,2,4-triazole-3-carboxamide}, which is incorporated into RNA but may direct misincorporation and function as a lethal mutagen instead of terminating RNA synthesis (51). Available evidence suggests that clevudine-TP, like other NRTIs, binds to the polymerase active site (or a site very close to it) to exert its effects on HP. First, clevudine-resistant HBV mutants have emerged after long-term therapy and harbor substitutions at the HP dNTP binding site (52). Second, clevudine-resistant HP mutants show cross-resistance to other NRTIs, such as lamivudine and emtricitabine (both deoxycytidine analogs) and telbivudine (a thymidine analog) (23, 52). Third, we have shown here that clevudine, like other NRTIs, required its TP form to inhibit HP protein priming. As clevudine-MP is not incorporated into the DNA products and functions in a noncompetitive manner, we suggest that its binding to the HP active site alters the HP structure such that HP is unable to bind or incorporate any of the natural dNMP substrates; i.e., clevudine-TP acts as a novel, noncompetitive inhibitor of HP similar to a nonnucleoside RT inhibitor (NNRTI) (50). Unlike other NNRTIs, however, clevudine-TP exerts its effect via binding to the HP active site instead of a distinct site, as has been defined for NNRTIs on the human immunodeficiency virus 1 (HIV-1) RT (50, 53). Although no high-resolution structure of HP is available, a previous molecular modeling study indeed suggested that clevudine-TP binding to the HP dNTP binding site induces distortion of the polymerase active site, precluding incorporation of clevudine into DNA (40). As other l-form NRTIs, such as FTC, 3TC, and telbivudine, act as competitive HP inhibitors (23), the l-configuration of clevudine is apparently insufficient for its novel, noncompetitive mechanism of HP inhibition.
Using our newly developed in vitro priming assay, we have verified here that entecavir-TP acted as a competitive (with respect to dGTP) inhibitor of authentic HP protein priming (Fig. 7). Furthermore, we have now demonstrated directly that entecavir could be used by HP to initiate protein priming, in place of dGTP, and following its covalent attachment to HP, entecavir allowed at least one dAMP incorporation during DNA polymerization, confirming that entecavir is not an obligate chain terminator (23, 27, 54). We have also formally demonstrated, for the first time, that the diphosphate form of tenofovir DF can inhibit the second step of the HP priming reaction and prevent formation of a functional dGAA primer (Fig. 7). Importantly, analysis of the products of HP priming reactions with tenofovir DF-DP indicated the presence of a novel product consistent with a tenofovir-terminated primer. While not unexpected, based on the knowledge that tenofovir terminates HIV-1 DNA elongation following incorporation by the HIV-1 RT (55), our data provide the most direct evidence to date that tenofovir is incorporated into newly synthesized HBV DNA.
As we have shown here that clevudine inhibited HBV replication through a fundamentally different mechanism than that of the other nucleoside analogs tested, clevudine may synergize with other nucleoside analogs to inhibit HBV replication. Indeed, when clevudine was combined with entecavir, lamivudine, adefovir, or tenofovir, a synergistic inhibitory effect was observed against HBV replication (56). In contrast, a synergistic effect was not observed when tenofovir was combined with lamivudine, entecavir, adefovir, or telbivudine (57). These results suggest that compounds with properties similar to those of clevudine are good candidates for combination therapy with currently approved NRTIs for HBV therapy. Efforts to develop clevudine in the United States were abandoned due to drug-related myopathy that occurred in a minority of patients (33). However, identification and development of compounds derived from or similar to clevudine, which retain the novel anti-HBV activity but lack the toxicity, may be useful in combination with approved NRTIs. Nucleoside analog-based therapy has been a mainstay in antiviral (HBV, HIV, hepatitis C virus, and herpesviruses) and anticancer chemotherapy (49, 58). Our discovery of the novel mechanism of action of clevudine should inspire further efforts to develop novel compounds to expand the current armaments for treatment of these deadly diseases.
Supplementary Material
ACKNOWLEDGMENTS
We thank Morgan Boyer and Fen Wan for excellent technical assistance and David Toft for purified chaperone proteins. We thank Kathryn Kitrinos for helpful scientific discussions and facilitating the transfer of antiviral compounds between institutions.
This work was supported by a Public Health Service grant (R01 AI074982 to J.H.) from the National Institutes of Health and by funding from Pharmasset (to J.H.). S.J. was supported by the training grant “Viruses and Cancer,” 2 T32 CA60395, from the National Cancer Institute.
E.M. and W.D. are current employees of Gilead Science and hold Gilead stock.
Footnotes
Published ahead of print 17 June 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.00599-13.
REFERENCES
- 1.El-Serag HB. 2012. Epidemiology of viral hepatitis and hepatocellular carcinoma. Gastroenterology 142:1264–1273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Summers J, Mason WS. 1982. Replication of the genome of a hepatitis B-like virus by reverse transcription of an RNA intermediate. Cell 29:403–415 [DOI] [PubMed] [Google Scholar]
- 3.Seeger C, Zoulim F, Mason WS. 2007. Hepadnaviruses, p 2977–3030 In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE. (ed), Fields virology, 5th ed. Lippincott, Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 4.Hu J, Seeger C. 1996. Expression and characterization of hepadnavirus reverse transcriptases. Methods Enzymol. 275:195–208 [DOI] [PubMed] [Google Scholar]
- 5.Lanford RE, Notvall L, Lee H, Beames B. 1997. Transcomplementation of nucleotide priming and reverse transcription between independently expressed TP and RT domains of the hepatitis B virus reverse transcriptase. J. Virol. 71:2996–3004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lanford RE, Notvall L, Beames B. 1995. Nucleotide priming and reverse transcriptase activity of hepatitis B virus polymerase expressed in insect cells. J. Virol. 69:4431–4439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jones SA, Boregowda R, Spratt TE, Hu J. 2012. In vitro epsilon RNA-dependent protein priming activity of human hepatitis B virus polymerase. J. Virol. 86:5134–5150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zoulim F, Seeger C. 1994. Reverse transcription in hepatitis B viruses is primed by a tyrosine residue of the polymerase. J. Virol. 68:6–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang GH, Seeger C. 1992. The reverse transcriptase of hepatitis B virus acts as a protein primer for viral DNA synthesis. Cell 71:663–670 [DOI] [PubMed] [Google Scholar]
- 10.Wang GH, Seeger C. 1993. Novel mechanism for reverse transcription in hepatitis B viruses. J. Virol. 67:6507–6512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hu J, Anselmo D. 2000. In vitro reconstitution of a functional duck hepatitis B virus reverse transcriptase: posttranslational activation by Hsp90. J. Virol. 74:11447–11455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hu J, Flores D, Toft D, Wang X, Nguyen D. 2004. Requirement of heat shock protein 90 for human hepatitis B virus reverse transcriptase function. J. Virol. 78:13122–13131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hu J, Seeger C. 1996. Hsp90 is required for the activity of a hepatitis B virus reverse transcriptase. Proc. Natl. Acad. Sci. U. S. A. 93:1060–1064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hu J, Toft DO, Seeger C. 1997. Hepadnavirus assembly and reverse transcription require a multi-component chaperone complex which is incorporated into nucleocapsids. EMBO J. 16:59–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bartenschlager R, Schaller H. 1992. Hepadnaviral assembly is initiated by polymerase binding to the encapsidation signal in the viral RNA genome. EMBO J. 11:3413–3420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nassal M, Rieger A. 1996. A bulged region of the hepatitis B virus RNA encapsidation signal contains the replication origin for discontinuous first-strand DNA synthesis. J. Virol. 70:2764–2773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tavis JE, Ganem D. 1996. Evidence for activation of the hepatitis B virus polymerase by binding of its RNA template. J. Virol. 70:5741–5750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tavis JE, Massey B, Gong Y. 1998. The duck hepatitis B virus polymerase is activated by its RNA packaging signal, epsilon. J. Virol. 72:5789–5796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang X, Hu J. 2002. Distinct requirement for two stages of protein-primed initiation of reverse transcription in hepadnaviruses. J. Virol. 76:5857–5865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lin L, Wan F, Hu J. 2008. Functional and structural dynamics of hepadnavirus reverse transcriptase during protein-primed initiation of reverse transcription: effects of metal ions. J. Virol. 82:5703–5714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Abraham TM, Loeb DD. 2007. The topology of hepatitis B virus pregenomic RNA promotes its replication. J. Virol. 81:11577–11584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Scaglione SJ, Lok AS. 2012. Effectiveness of hepatitis B treatment in clinical practice. Gastroenterology 142:1360–1368 [DOI] [PubMed] [Google Scholar]
- 23.De Clercq E, Ferir G, Kaptein S, Neyts J. 2010. Antiviral treatment of chronic hepatitis B virus (HBV) infections. Viruses 2:1279–1305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zoulim F, Locarnini S. 2009. Hepatitis B virus resistance to nucleos(t)ide analogues. Gastroenterology 137:1593–1608 [DOI] [PubMed] [Google Scholar]
- 25.Innaimo SF, Seifer M, Bisacchi GS, Standring DN, Zahler R, Colonno RJ. 1997. Identification of BMS-200475 as a potent and selective inhibitor of hepatitis B virus. Antimicrob. Agents Chemother. 41:1444–1448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tenney DJ, Rose RE, Baldick CJ, Pokornowski KA, Eggers BJ, Fang J, Wichroski MJ, Xu D, Yang J, Wilber RB, Colonno RJ. 2009. Long-term monitoring shows hepatitis B virus resistance to entecavir in nucleoside-naive patients is rare through 5 years of therapy. Hepatology 49:1503–1514 [DOI] [PubMed] [Google Scholar]
- 27.Seifer M, Hamatake RK, Colonno RJ, Standring DN. 1998. In vitro inhibition of hepadnavirus polymerases by the triphosphates of BMS-200475 and lobucavir. Antimicrob. Agents Chemother. 42:3200–3208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Marcellin P, Gane E, Buti M, Afdhal N, Sievert W, Jacobson IM, Washington MK, Germanidis G, Flaherty JF, Schall RA, Bornstein JD, Kitrinos KM, Subramanian GM, McHutchison JG, Heathcote EJ. 2013. Regression of cirrhosis during treatment with tenofovir disoproxil fumarate for chronic hepatitis B: a 5-year open-label follow-up study. Lancet 381:468–475 [DOI] [PubMed] [Google Scholar]
- 29.Delaney WE, Ray AS, Yang H, Qi X, Xiong S, Zhu Y, Miller MD. 2006. Intracellular metabolism and in vitro activity of tenofovir against hepatitis B virus. Antimicrob. Agents Chemother. 50:2471–2477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Staschke KA, Colacino JM. 1994. Priming of duck hepatitis B virus reverse transcription in vitro: premature termination of primer DNA induced by the 5′-triphosphate of fialuridine. J. Virol. 68:8265–8269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chu CK, Ma T, Shanmuganathan K, Wang C, Xiang Y, Pai SB, Yao GQ, Sommadossi JP, Cheng YC. 1995. Use of 2′-fluoro-5-methyl-beta-l-arabinofuranosyluracil as a novel antiviral agent for hepatitis B virus and Epstein-Barr virus. Antimicrob. Agents Chemother. 39:979–981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Balakrishna Pai S, Liu SH, Zhu YL, Chu CK, Cheng YC. 1996. Inhibition of hepatitis B virus by a novel L-nucleoside, 2′-fluoro-5-methyl-beta-l-arabinofuranosyl uracil. Antimicrob. Agents Chemother. 40:380–386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jang JH, Kim JW, Jeong SH, Myung HJ, Kim HS, Park YS, Lee SH, Hwang JH, Kim N, Lee DH. 2011. Clevudine for chronic hepatitis B: antiviral response, predictors of response, and development of myopathy. J. Viral Hepat. 18:84–90 [DOI] [PubMed] [Google Scholar]
- 34.Korba BE, Furman PA, Otto MJ. 2006. Clevudine: a potent inhibitor of hepatitis B virus in vitro and in vivo. Expert Rev. Anti Infect. Ther. 4:549–561 [DOI] [PubMed] [Google Scholar]
- 35.Lee HS, Chung YH, Lee K, Byun KS, Paik SW, Han JY, Yoo K, Yoo HW, Lee JH, Yoo BC. 2006. A 12-week clevudine therapy showed potent and durable antiviral activity in HBeAg-positive chronic hepatitis B. Hepatology 43:982–988 [DOI] [PubMed] [Google Scholar]
- 36.Peek SF, Cote PJ, Jacob JR, Toshkov IA, Hornbuckle WE, Baldwin BH, Wells FV, Chu CK, Gerin JL, Tennant BC, Korba BE. 2001. Antiviral activity of clevudine [L-FMAU, (1-(2-fluoro-5-methyl-beta, L-arabinofuranosyl) uracil)] against woodchuck hepatitis virus replication and gene expression in chronically infected woodchucks (Marmota monax). Hepatology 33:254–266 [DOI] [PubMed] [Google Scholar]
- 37.Kaplan PM, Greenman RL, Gerin JL, Purcell RH, Robinson WS. 1973. DNA polymerase associated with human hepatitis B antigen. J. Virol. 12:995–1005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yao GQ, Liu SH, Chou E, Kukhanova M, Chu CK, Cheng YC. 1996. Inhibition of Epstein-Barr virus replication by a novel L-nucleoside, 2′-fluoro-5-methyl-beta-l-arabinofuranosyluracil. Biochem. Pharmacol. 51:941–947 [DOI] [PubMed] [Google Scholar]
- 39.Lewis W, Meyer RR, Simpson JF, Colacino JM, Perrino FW. 1994. Mammalian DNA polymerases alpha, beta, gamma, delta, and epsilon incorporate fialuridine (FIAU) monophosphate into DNA and are inhibited competitively by FIAU triphosphate. Biochemistry 33:14620–14624 [DOI] [PubMed] [Google Scholar]
- 40.Chong Y, Chu CK. 2002. Understanding the unique mechanism of L-FMAU (clevudine) against hepatitis B virus: molecular dynamics studies. Bioorg. Med. Chem. Lett. 12:3459–3462 [DOI] [PubMed] [Google Scholar]
- 41.Jones SA, Hu J. 2013. Protein-primed terminal transferase activity of hepatitis B virus polymerase. J. Virol. 87:2563–2576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lin L, Hu J. 2008. Inhibition of hepadnavirus reverse transcriptase-epsilon RNA interaction by porphyrin compounds. J. Virol. 82:2305–2312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hu J, Boyer M. 2006. Hepatitis B virus reverse transcriptase and epsilon RNA sequences required for specific interaction in vitro. J. Virol. 80:2141–2150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ladner SK, Otto MJ, Barker CS, Zaifert K, Wang GH, Guo JT, Seeger C, King RW. 1997. Inducible expression of human hepatitis B virus (HBV) in stably transfected hepatoblastoma cells: a novel system for screening potential inhibitors of HBV replication. Antimicrob. Agents Chemother. 41:1715–1720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nguyen DH, Gummuluru S, Hu J. 2007. Deamination-independent inhibition of hepatitis B virus reverse transcription by APOBEC3G. J. Virol. 81:4465–4472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cortes Ledesma F, El Khamisy SF, Zuma MC, Osborn K, Caldecott KW. 2009. A human 5′-tyrosyl DNA phosphodiesterase that repairs topoisomerase-mediated DNA damage. Nature 461:674–678 [DOI] [PubMed] [Google Scholar]
- 47.Levine S, Hernandez D, Yamanaka G, Zhang S, Rose R, Weinheimer S, Colonno RJ. 2002. Efficacies of entecavir against lamivudine-resistant hepatitis B virus replication and recombinant polymerases in vitro. Antimicrob. Agents Chemother. 46:2525–2532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Langley DR, Walsh AW, Baldick CJ, Eggers BJ, Rose RE, Levine SM, Kapur AJ, Colonno RJ, Tenney DJ. 2007. Inhibition of hepatitis B virus polymerase by entecavir. J. Virol. 81:3992–4001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.De Clercq E. 2010. In search of a selective therapy of viral infections. Antiviral Res. 85:19–24 [DOI] [PubMed] [Google Scholar]
- 50.Sarafianos SG, Marchand B, Das K, Himmel DM, Parniak MA, Hughes SH, Arnold E. 2009. Structure and function of HIV-1 reverse transcriptase: molecular mechanisms of polymerization and inhibition. J. Mol. Biol. 385:693–713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Crotty S, Cameron C, Andino R. 2002. Ribavirin's antiviral mechanism of action: lethal mutagenesis? J. Mol. Med. (Berlin) 80:86–95 [DOI] [PubMed] [Google Scholar]
- 52.Kwon SY, Park YK, Ahn SH, Cho ES, Choe WH, Lee CH, Kim BK, Ko SY, Choi HS, Park ES, Shin GC, Kim KH. 2010. Identification and characterization of clevudine-resistant mutants of hepatitis B virus isolated from chronic hepatitis B patients. J. Virol. 84:4494–4503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Das K, Martinez SE, Bauman JD, Arnold E. 2012. HIV-1 reverse transcriptase complex with DNA and nevirapine reveals non-nucleoside inhibition mechanism. Nat. Struct. Mol. Biol. 19:253–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tchesnokov EP, Obikhod A, Schinazi RF, Gotte M. 2008. Delayed chain termination protects the anti-hepatitis B virus drug entecavir from excision by HIV-1 reverse transcriptase. J. Biol. Chem. 283:34218–34228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tuske S, Sarafianos SG, Clark AD, Jr, Ding J, Naeger LK, White KL, Miller MD, Gibbs CS, Boyer PL, Clark P, Wang G, Gaffney BL, Jones RA, Jerina DM, Hughes SH, Arnold E. 2004. Structures of HIV-1 RT-DNA complexes before and after incorporation of the anti-AIDS drug tenofovir. Nat. Struct. Mol. Biol. 11:469–474 [DOI] [PubMed] [Google Scholar]
- 56.Niu C, Bao H, Tolstykh T, Micolochick Steuer HM, Murakami E, Korba B, Furman PA. 2010. Evaluation of the in vitro anti-HBV activity of clevudine in combination with other nucleoside/nucleotide inhibitors. Antivir. Ther. 15:401–412 [DOI] [PubMed] [Google Scholar]
- 57.Zhu Y, Curtis M, Qi X, Miller MD, Borroto-Esoda K. 2009. Anti-hepatitis B virus activity in vitro of combinations of tenofovir with nucleoside/nucleotide analogues. Antivir. Chem. Chemother. 19:165–176 [DOI] [PubMed] [Google Scholar]
- 58.Galmarini CM, Mackey JR, Dumontet C. 2002. Nucleoside analogues and nucleobases in cancer treatment. Lancet Oncol. 3:415–424 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.