

Abstract
BMS-626529 is a novel small-molecule HIV-1 attachment inhibitor active against both CCR5- and CXCR4-tropic viruses. BMS-626529 functions by preventing gp120 from binding to CD4. A prodrug of this compound, BMS-663068, is currently in clinical development. As a theoretical resistance pathway to BMS-663068 could be the development of a CD4-independent phenotype, we examined the activity of BMS-626529 against CD4-independent viruses and investigated whether resistance to BMS-626529 could be associated with a CD4-independent phenotype. Finally, we evaluated whether cross-resistance exists between BMS-626529 and other HIV-1 entry inhibitors. Two laboratory-derived envelopes with a CD4-independent phenotype (one CXCR4 tropic and one CCR5 tropic), five envelopes from clinical isolates with preexisting BMS-626529 resistance, and several site-specific mutant BMS-626529-resistant envelopes were examined for their dependence on CD4 for infectivity or susceptibility to BMS-626529. Viruses resistant to other entry inhibitors (enfuvirtide, maraviroc, and ibalizumab) were also examined for susceptibility to BMS-626529. Both CD4-independent laboratory isolates retained sensitivity to BMS-626529 in CD4− cells, while HIV-1 envelopes from viruses resistant to BMS-626529 exhibited no evidence of a CD4-independent phenotype. BMS-626529 also exhibited inhibitory activity against ibalizumab- and enfuvirtide-resistant envelopes. While there appeared to be some association between maraviroc resistance and reduced susceptibility to BMS-626529, an absolute correlation cannot be presumed, since some CCR5-tropic maraviroc-resistant envelopes remained sensitive to BMS-626529. Clinical use of the prodrug BMS-663068 is unlikely to promote resistance via generation of CD4-independent virus. No cross-resistance between BMS-626529 and other HIV entry inhibitors was observed, which could allow for sequential or concurrent use with different classes of entry inhibitors.
INTRODUCTION
A continuing need exists for development of novel antiretroviral drugs and regimens in order to address the tolerability and long-term safety concerns associated with current treatment options, the immune dysfunction induced by HIV infection, and the emergence of drug resistance (1, 2). Entry of HIV into host cells is now well characterized as a multistep process beginning with the attachment of gp120, the surface subunit of the viral envelope, to the CD4 receptor on the cell surface. CD4 binding triggers exposure of structural elements within gp120 that bind to one of two coreceptors (either C-C chemokine receptor 5 [CCR5] or C-X-C chemokine receptor type 4 [CXCR4]), allowing insertion of the transmembrane subunit gp41 into the target cell membrane. This in turn results in fusion of the cell and virus membranes (3, 4).
A number of agents have been developed to target the inhibition of the entry process. These include maraviroc (MVC), which targets the interaction of gp120 with the CCR5 coreceptor (5), and enfuvirtide (ENF), an injectable peptide that prevents gp41-mediated fusion of the viral and host cell membranes (6). Additionally, ibalizumab, a CD4 binding monoclonal antibody that blocks CD4-dependent virus entry, is currently in clinical development (7, 8).
HIV-1 attachment inhibitors (AIs) represent a novel class of entry inhibitors that bind to gp120 and selectively inhibit the successful interaction between the virus and CD4, thereby preventing viral entry into host cells (9). Proof of concept for the AI class was achieved in an 8-day monotherapy trial of the progenitor AI BMS-488043 (10). Subsequently, efforts to increase the inhibitory potency of the AI class against specific HIV-1 isolates resulted in the discovery of BMS-626529 (11). The generally low solubility and poor intrinsic dissolution properties of this compound were addressed through development of a phosphonooxymethyl prodrug, BMS-663068, which has demonstrated clinical antiviral activity in a proof-of-concept study (12). In a monotherapy study of HIV-1 subtype B-infected subjects, correlates of nonresponse mapped to amino acid changes in gp120, previously demonstrated to confer in vitro resistance to BMS-626529 (13, 14). In that study, the envelope substitution M426L was found to be strongly, although not exclusively, associated with low susceptibility to BMS-626529 (13). The overall prevalence of the M426L substitution in HIV-1-infected individuals differs according to subtype; in subjects with subtype B infection, the prevalence is 7.3% (15, 16). Other envelope amino acid changes that were shown to encode reduced susceptibility to BMS-626529 in this cohort included S375M/T, M434I, and M475I (14). In addition, for the CRF01_AE viruses, the S375H and M475I changes were found to contribute to resistance to BMS-626529 for all viruses in this subtype (14, 17).
While most HIV-1 viruses are dependent on the CD4 receptor for entry into cells, viruses that can infect CD4-negative cells have been derived by virus passage on CD4-negative, coreceptor-positive cells in tissue culture (18). Entry of such viruses into host cells is mediated by increased exposure of the coreceptor binding site through changes in the site itself or in the protein loops that in CD4-dependent viruses mask this region until bound to CD4 (18). As the putative mode of action of BMS-626529 is blocking of the gp120-CD4 interaction (although differing modes of action have been proposed for the earlier AIs BMS-378806 and BMS-488043) (19, 20), it is possible that the AI may not inhibit CD4-independent virus entry. Furthermore, it is theoretically possible that resistance to AIs may occur through selection of CD4-independent virus; however, such viruses have rarely been isolated in vivo, which may in part be related to their increased sensitivity to antibody neutralization compared to CD4-dependent viruses (18, 21–24). Studies of the activity of an earlier AI, BMS-378806, against CD4-independent virus produced conflicting results. One group reported that the drug was unable to inhibit CD4-independent virus (25), while a second report presented evidence of activity (20). The later AI, BMS-488043, has been reported to retain activity against CD4-independent virus (25). The current analyses examined the potential for BMS-626529 to inhibit CD4-independent virus and whether BMS-626529-resistant viruses can have a CD4-independent phenotype. Additionally, the susceptibility to BMS-626529 of viruses selected for resistance to the HIV-1 entry inhibitors ibalizumab, MVC, and ENF was determined.
(This work was presented in part at the XIX International AIDS Conference, Washington, DC, 22 to 27 July 2012 [26].)
MATERIALS AND METHODS
Compounds.
BMS-626529 was synthesized at Bristol-Myers Squibb, and MVC was synthesized by using reported or known reactions (27). The gp41 inhibitor ENF (catalog number 101422-654; VWR) and the CXCR4 antagonist AMD3100 (28) (catalog number A5602-5MG; Sigma Corp.) were purchased from commercial sources. Ibalizumab, an anti-CD4 monoclonal antibody with anti-HIV activity (7, 8), was cloned, expressed, and purified in-house (29).
Viruses and cells.
Infection assays were performed by using ACTOne cell lines (modified HEK293T cell lines; Codex BioSolutions, Inc.). ACTOne cells expressing either CCR5 or CXCR4 without CD4 (ACTOne-CCR5 and ACTOne-CXCR4, respectively) were used. For comparative assays, HeLa cells expressing CD4/CCR5/CXCR4 proteins or a HeLa cell line with a CD4−/CCR5−/CXCR4− phenotype were used.
HIV-1 ADA and LAI viruses were obtained through the NIH AIDS Research and Reference Reagent Program. A CD4-independent CCR5-tropic virus was constructed by introducing the S190R and N197S substitutions into the HIV ADA envelope and then subcloning the mutant envelope into an NL4-3 proviral vector to generate replication-competent virus containing the luciferase gene (30, 31). CD4-independent entry of the newly constructed virus was confirmed by using a luciferase reporting system following infection of both ACTOne-CCR5 and ACTOne-CD4/CCR5 cells (see Fig. S1 in the supplemental material).
Five clinical envelope clones were gifts from Robert Doms (University of Pennsylvania). The S1 and S2 clones (pretreatment and sensitive to MVC) and clones R3 and R4 (day 224 of treatment and resistant to MVC) were all isolated from a subject with virologic failure on MVC treatment (32). The envelope clone Post 5.1 was isolated at week 12 of treatment from a different subject, who was treated with the CCR5 antagonist aplaviroc. This envelope was resistant to both aplaviroc and MVC (33). CXCR4-tropic, CD4-independent HIV-1-8x (HIV-8x) envelope was also a gift from Robert Doms (32–34). The HIV-8x envelope sequence was altered by using site-directed mutagenesis (SDM).
BMS-626529-resistant viruses were obtained from clinical samples obtained from subjects enrolled in the proof-of-concept monotherapy study (12). Further BMS-626529-resistant viruses were constructed via introduction of the M426L substitution into various envelopes or introduction of other substitutions determined in vitro to reduce susceptibility to BMS-626529 into the NL4-3 proviral vector containing the luciferase gene. The proviral vector, in which the env gene was deleted and a section of the nef gene from NL4-3 was replaced with the Renilla luciferase gene, was constructed at Bristol-Myers Squibb.
ENF-resistant viruses were constructed through the introduction of five different substitutions, known to induce ENF resistance, into an NL4-3 envelope via SDM and used to make pseudotype viruses. Pseudotype viruses were prepared as previously described (35). Ibalizumab-resistant NL4-3 virus was selected under increasing concentrations of ibalizumab. Briefly, MT-2 cells were infected with NL4-3 virus in the presence of a 2× half-maximal effective concentration (EC50) (2 nM) of ibalizumab and incubated at 37°C. Cells were monitored daily until virus breakthrough was observed by the appearance of syncytia (usually 3 to 7 days). Supernatants were removed and used to infect fresh MT-2 cells with an increased concentration (twice the previous concentration) of ibalizumab, and the process was repeated. Significant virus growth was observed at ∼4,000× the EC50 of ibalizumab (4 μM) at passage 12, and the virus was passaged twice more at 4 μM before harvesting. Virus from passage 14 was shown to be resistant to ibalizumab in antiviral assays, and the env gene was sequenced to identify substitutions that may encode resistance. In addition, a clinical envelope with partial resistance to ibalizumab was selected from cloned envelopes that were obtained through Bristol-Myers Squibb clinical trials (12). Previous studies have suggested that resistance can map to the loss of N-linked glycosylation sites in the V5 region of gp120 (36, 37). An envelope with one potential N-linked glycosylation site in V5 was identified and shown to be partially resistant to ibalizumab through its lack of complete inhibition at high ibalizumab concentrations (low maximum percent inhibition [MPI]).
Drug susceptibility assays.
All susceptibility assays were performed in duplicate or triplicate within each experiment, and each experiment was repeated at least twice. Standard deviations were calculated and are presented.
Cell-cell fusion.
A cell-cell fusion assay was used to evaluate the sensitivity of HIV-1 envelopes to BMS-626529 (14). Briefly, env clones were amplified by reverse transcriptase PCR (RT-PCR) and verified by sequencing, and the PCR products were cloned into the pCMV-HA expression vector (Clontech) by using In-Fusion HD EcoDry technology (Clontech) and transfected into cells to create effector cells. Cells were transfected with equal amounts of envelope-expressing plasmid DNA (50 ng/well in 6-well plates). Effector cells were incubated with target cells, expressing mixtures of CD4, CCR5, or CXCR4, in the presence of HIV-1 entry inhibitors, and the extent of cell fusion was monitored after 18 h of incubation by using the Steady-Glo luciferase assay (Promega, Madison, WI).
For the experiments with the HIV-8x envelope, the gp160 genes of LAI and HIV-8x were cloned into ApaI and NotI sites of the pCMV-HA expression vector (Clontech, Mountain View, CA) by using an In-Fusion HD EcoDry kit (Clontech). The mutant envelopes were generated through SDM on the cloned LAI envelope by using a QuikChange II XL site-directed mutagenesis kit (Stratagene, La Jolla, CA). Effector cells were then prepared by cotransfecting HeLa cells with the envelope plasmid DNAs along with a pTet-off plasmid (Clontech). ACTOne cells expressing CXCR4 were transfected with pTRE2Hyg-Luc plasmids (Clontech) and used as target cells for CD4-independent cell fusion. A HeLa cell expressing CD4/CCR5/CXCR4 and containing an integrated copy of pTRE2Hyg-Luc was used as a target cell for CD4-dependent cell fusion control. The effector and target cells were trypsinized, mixed at a ratio of 1:2, and seeded into a 96-well plate. After 18 h of incubation, the extent of luciferase expression was measured by using the Steady-Glo luciferase assay (Promega).
Virus assays.
Pseudotype viruses containing a luciferase gene were used to infect cells in the presence of HIV-1 entry inhibitors, as previously described (35). Viral inputs for the pseudotype assay were normalized by luciferase activity (light), targeting 10,000 relative light units (RLU) per infection. For ACTOne cells expressing either CCR5 or CXCR4, 3,000 RLU were targeted per infection. Following infection of cell lines expressing CD4/CCR5/CXCR4, drug susceptibility was measured at 3 days postinfection by comparison of luciferase activities in the presence and absence of drug. In addition, the PhenoSense Entry assay (38) was used to determine the drug susceptibilities of certain clinical envelopes, as previously described, and data are presented as 50% inhibitory concentrations (IC50s), in line with Monogram reporting (11). In some experiments, fully replicating viruses containing different envelopes (LAI and ADA) were used. These env genes were inserted into a replication-competent NL4-3 proviral vector in which a section of the nef gene was replaced with the Renilla luciferase gene.
RESULTS
BMS-626529 retains activity against CD4-independent virus.
The effects of BMS-626529 and other entry inhibitors on CD4-dependent and -independent entry were evaluated in a CCR5- and CXCR4-tropic context. It was previously shown that glycosylation site substitutions in the CCR5-tropic ADA envelope enable virus to infect cells in a CD4-independent fashion (30, 31) and that earlier AIs were able to inhibit this virus infection in a CD4-independent fashion, although there were also conflicting results (20, 25). Thus, ADA wild-type (CD4- and CCR5-dependent) and ADA S190R/N197S-containing (CD4-independent and CCR5-dependent) viruses (30, 31) were used to infect both ACTOne-CCR5 and HeLa CD4/CCR5/CXCR4 cells. Entry inhibition by BMS-626529 was compared with those by ibalizumab, MVC, and ENF. The results are shown in Table 1. As expected, wild-type ADA virus was not infectious in the ACTOne-CCR5 cells due to the lack of CD4 expression in these cells. The double mutant ADA virus was infectious in both cell lines, and MVC and ENF were able to inhibit the ADA S190R/N197S virus in both cases. The EC50s differed by 3.4- to 7.5-fold between the cell lines, presumably due to cell line differences, but it is clear that both MVC and ENF are active against the virus under both conditions. On the other hand, ibalizumab inhibited entry of ADA S190/N197S virus into CD4-expressing HeLa CD4/CCR5/CXCR4 cells only and could not inhibit infection in a CD4-independent fashion, as expected. The AI BMS-626529 was able to inhibit infection of the ADA S190/N197S virus in both cell lines, demonstrating that BMS-626529 inhibits entry of both a CD4-dependent and CD4-independent CCR5-tropic virus.
Table 1.
Effects of HIV-1 entry inhibitors on CD4-dependent and CD4-independent, CCR5-dependent entrya
| HIV-1 entry inhibitor | Target | Inhibition | Mean EC50 (nM) ± SDb |
|||
|---|---|---|---|---|---|---|
| HeLa CD4/CCR5/CXCR4 |
ACTOne-CCR5 |
|||||
| ADA | ADA S190R/N197S | ADA | ADA S190R/N197S | |||
| BMS-626529 | gp120 | gp120-CD4 interaction | 0.4 ± 0.3 | 0.8 ± 0.1 | No infection | 0.2 ± 0.1 |
| Ibalizumab | CD4 | gp120-coreceptor interaction | 16.6 ± 5.9 | 16.0 ± 1.5 | No infection | >4,950 |
| MVC | CCR5 | gp120-CCR5 interaction | 1.9 ± 0.1 | 0.7 ± 0.7 | No infection | 0.2 ± 0.2 |
| ENF | gp41 | gp41 hairpin formation | 6.5 ± 2.0 | 21.9 ± 7.7 | No infection | 24.1 ± 26.4 |
ADA, CD4 dependent, CCR5 dependent; ADA S190R/N197S, CD4 independent, CCR5 dependent; ENF, enfuvirtide; MVC, maraviroc.
Mean values of three independent experiments.
To exclude the possibility that BMS-626529 inhibition of CD4-independent viruses is specific to CCR5-tropic viruses, a similar experiment was conducted by using LAI wild-type (CD4- and CXCR4-dependent) and HIV-8x (CD4-independent and CXCR4-dependent) viruses (34). A functional HXBc2 clone was not available, so the LAI strain was selected based on its high degree of relatedness to HXBc2 (39), from which HIV-8x was derived (21). These viruses were used to infect ACTOne-CXCR4 and HeLa CD4/CCR5/CXCR4 cells in the presence of BMS-626529, the CXCR4 antagonist AMD3100, or ENF. The results are shown in Table 2. BMS-626529 inhibited CD4-dependent entry of wild-type virus and CD4-independent CXCR4-dependent entry of HIV-8x envelope. Thus, BMS-626529 is also able to inhibit infection of a CXCR4-tropic virus in a CD4-independent manner. However, the HIV-8x envelope exhibited decreased susceptibility to entry inhibition by BMS-626529 compared with the related LAI virus (Table 2). HIV-8x envelope has a deletion of nucleotide (nt) 2118, which results in a frameshift and consequent loss of the cytoplasmic domain. Decreased susceptibility of envelopes without a cytoplasmic domain was previously reported for an earlier AI (9). Table 3 shows that restoration of the cytoplasmic domain by SDM resulted in increased susceptibility to BMS-626529 and loss of CD4-independent entry. Furthermore, introduction of the G431E substitution into HIV-8x, which also abrogates CD4-independent entry (40), did not alter susceptibility to BMS-626529. This suggests that the reduced susceptibility of HIV-8x to BMS-626529 is not due to its CD4-independent phenotype.
Table 2.
Effects of HIV-1 entry inhibitors on CD4-dependent and CD4-independent, CXCR4-dependent entrya
| HIV-1 entry inhibitor | Target | Inhibition | Mean EC50 (nM) ± SDb |
|||
|---|---|---|---|---|---|---|
| HeLa CD4/CCR5/CXCR4 |
ACTOne-CXCR4 |
|||||
| LAI | HIV-8x | LAI | HIV-8x | |||
| BMS-626529 | gp120 | gp120-CD4 interaction | 0.9 ± 0.4 | 97 ± 14 | No infection | 17 ± 6 |
| Ibalizumab | CD4 | gp120-coreceptor interaction | 1.6 ± 0.2 | >4,990 | No infection | >4,950 |
| AMD-3100 | CXCR4 | gp120-CXCR4 interaction | 16 ± 2.0 | 79 ± 18 | No infection | 35 ± 18 |
| ENF | gp41 | gp41 hairpin formation | 9.7 ± 2.2 | 650 ± 123 | No infection | 125 ± 44.5 |
LAI, CD4 dependent, CXCR4 dependent; HIV-8x, CD4 independent, CXCR4 dependent.
Mean values of three independent experiments.
Table 3.
Effects of restoration of the cytoplasmic tail and G431E substitution on the susceptibility of HIV-8x to BMS-626529
| HIV-1 entry inhibitor | Mean EC50 (nM) ± SDa |
|||||
|---|---|---|---|---|---|---|
| HeLa CD4/CCR5/CXCR4 |
ACTOne-CXCR4 |
|||||
| HIV-8x wild type | HIV-8x with tail | HIV-8x G431E | HIV-8x wild type | HIV-8x with tail | HIV-8x G431E | |
| BMS-626529 | 97 ± 14 | 12 ± 0.6 | 131 ± 33 | 106 ± 25 | No infection | No infection |
| AMD-3100 | 79 ± 18 | 15 ± 11 | 12 ± 6 | 30 ± 15 | No infection | No infection |
Mean values of two independent experiments.
To provide further confirmation that BMS-626529 inhibits CD4-independent entry, the BMS-626529 resistance substitution M426L (13, 14, 41) was introduced into ADA wild-type and ADA S190R/N197S envelopes. In both cases, the potency of BMS-626529 against L426 virus was greatly reduced compared with the potency against M426 virus, confirming that BMS-626529 acts directly on gp120 and inhibits both CD4-dependent and CD4-independent entry (Table 4).
Table 4.
Confirmation that BMS-626529 inhibits ADA S190R/N197S virus infection of ACTOne-CCR5 cells by a mechanism consistent with an AI
| HIV-1 entry inhibitor | Mean EC50 (nM) ± SDa |
|||||||
|---|---|---|---|---|---|---|---|---|
| HeLa CD4/CCR5/CXCR4 |
ACTOne-CCR5 |
|||||||
| ADA |
ADA S190R/N197S |
ADA |
ADA S190R/N197S |
|||||
| M426 | L426 | M426 | L426 | M426 | L426 | M426 | L426 | |
| BMS-626529 | 0.2 ± 0.0 | 36 ± 3 | 0.9 ± 0.2 | 78 ± 5 | No infection | No infection | 1 ± 0.4 | 57 ± 10 |
| MVC | 0.8 ± 0.5 | 0.5 ± 0.0 | 0.9 ± 0.2 | 0.7 ± 0.1 | No infection | No infection | 1 ± 0.6 | 0.4 ± 0.1 |
Mean values of two independent experiments.
BMS-626259-resistant viruses retain CD4-dependent entry.
A potential mechanism for resistance to BMS-626529 could be the selection of virus that no longer requires CD4 interactions for infection. Thus, envelopes with site-directed substitutions conferring resistance to BMS-626529 were examined for their ability to grow in cells devoid of CD4. Functional LAI envelopes containing BMS-626529 resistance substitutions selected by in vitro passage with increasing concentrations of the drug (for L116P, EC50 of >10,000 nM; for M426L, EC50 of 71 nM; for M475I, EC50 of 15 nM; for M426L/M475I, EC50 of >10,000 nM [determined by a virus infectivity assay {14}]) were transfected into HeLa CD4−/CCR5−/CXCR4− cells and investigated in a cell fusion assay with both HeLa CD4/CCR5/CXCR4 and ACTOne-CXCR4 target cells. The HIV-8x envelope was used as a control and exhibited fusion activity in ACTOne-CXCR4 cells, albeit less than that observed with HeLa CD4/CCR5/CXCR4 cells. However, the efficiency of fusion of cells containing BMS-626529-resistant clones with ACTOne-CXCR4 cells was at or below background levels, indicating that these clones retained a CD4-dependent phenotype (Fig. 1).
Fig 1.
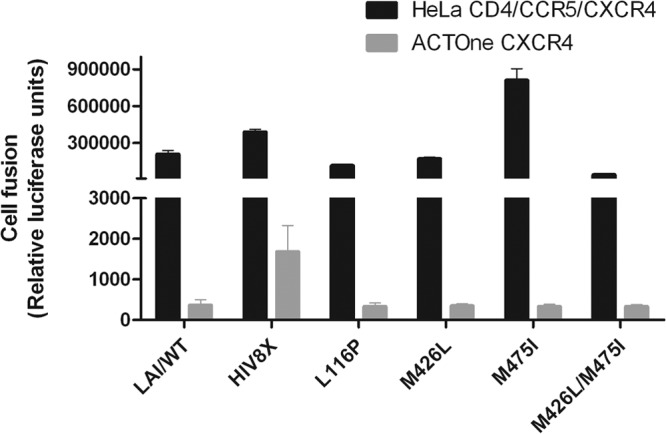
CD4-dependent and -independent fusion entry of wild-type and mutant LAI envelopes. Emission of light based on luciferase levels following fusion of wild-type LAI (LAI/WT) with ACTOne-CXCR4 cells was ∼200 RLU (negative controls using mock-transfected cells did not produce a measurable signal) and was equivalent to that following fusion of L116P virus with ACTOne-CXCR4 cells. The bars represent the mean values of two independent experiments. Standard deviations are shown as error bars.
It has been observed that susceptibility to BMS-626529 is variable among virus strains, with a fraction of naive subtype B envelopes exhibiting little to no sensitivity to the compound (11, 12). Thus, envelopes from seven subjects in the proof-of-concept study for BMS-663068 were found to have high IC50s (>100 nM) of BMS-626529, and the majority of these subjects did not respond to the compound during the monotherapy phase of the study (12). In order to test whether these envelopes could have a CD4-independent phenotype, cloned envelopes from baseline samples from five of these subjects were incorporated into pseudoviruses and examined for their ability to infect CD4− cells. The BMS-626529 IC50s of the viruses obtained from these samples, as determined by the PhenoSense Entry assay, were 310 nM (one subject), 3,043 nM (one subject), and >20,000 nM (three subjects). Virus from all subjects was confirmed to be CCR5 tropic by using the Trofile assay (Monogram Biosciences). None of the pseudotype viruses were able to efficiently infect ACTOne-CCR5 cells (Fig. 2), confirming that these envelopes with intrinsically high IC50s of BMS-626529 remain CD4 dependent. One pseudotype virus containing a gp160 protein from subject 5 (clone 5-14; Fig. 2) did produce slightly higher luciferase units in ACTOne-CCR5 cells than the other four viruses. However, the addition of MVC to these virus-infected ACTOne-CCR5 cells had no effect on luciferase levels, indicating that the extremely low level of fluorescence observed is due to nonspecific background (not shown).
Fig 2.
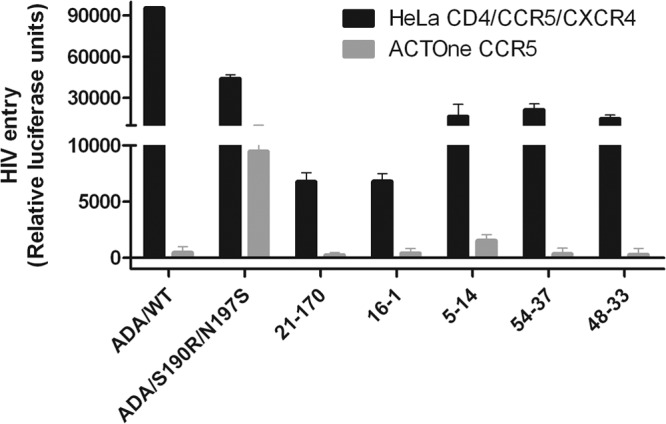
CD4-dependent and -independent entry of pseudotype viruses with envelopes with decreased susceptibility to BMS-626529. The addition of MVC to 5-14 pseudotype virus-infected ACTOne-CCR5 cells had no effect on luciferase levels, indicating that the low level of fluorescence observed is due to nonspecific background. The bars represent the mean values of three independent experiments. Standard deviations are shown as error bars. WT, wild type.
Lack of cross-resistance between BMS-626529 and other HIV-1 entry inhibitors.
BMS-626529, MVC, ENF, and ibalizumab all target the entry process, and resistance to each of these compounds maps to amino acids within gp120 or gp41 (14, 42–45). Thus, it is possible that specific amino acids selected by one agent can encode cross-resistance to BMS-626529. As a means toward probing this question, four functional envelope clones with known BMS-626529 resistance substitutions were examined in a cell-cell fusion assay for susceptibility to MVC, ENF, and ibalizumab. The envelope clones were derived from baseline samples from subjects in the proof-of-concept study (12) and contain either the M426L, M426L/M475I, or S375M/M434I substitutions. Two envelope clones came from a dual-mixed subject and possessed only the M426L substitution, but one envelope clone was CCR5 tropic, while the other was CXCR4 tropic (Table 5). When examined against ENF and ibalizumab, the four envelope clones exhibited high susceptibility within a tight range. Against MVC, 3/4 envelope clones exhibited high susceptibility within a tight range, while the CXCR4-tropic envelope clone, as expected, was not inhibited by MVC. Thus, functional envelope clones from BMS-626529-resistant clinical samples retain susceptibility to other HIV-1 entry inhibitors.
Table 5.
Susceptibilities of BMS-626529-resistant clinical samples to other entry inhibitors
| Subject (clone) | Tropism | BMS-626529 resistance substitution(s) | Mean fold change in EC50 ± SDa |
Mean EC50 (nM) ± SD of MVCa | ||
|---|---|---|---|---|---|---|
| BMS-626529 | ENF | Ibalizumab | ||||
| 16 (1) | CCR5 | M426L/M475I | 2,932 ± 404 | 4.2 ± 2.3 | 0.6 ± 0.1 | 7.4 ± 1.3 |
| 21 (170) | CCR5 | M426L | 386 ± 95 | 13.4 ± 2.8 | 1.6 ± 0.9 | 9.2 ± 1.8 |
| 21 (169) | CXCR4 | M426L | 215 ± 76 | 1.9 ± 0.6 | 0.5 ± 0.1 | >5,000 |
| 41 (33) | CCR5 | S375M/M434I | >19,418 | 1.5 ± 0.3 | 0.7 ± 0.1 | 4.0 ± 0.1 |
Mean values of two independent experiments.
Viruses resistant to other HIV-1 entry inhibitors retain susceptibility to BMS-626529.
As a corollary to the above-mentioned study, susceptibility of BMS-626529 to viruses resistant to other entry inhibitors was examined. To that end, an NL4-3 virus resistant to the anti-CD4 monoclonal antibody ibalizumab was selected through sequential passage with increasing concentrations of antibody. Sequencing of the ibalizumab-resistant virus identified S162N and A605T/A substitutions in the envelope. Although A162N is a substitution that has not yet been reported, it destroys a potential glycosylation site outside the V5 region (after V1/V2 but before V3). This virus is highly resistant to ibalizumab, with an EC50 >748-fold higher than that of wild-type NL4-3 (Table 6). Consequently, the MPI for this virus never reached 50% at the highest concentration tested (995 nM). Additionally, a clinical envelope with reduced susceptibility to ibalizumab was identified by analyzing sequences around the V5 region for clones missing potential N-linked glycosylation sites (37). One envelope, termed ibal-R, was identified with one potential N-linked glycosylation site in this region. The EC50 of this clone in the cell-cell fusion assay (ibal-R) is only ∼4-fold higher than that of a sensitive clinical envelope (ibal-S), which contains two N-linked glycosylation sites in the V5 region (2.7 versus 0.63 nM, respectively). However, ibal-R exhibits a lower MPI of ∼82%, which is a characteristic of ibalizumab-resistant envelopes (36, 37). An M426L site-directed mutant of the ibal-R clone was also made, and these three envelopes were analyzed for susceptibility to BMS-626529 in a cell-cell fusion assay (Table 6). Both the ibal-S and ibal-R envelopes were sensitive to BMS-626529 and exhibited ∼100% MPI. As expected, the ibal-RM426L envelope exhibited decreased susceptibility to BMS-626529 (∼100-fold) but still showed 100% MPI, while it retained resistance to ibalizumab through a lower MPI. Thus, ibalizumab-resistant viruses retained susceptibility to BMS-626529, suggesting a lack of cross-resistance between these agents.
Table 6.
Susceptibility of ibalizumab-resistant virus to BMS-626529a
| HIV-1 entry inhibitor | Mean EC50 (nM) ± SD for NL4-3 |
Clinical envelope |
||||||
|---|---|---|---|---|---|---|---|---|
| Wild type | NL4-3(S162N,A605T/A) | Mean EC50 (nM) ± SD |
Mean MPI ± SD |
|||||
| Ibal-S (2 PNGSs) | Ibal-R | Ibal-R M426L | Ibal-S | Ibal-R | Ibal-R M426L | |||
| BMS-626529 | 0.12 ± 0.0 | 0.082 ± 0.081 | 11.4 ± 2.9 | 3.9 ± 0.8 | 455 ± 87 | 101.7 ± 0.03 | 101.2 ± 0.0 | 100.7 ± 0.5 |
| Ibalizumab | 1.33 ± 0.1 | >995 | 0.45 ± 0.1 | 3.2 ± 0.1 | 5.5 ± 3.2 | 102.6 ± 0.6 | 80.0 ± 12.0 | 83.2 ± 1.8 |
Ibal-R, ibalizumab-resistant clinical envelope; ibal-S, ibalizumab-sensitive clinical envelope; MPI, maximum percent inhibition; PNGSs, potential N-linked glycosylation sites in V5. Mean values of two independent experiments are shown.
Analysis of cross-reactivity to ENF-resistant envelopes was performed in two ways. A series of five clinical envelopes resistant to ENF and present in the Monogram collection (sequences unknown) were examined for susceptibility to BMS-626529 in the PhenoSense Entry assay. All five envelopes exhibited sensitivity (fold change in the IC50 [FC-IC50] of 0.49 to 2.9) to the AI and resistance to ENF (Table 7). In addition, a series of single-amino-acid changes in the gp41 region of NL4-3, which are known to result in resistance to ENF, were made by SDM, and the envelopes were incorporated into pseudovirus particles (42, 43). These pseudoviruses were used to infect MT-2 cells, and their susceptibility to BMS-626529 and ENF was assayed (Table 8). The point mutants exhibited differing levels of resistance to ENF, but all viruses were highly sensitive to BMS-626529. This suggests that there is no cross-resistance between ENF and BMS-626529.
Table 7.
Susceptibility of ENF-resistant virus and clinical envelopes to BMS-626529 determined by the PhenoSense Entry assaya
| Resistant envelope | FC-IC50 |
|
|---|---|---|
| BMS-626529 | Enfuvirtide | |
| 1 | 2.9 | 30–100 |
| 5 | 0.17 | 30–100 |
| 8 | 0.32 | 30–100 |
| 10 | 0.18 | >100 |
| 14 | 0.49 | >100 |
FC-IC50, fold change in IC50 compared with control envelope. Mean values of two independent experiments are shown.
Table 8.
Susceptibility of ENF-resistant virus and clinical envelopes to BMS-626529 determined by a pseudotype virus assaya
| Substitution | BMS-626529 |
Enfuvirtide |
||
|---|---|---|---|---|
| Mean IC50 (nM) ± SD | FC-IC50 | Mean IC50 (nM) ± SD | FC-IC50 | |
| Wild type | 0.1 ± 0.06 | 1 | 2 ± 0.2 | 1 |
| G36D | 0.2 ± 0.03 | 2 | 154 ± 15 | 77 |
| V38A | 0.1 ± 0.004 | 1 | 481 ± 75 | 240 |
| Q40H | 0.1 ± 0.02 | 1 | 11 ± 2 | 5.5 |
| N42T | 0.1 ± 0.01 | 1 | 26 ± 0.4 | 13 |
| N43D | 0.1 ± 0.09 | 1 | 302 ± 20 | 151 |
Mean values of two independent experiments are shown.
Finally, the potential for cross-resistance of BMS-626529 to MVC-resistant viruses was examined. Previously, it was shown that BMS-626529 exhibits equivalent activity against CCR5- or CXCR4-tropic viruses (11, 13, 14). Thus, CXCR4-tropic viruses, which inherently exhibit resistance to MVC, are not innately cross-resistant to BMS-626529. This leaves the much rarer CCR5-tropic MVC-resistant envelopes. Two viruses of this phenotype (sequences unknown) from the Monogram collection were examined by using the PhenoSense Entry assay (Table 9). Both viruses exhibited a low MPI against MVC, indicating resistance, while one (MVC res 3) was sensitive to BMS-626529 and the other (MVC res 2) was resistant, with an FC-IC50 of >5,000. This suggests that resistance to MVC in a CCR5-tropic envelope does not necessarily also encode resistance to BMS-626529. To probe this further, a series of CCR5-tropic envelope clones obtained from two subjects treated with either MVC or the investigational CCR5 antagonist aplaviroc was obtained from Robert Doms (University of Pennsylvania). All five of these envelope clones are CCR5 tropic (32, 33). These envelopes were evaluated in a cell-cell fusion assay for susceptibility to both BMS-626529 and MVC (Table 9). In the one subject treated with MVC, four sequential envelope clones (S1, S2, R3, and R4) were tested (32). Sensitivities to both MVC and BMS-626529 were observed by using the S1 and S2 envelopes, while resistance to both compounds was observed in the R4 envelope. Resistance to both compounds was also observed in the Post 5.1 sample from a second subject who was treated with aplaviroc (33). However, in the R3 envelope from the first subject, resistance to MVC was clearly observed, with an MPI of 56.2%, while the envelope retained complete sensitivity to BMS-626529. Evaluation of the sequences of R4 and Post 5.1 for any of the known genetic correlates of resistance to BMS-626529 (13, 14) showed that the R4 clone contains a threonine substitution at amino acid position 434. It was previously shown that this substitution results in high-level resistance to the early AI BMS-378806 (46). Site-directed mutagenesis was performed to confirm the role of the substitution in resistance to BMS-626529; however, the T434M change in R4 resulted in an inactive gp160 molecule. Thus, an M434T change was made in the LAI envelope, and this virus exhibited ∼16-fold-decreased susceptibility to BMS-626529 (data not shown). No known resistance correlates of BMS-626529 were identified in the Post 5.1 sample. In conclusion, the data strongly suggest that although some envelopes did exhibit cross-resistance to both compounds, resistance to BMS-626529 in an R5-tropic virus is not necessarily linked with MVC resistance.
Table 9.
Susceptibility of MVC-resistant envelopes to BMS-626529a
| Assay and clone | BMS-626529 |
MVC |
||
|---|---|---|---|---|
| Mean EC50 (nM) ± SD | Mean MPI ± SD | Mean EC50 (nM) ± SD | Mean MPI ± SD | |
| PhenoSense Entry assay | ||||
| MVC res 2 | >5,000 | NA | NA | <66 |
| MVC res 3 | 6.7 | 100 | NA | <33 |
| Cell fusion assay | ||||
| S1 | 3.5 ± 0.3 | 100 ± 0.1 | 1.3 ± 0.1 | 100 ± 0.1 |
| S2 | 9.6 ± 0.6 | 100 ± 0.1 | 3.2 ± 0.2 | 100 ± 0.1 |
| R3 | 5.0 ± 1.8 | 100 ± 0.2 | 11 ± 2.2 | 56 ± 2.2 |
| R4 | 101 ± 31 | 93 ± 7.1 | >3,335 | 39 ± 18 |
| Post 5.1 | 209 ± 5.1 | 98 ± 1.7 | >5,000 | 15 ± 19 |
Resistance to either BMS-626529 or MVC is indicated by boldface type. S1 and S2 (isolated pretreatment) and R3 and R4 (isolated on day 224) cloned envelopes were isolated from a patient who experienced virologic failure on MVC treatment (33). The Post 5.1 envelope was isolated from a patient who was treated with the CCR5 antagonist aplaviroc and was resistant to both aplaviroc and MVC (50). Data are the mean values of two independent experiments. NA, not applicable because inhibition did not reach high enough levels.
DISCUSSION
The HIV-1 AI BMS-626529 was previously shown in a proof-of-concept 8-day clinical monotherapy study in subjects receiving the prodrug BMS-663068 to induce substantial declines in plasma HIV-1 RNA levels in both antiretroviral-naive and -experienced subjects (the median maximum decline in plasma HIV-1 RNA load ranged from 1.21 to 1.73 log10 copies/ml) (12). BMS-626529 targets the interaction between gp120 and the host receptor CD4. This mode of action raises the possibility that CD4-independent viruses may be resistant to BMS-626529 or that treatment with BMS-663068 may promote the emergence of CD4-independent viruses. In the present study, we have performed a number of in vitro experiments to further characterize BMS-626529, particularly with regard to its activity against CD4-independent HIV-1 infection.
Laboratory-derived CD4-independent viruses retained susceptibility to BMS-626529, regardless of their coreceptor tropism (CCR5 tropic, ADA S190R/N197S; CXCR4 tropic, HIV-8x). While of limited clinical relevance, given that such viruses have been isolated in vivo only on rare occasions (23, 24), possibly because these viruses have been shown to be more sensitive to antibody neutralization than CD4-dependent viruses (18, 21, 22), these data indicate that clinical use of BMS-663068 is unlikely to promote resistance via selective pressure for the generation of CD4-independent viruses. This is supported by the fact that all envelopes with known in vitro-derived BMS-626529 resistance substitutions remained CD4 dependent. Moreover, clinical isolates with decreased susceptibility to BMS-626529 also remained CD4 dependent. The activity of BMS-626529 in CD4-independent virus strains, regardless of coreceptor tropism, suggests that the compound has broad effects, preventing binding not just to CD4 but also to both CCR5 and CXCR4.
A new model developed by Langley et al. helps to explain these findings. In this model, BMS-626529 binds gp120 and prevents the gp120 conformation change required for CD4 binding and exposure of the coreceptor binding site (D. R. Langley, S. R. Kimura, P. Sivaprakasam, N. Zhou, I. Dicker, T. Wang, J. Kadow, N. A. Meanwell, and M. Krystal, presented at the 20th Conference on Retroviruses and Opportunistic Infections, Atlanta, GA, 3 to 6 March 2013). In CD4-independent virus, gp120 can adopt a conformation that presents the coreceptor binding site in the absence of CD4 while still allowing BMS-626529 to bind. The model predicts that BMS-626529 blocks both wild-type and CD4-independent gp120 from adopting a CD4-bound conformation and presenting the coreceptor binding site. This unique mechanism of action prevents CD4-dependent gp120 from binding CD4 and coreceptor- and CD4-independent gp120 from directly binding the coreceptor. This model may also reconcile differences from the mechanism of action that was proposed for an earlier prototype inhibitor, BMS-378806 (20). We have previously demonstrated mutually exclusive binding of AIs BMS-488043 and BMS-378806 and soluble CD4 (sCD4) to gp120; order-of-addition studies showed that prior inhibitor-gp120 binding was required for sCD4 exclusion (9). These findings are in contrast to work by Si et al. (20), which showed a lack of sCD4 binding inhibition by BMS-378806. There are several possible explanations for the differences between our study and that of Si et al. Si et al. added sCD4 prior to the addition of BMS-378806, whereas we added sCD4 after the addition of BMS-378806. In addition, the relatively high concentration of sCD4 used by Si et al. compared with that used in our study may have helped outcompete the ability of BMS-378806 to block sCD4-gp120 binding. Also, the use of a cytoplasmic-tail-truncated (ΔCT) envelope variant that exhibited changed envelope conformations with enhanced exposure of CD4 and coreceptor binding regions in the study by Si et al. may have resulted in the lack of inhibition of fluorescein isothiocyanate (FITC)-CD4 binding to the cell surface envelope by BMS-378806 (20). We have previously shown that virions pseudotyped with ΔCT envelope variants displayed a diminished susceptibility to AI inhibition of sCD4-virion binding or sCD4-induced 17b-virion binding compared with wild-type virions (9). Regardless of this, even if there are some envelopes that can still bind CD4 in the presence of bound BMS-626529, the current data suggest that these attachment events do not produce the downstream conformational changes in gp120 required for coreceptor binding. This suggests a robust mode of action for BMS-626529, which may have implications in terms of its clinical activity.
We have previously reported synergistic in vitro antiviral activity for BMS-626529 in combination with either MVC or ENF and additive to synergistic effects in combination with AMD3100 (11). Since BMS-663068 may be used either after virologic failure or in combination with other entry inhibitors, it is imperative to understand whether there is potential for cross-resistance of the AI with these inhibitors. The possibility that development of resistance to BMS-626529 may result in decreased susceptibility to other HIV-1 entry inhibitors was investigated in various in vitro antiviral assays. Most of the BMS-626529-resistant clones examined (many of which were derived from clinical samples from the monotherapy study) remained susceptible to other entry inhibitors, including ibalizumab, MVC, and ENF. The only exception was a CXCR4-tropic clone that was highly resistant to MVC, as would be expected.
In addition, the possibility that resistance to a different HIV-1 entry inhibitor would also result in reduced susceptibility to BMS-626529 was investigated. Ibalizumab- and ENF-resistant viruses remained susceptible to BMS-626529. However, while some CCR5-tropic MVC-resistant envelopes were susceptible to BMS-626529, others were not, although resistance to BMS-626529 did not appear to be linked directly to MVC resistance. Resistance to MVC in clinical treatment usually develops as a result of outgrowth of a preexisting CXCR4-tropic population (47). In contrast, MVC-resistant viruses generated in the laboratory display the ability to recognize drug-bound CCR5 (32), but development of resistance to MVC via a CCR5-tropic virus is extremely rare in the clinical setting. In any case, the identification of multiple CCR5-tropic envelopes (one from a series of MVC-treated longitudinal samples) that retain susceptibility to BMS-626529 but are MVC resistant suggests that resistance to each of the compounds is independent of the other.
The lack of cross-resistance observed in this study may be explained by the differing mechanisms of action of the four compounds and known polymorphisms in gp120 that are present in naive samples that can encode resistance to BMS-626529. A similar lack of cross-resistance between MVC and ENF was noted previously (48).
Recently, Ratcliff et al. reported the isolation of an MVC-resistant variant from a subtype A HIV-1 primary isolate selected through dose escalation of the inhibitor (49). Resistance of this virus to MVC appeared to be competitive and to map to an N425K change in gp120. Considering that this is adjacent to M426, we sought to investigate whether 425K also conferred cross-resistance to BMS-626529. As noted by Ratcliff et al. (49), making this substitution within the context of a subtype B clone did not produce a functional protein. Thus, mutagenesis of an envelope clone from a subtype A clade primary isolate was performed, and a functional protein that could be used in a drug susceptibility assay was able to be produced. However, the 425K envelope did not exhibit decreased susceptibility to either BMS-626529 or MVC (data not shown), showing that the MVC-resistant phenotype is HIV-1 strain dependent.
The data presented here have the obvious limitation of being conducted in vitro, with some experiments performed using laboratory virus strains not encountered in clinical practice. Therefore, the generalization of these findings to clinical situations will need confirmation in long-term clinical trials.
In summary, these data indicate that the AI BMS-626529 possesses a robust mechanism of action conferring activity against CD4-dependent HIV-1 viruses and both CCR5- and CXCR4-tropic CD4-independent viruses. No notable cross-resistance with other HIV-1 entry inhibitors was observed. These results suggest that AIs may be suitable for use sequentially or concurrently with other classes of HIV-1 entry inhibitors and also support the continued clinical development of BMS-663068. A phase 2b study of BMS 663068 in treatment-experienced HIV-1-infected subjects is ongoing (registration number NCT01384734).
Supplementary Material
ACKNOWLEDGMENTS
We thank Robert Doms, who provided key CD4-independent and CCR5 inhibitor-resistant viruses that were used in this analysis.
This study was funded by Bristol-Myers Squibb. Editorial assistance was provided by Ben Caldwell of MediTech Media and was funded by Bristol-Myers Squibb.
Footnotes
Published ahead of print 17 June 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.00513-13.
REFERENCES
- 1.Dau B, Holodniy M. 2009. Novel targets for antiretroviral therapy: clinical progress to date. Drugs 69:31–50 [DOI] [PubMed] [Google Scholar]
- 2.Nixon DE, Landay AL. 2010. Biomarkers of immune dysfunction in HIV. Curr. Opin. HIV AIDS 5:498–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kuritzkes DR. 2009. HIV-1 entry inhibitors: an overview. Curr. Opin. HIV AIDS 4:82–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tilton JC, Doms RW. 2010. Entry inhibitors in the treatment of HIV-1 infection. Antiviral Res. 85:91–100 [DOI] [PubMed] [Google Scholar]
- 5.Gulick RM, Lalezari J, Goodrich J, Clumeck N, DeJesus E, Horban A, Nadler J, Clotet B, Karlsson A, Wohlfeiler M, Montana JB, McHale M, Sullivan J, Ridgway C, Felstead S, Dunne MW, van der Ryst E, Mayer H. 2008. Maraviroc for previously treated patients with R5 HIV-1 infection. N. Engl. J. Med. 359:1429–1441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lalezari JP, Henry K, O'Hearn M, Montaner JS, Piliero PJ, Trottier B, Walmsley S, Cohen C, Kuritzkes DR, Eron JJ, Jr, Chung J, DeMasi R, Donatacci L, Drobnes C, Delehanty J, Salgo M. 2003. Enfuvirtide, an HIV-1 fusion inhibitor, for drug-resistant HIV infection in North and South America. N. Engl. J. Med. 348:2175–2185 [DOI] [PubMed] [Google Scholar]
- 7.Bruno CJ, Jacobson JM. 2010. Ibalizumab: an anti-CD4 monoclonal antibody for the treatment of HIV-1 infection. J. Antimicrob. Chemother. 65:1839–1841 [DOI] [PubMed] [Google Scholar]
- 8.Song R, Franco D, Kao CY, Yu F, Huang Y, Ho DD. 2010. Epitope mapping of ibalizumab, a humanized anti-CD4 monoclonal antibody with anti-HIV-1 activity in infected patients. J. Virol. 84:6935–6942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ho HT, Fan L, Nowicka-Sans B, McAuliffe B, Li CB, Yamanaka G, Zhou N, Fang H, Dicker I, Dalterio R, Gong YF, Wang T, Yin Z, Ueda Y, Matiskella J, Kadow J, Clapham P, Robinson J, Colonno R, Lin PF. 2006. Envelope conformational changes induced by human immunodeficiency virus type 1 attachment inhibitors prevent CD4 binding and downstream entry events. J. Virol. 80:4017–4025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hanna GJ, Lalezari J, Hellinger JA, Wohl DA, Nettles R, Persson A, Krystal M, Lin P, Colonno R, Grasela DM. 2011. Antiviral activity, pharmacokinetics, and safety of BMS-488043, a novel oral small-molecule HIV-1 attachment inhibitor, in HIV-1-infected subjects. Antimicrob. Agents Chemother. 55:722–728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nowicka-Sans B, Gong YF, McAuliffe B, Dicker I, Ho HT, Zhou N, Eggers B, Lin PF, Ray N, Wind-Rotolo M, Zhu L, Majumdar A, Stock D, Lataillade M, Hanna GJ, Matiskella JD, Ueda Y, Wang T, Kadow JF, Meanwell NA, Krystal M. 2012. In vitro antiviral characteristics of HIV-1 attachment inhibitor BMS-626529, the active component of the prodrug BMS-663068. Antimicrob. Agents Chemother. 56:3498–3507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nettles R, Schurmann D, Zhu L, Stonier M, Huang S-P, Chang I, Chien C, Krystal M, Wind-Rotolo M, Ray N, Hanna GJ, Bertz R, Grasela DM. 2012. Pharmacodynamics, safety, and pharmacokinetics of BMS-663068, an oral HIV-1 attachment inhibitor in HIV-1-infected subjects. J. Infect. Dis. 206:1002–1011 [DOI] [PubMed] [Google Scholar]
- 13.Ray N, Hwang C, Healy MD, Whitcomb J, Lataillade M, Wind-Rotolo M, Krystal M, Hanna GJ. 22 April 2013. Prediction of virologic response and assessment of resistance emergence to the HIV-1 attachment inhibitor BMS-626529 during 8-day monotherapy with its prodrug BMS-663068. J. Acquir. Immune Defic. Syndr. [Epub ahead of print.] 10.1097/QAI.0b013e31829726f3 [DOI] [PubMed] [Google Scholar]
- 14.Zhou N, Ray N, Healy M, Langley D, Hwang C, Lataillade M, Hanna G, Krystal M. 2012. Genotypic and phenotypic correlates of virologic response to the attachment inhibitor BMS-626529 in a short-term monotherapy study with its prodrug BMS-663068, abstr 6, p A14. Abstr. 2012 Int. Wkshp. HIV Hepatitis Virus Drug Resist., Sitges, Spain [Google Scholar]
- 15.Charpentier C, Larrouy L, Visseaux B, Landman R, Levittas M, Storto A, Damond F, Yazdanpanah Y, Yeni P, Brun-Vezinet F, Descamps D. 2012. Prevalence of subtype-related polymorphisms associated with in vitro resistance to attachment inhibitor BMS-626529 in HIV-1 ‘non-B'-infected patients. J. Antimicrob. Chemother. 67:1459–1461 [DOI] [PubMed] [Google Scholar]
- 16.Soulie C, Lambert-Niclot S, Fofana DB, Fourati S, Ait-Arkoub Z, Sayon S, Simon A, Katlama C, Calvez V, Marcelin AG. 8 February 2013. Frequency of amino acid changes associated with resistance to attachment inhibitor BMS-626529 in R5- and X4-tropic HIV-1 subtype B. J. Antimicrob. Chemother. [Epub ahead of print.] 10.1093/jac/dkt018 [DOI] [PubMed] [Google Scholar]
- 17.Schader SM, Colby-Germinario SP, Quashie PK, Oliveira M, Ibanescu RI, Moisi D, Mesplede T, Wainberg MA. 2012. HIV gp120 H375 is unique to HIV-1 subtype CRF01_AE and confers strong resistance to the entry inhibitor BMS-599793, a candidate microbicide drug. Antimicrob. Agents Chemother. 56:4257–4267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Haim H, Strack B, Kassa A, Madani N, Wang L, Courter JR, Princiotto A, McGee K, Pacheco B, Seaman MS, Smith AB, Sodroski J., III 2011. Contribution of intrinsic reactivity of the HIV-1 envelope glycoproteins to CD4-independent infection and global inhibitor sensitivity. PLoS Pathog. 7:e1002101. 10.1371/journal.ppat.1002101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Da LT, Quan JM, Wu YD. 2011. Understanding the binding mode and function of BMS-488043 against HIV-1 viral entry. Proteins 79:1810–1819 [DOI] [PubMed] [Google Scholar]
- 20.Si Z, Madani N, Cox JM, Chruma JJ, Klein JC, Schon A, Phan N, Wang L, Biorn AC, Cocklin S, Chaiken I, Freire E, Smith AB, Sodroski JG., III 2004. Small-molecule inhibitors of HIV-1 entry block receptor-induced conformational changes in the viral envelope glycoproteins. Proc. Natl. Acad. Sci. U. S. A. 101:5036–5041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Edwards TG, Hoffman TL, Baribaud F, Wyss S, Labranche CC, Romano J, Adkinson J, Sharron M, Hoxie JA, Doms RW. 2001. Relationships between CD4 independence, neutralization sensitivity, and exposure of a CD4-induced epitope in a human immunodeficiency virus type 1 envelope protein. J. Virol. 75:5230–5239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kolchinsky P, Kiprilov E, Sodroski J. 2001. Increased neutralization sensitivity of CD4-independent human immunodeficiency virus variants. J. Virol. 75:2041–2050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xiao P, Usami O, Suzuki Y, Ling H, Shimizu N, Hoshino H, Zhuang M, Ashino Y, Gu H, Hattori T. 2008. Characterization of a CD4-independent clinical HIV-1 that can efficiently infect human hepatocytes through chemokine (C-X-C motif) receptor 4. AIDS 22:1749–1757 [DOI] [PubMed] [Google Scholar]
- 24.Zerhouni B, Nelson JA, Saha K. 2004. Isolation of CD4-independent primary human immunodeficiency virus type 1 isolates that are syncytium inducing and acutely cytopathic for CD8+ lymphocytes. J. Virol. 78:1243–1255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guo Q, Ho HT, Dicker I, Fan L, Zhou N, Friborg J, Wang T, McAuliffe BV, Wang HG, Rose RE, Fang H, Scarnati HT, Langley DR, Meanwell NA, Abraham R, Colonno RJ, Lin PF. 2003. Biochemical and genetic characterizations of a novel human immunodeficiency virus type 1 inhibitor that blocks gp120-CD4 interactions. J. Virol. 77:10528–10536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li Z, Zhou N, Sun Y, Ray N, Lataillade M, Hanna GJ, Krystal M. 2012. XIX Int. AIDS Conf., Washington, DC, 22 to 27 July 2012, poster TUPE015 [Google Scholar]
- 27.Lou S, Moquist PN, Schaus SE. 2007. Asymmetric allylboration of acyl imines catalyzed by chiral diols. J. Am. Chem. Soc. 129:15398–15404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.De Clercq E. 2009. The AMD3100 story: the path to the discovery of a stem cell mobilizer (Mozobil). Biochem. Pharmacol. 77:1655–1664 [DOI] [PubMed] [Google Scholar]
- 29.Burkly LC, Chisholm PL, Thomas DW, Rosa MD, Rosa JJ. 5,871,732 US patent. 1999 Feb;
- 30.Kolchinsky P, Mirzabekov T, Farzan M, Kiprilov E, Cayabyab M, Mooney LJ, Choe H, Sodroski J. 1999. Adaptation of a CCR5-using, primary human immunodeficiency virus type 1 isolate for CD4-independent replication. J. Virol. 73:8120–8126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kolchinsky P, Kiprilov E, Bartley P, Rubinstein R, Sodroski J. 2001. Loss of a single N-linked glycan allows CD4-independent human immunodeficiency virus type 1 infection by altering the position of the gp120 V1/V2 variable loops. J. Virol. 75:3435–3443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tilton JC, Wilen CB, Didigu CA, Sinha R, Harrison JE, Agrawal-Gamse C, Henning EA, Bushman FD, Martin JN, Deeks SG, Doms RW. 2010. A maraviroc-resistant HIV-1 with narrow cross-resistance to other CCR5 antagonists depends on both N-terminal and extracellular loop domains of drug-bound CCR5. J. Virol. 84:10863–10876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tilton JC, Amrine-Madsen H, Miamidian JL, Kitrinos KM, Pfaff J, Demarest JF, Ray N, Jeffrey JL, Labranche CC, Doms RW. 2010. HIV type 1 from a patient with baseline resistance to CCR5 antagonists uses drug-bound receptor for entry. AIDS Res. Hum. Retroviruses 26:13–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hoffman TL, Labranche CC, Zhang W, Canziani G, Robinson J, Chaiken I, Hoxie JA, Doms RW. 1999. Stable exposure of the coreceptor-binding site in a CD4-independent HIV-1 envelope protein. Proc. Natl. Acad. Sci. U. S. A. 96:6359–6364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lin PF, Nowicka-Sans B, Terry B, Zhang S, Wang C, Fan L, Dicker I, Gali V, Higley H, Parkin N, Tenney D, Krystal M, Colonno R. 2008. Entecavir exhibits inhibitory activity against human immunodeficiency virus under conditions of reduced viral challenge. Antimicrob. Agents Chemother. 52:1759–1767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Toma J, Weinheimer SP, Stawiski E, Whitcomb JM, Lewis ST, Petropoulos CJ, Huang W. 2011. Loss of asparagine-linked glycosylation sites in variable region 5 of human immunodeficiency virus type 1 envelope is associated with resistance to CD4 antibody ibalizumab. J. Virol. 85:3872–3880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pace CS, Fordyce MW, Franco D, Kao CY, Seaman MS, Ho DD. 2013. Anti-CD4 monoclonal antibody ibalizumab exhibits breadth and potency against HIV-1, with natural resistance mediated by the loss of a V5 glycan in envelope. J. Acquir. Immune Defic. Syndr. 62:1–9 [DOI] [PubMed] [Google Scholar]
- 38.Coakley E, Petropoulos CJ, Whitcomb JM. 2005. Assessing chemokine co-receptor usage in HIV. Curr. Opin. Infect. Dis. 18:9–15 [DOI] [PubMed] [Google Scholar]
- 39.Limon A, Nakajima N, Lu R, Ghory HZ, Engelman A. 2002. Wild-type levels of nuclear localization and human immunodeficiency virus type 1 replication in the absence of the central DNA flap. J. Virol. 76:12078–12086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.LaBranche CC, Hoffman TL, Romano J, Haggarty BS, Edwards TG, Matthews TJ, Doms RW, Hoxie JA. 1999. Determinants of CD4 independence for a human immunodeficiency virus type 1 variant map outside regions required for coreceptor specificity. J. Virol. 73:10310–10319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhou N, Nowicka-Sans B, Zhang S, Fan L, Fang J, Fang H, Gong YF, Eggers B, Langley DR, Wang T, Kadow J, Grasela D, Hanna GJ, Alexander L, Colonno R, Krystal M, Lin PF. 2011. In vivo patterns of resistance to the HIV attachment inhibitor BMS-488043. Antimicrob. Agents Chemother. 55:729–737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Baldwin C, Berkhout B. 2007. HIV-1 drug-resistance and drug-dependence. Retrovirology 4:78. 10.1186/1742-4690-4-78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lu J, Sista P, Giguel F, Greenberg M, Kuritzkes DR. 2004. Relative replicative fitness of human immunodeficiency virus type 1 mutants resistant to enfuvirtide (T-20). J. Virol. 78:4628–4637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pugach P, Marozsan AJ, Ketas TJ, Landes EL, Moore JP, Kuhmann SE. 2007. HIV-1 clones resistant to a small molecule CCR5 inhibitor use the inhibitor-bound form of CCR5 for entry. Virology 361:212–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Westby M, Smith-Burchnell C, Mori J, Lewis M, Mosley M, Stockdale M, Dorr P, Ciaramella G, Perros M. 2007. Reduced maximal inhibition in phenotypic susceptibility assays indicates that viral strains resistant to the CCR5 antagonist maraviroc utilize inhibitor-bound receptor for entry. J. Virol. 81:2359–2371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lin PF, Blair W, Wang T, Spicer T, Guo Q, Zhou N, Gong YF, Wang HG, Rose R, Yamanaka G, Robinson B, Li CB, Fridell R, Deminie C, Demers G, Yang Z, Zadjura L, Meanwell N, Colonno R. 2003. A small molecule HIV-1 inhibitor that targets the HIV-1 envelope and inhibits CD4 receptor binding. Proc. Natl. Acad. Sci. U. S. A. 100:11013–11018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Moore JP, Kuritzkes DR. 2009. A piece de resistance: how HIV-1 escapes small molecule CCR5 inhibitors. Curr. Opin. HIV AIDS 4:118–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.MacArthur RD, Novak RM. 2008. Reviews of anti-infective agents. Maraviroc: the first of a new class of antiretroviral agents. Clin. Infect. Dis. 47:236–241 [DOI] [PubMed] [Google Scholar]
- 49.Ratcliff AN, Shi W, Arts EJ. 2013. HIV-1 resistance to maraviroc conferred by a CD4 binding site mutation in the envelope glycoprotein gp120. J. Virol. 87:923–934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Johnson VA, Calvez V, Gunthard HF, Paredes R, Pillay D, Shafer R, Wensing AM, Richman DD. 2011. 2011 update of the drug resistance mutations in HIV-1. Top. Antivir. Med. 19:156–164 [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.