

Abstract
We report that the artemisinin-derived dimer diphenyl phosphate (DPP; dimer 838) is the most selective inhibitor of human cytomegalovirus (CMV) replication among a series of artemisinin-derived monomers and dimers. Dimer 838 was also unique in being an irreversible CMV inhibitor. The peroxide unit within artemisinins' chemical structures is critical to their activities, and its absence results in loss of anti-CMV activities. Surprisingly, the deoxy dimer of 838 retained modest anti-CMV activity, suggesting that the DPP moiety of dimer 838 contributes to its anti-CMV activities. DPP alone did not inhibit CMV replication, but triphenyl phosphate (TPP) had modest CMV inhibition, although its selectivity index was low. Artemisinin DPP derivatives dimer 838 and monomer diphenyl phosphate (compound 558) showed stronger CMV inhibition and a higher selectivity index than their analogs lacking the DPP unit. An add-on and removal assay revealed that removing DPP derivatives (compounds 558 and 838) but not the non-DPP backbones (artesunate and compound 606) at 24 h postinfection (hpi) already resulted in dominant CMV inhibition. CMV inhibition was fully irreversible with 838 and partially irreversible with 558, while non-DPP artemisinins were reversible inhibitors. While all artemisinin derivatives and TPP reduced the expression of the CMV immediate early 2 (IE2), UL44, and pp65 proteins at or after 48 hpi, only TPP inhibited the expression of both IE1 and IE2. Combination of a non-DPP dimer (compound 606) with TPP was synergistic in CMV inhibition, while ganciclovir and TPP were additive. Although TPP shared structural similarity with monomer DPP (compound 558) and dimer DPP (compound 838), its pattern of CMV inhibition was significantly different from the patterns of the artemisinins. These findings demonstrate that the DPP group contributes to the unique activities of compound 838.
INTRODUCTION
Artemisinins, compounds that became first-choice therapy for malaria, have recently received gained interest because of their antiviral activities (1, 2). Artesunate (AS) and the parent compound artemisinin were reported to inhibit human cytomegalovirus (CMV) replication in vitro and in vivo (3, 4). These findings were followed by a case report of AS therapy in an immunocompromised patient who had ganciclovir (GCV)-resistant CMV disease as well as preliminary data on CMV kinetics following treatment with AS (5, 6). Although AS inhibited GCV-resistant CMV strains in vitro, therapy was ineffective in controlling GCV-resistant CMV in a renal transplant recipient (7, 8). Since the serum concentrations achieved with the current dosing regimen of AS are unlikely to fully inhibit CMV replication and because higher doses may result in significant toxicities, improved and safe artemisinins with enhanced anti-CMV activity are desired (9). We have reported that artemisinin-derived dimers are highly selective inhibitors of CMV replication in vitro and are significantly more selective than their monomeric counterparts but do not have increased toxicity in human fibroblasts (10, 11). We identified the best dimer in CMV inhibition to be the dimer diphenyl phosphate (DPP; molecular weight [MW], 838; referred to as dimer 838). Dimer 838 proved to have unique anti-CMV activities even compared to those of other artemisinin-derived dimers, including irreversible inhibition of CMV replication and the ability to inhibit virus replication even when added to human fibroblasts prior to infection (12). Our continued work further reveals the characteristics of dimer 838 and delineates its improved anti-CMV activities compared to those of other artemisinin-derived dimers.
MATERIALS AND METHODS
Compounds.
GCV, monophenyl phosphate (MPP; MW, 174), DPP (MW, 250), and triphenyl phosphate (TPP; MW, 326) were purchased from Sigma Chemical Co. (St. Louis, MO). Monomers AS, monophenyl phosphate (compound 482), and diphenyl phosphate (compound 558) and dimers primary alcohol (compound 606), deoxy dimer of compound 606 (compound 574), monophenyl phosphate (compound 762), diphenyl phosphate (compound 838), and deoxy dimer of compound 838 (compound 806) were synthesized at Johns Hopkins University and solubilized in 100% dimethyl sulfoxide (DMSO) (Fig. 1) (13). Stocks of 10 mM (100 mM for TPP) were stored at −80°C. Synthetic compounds were at least 98% pure on the basis of proton nuclear magnetic resonance spectroscopy. Unless otherwise specified, the concentrations used for the experiments were 30 μM for AS, 1 μM for 558, 0.3 μM for 606, 0.1 μM for 838, 100 μM for TPP, and 10 μM for GCV. These concentrations were chosen on the basis of full virus inhibition by plaque reduction assay and more than 90% inhibition of pp28-luciferase expression while not causing significant cytotoxicity.
Fig 1.

Chemical structures of phosphate derivatives of artemisinin-derived monomers and dimers: the parent artemisinin-derived monomer and dimer (AS and compound 606, respectively), their MPP and DPP derivatives, and the nonartemisinins MPP, DPP, and TPP.
Viruses and antiviral assays.
The pp28-luciferase Towne strain was constructed as previously described (14). The expression of pp28-luciferase is strongly activated by 48 to 72 h postinfection (hpi) and is inhibited by DNA polymerase inhibitors (14). We reported that the pp28-luciferase assay is sensitive and reproducible and that its results highly correlate with those of the classic plaque reduction assay (14). Human foreskin fibroblasts (HFFs) were used for infection with CMV. Infection of HFFs and luciferase assays have been previously described (11). Unless otherwise specified, all CMV infections were carried out at a multiplicity of infection (MOI) of 1 PFU/cell. For each compound, the drug concentration resulting in 50% inhibition of virus growth (i.e., the 50% effective concentration [EC50]), the drug concentration resulting in 50% cell toxicity (i.e., the 50% cytotoxic concentration [CC50]), and the selectivity index (SI; CC50/EC50) were calculated. The quantitative CMV real-time PCR assay is based on detection of a 151-bp region from the highly conserved US17 gene (15). The limit of detection of the assay is 100 copies/ml (3.0 copies/reaction), and the measureable range is 2.4 to 8.0 log10 copies/ml. The PCR was performed using a total reaction volume of 50 μl including TaqMan 2× Universal PCR master mix (Applied Biosystems, Foster City, CA), US17 forward primer 5′-GCGTGCTTTTTAGCCTCTGCA-3′, US17 reverse primer 5′-AAAAGTTTGTGCCCCAACGGTA-3′ (final concentration for both primers, 300 nM), 6-carboxyfluorescein (FAM)-labeled probe FAM-TGATCGGCGTTATCGCGTTCTTGATC-TAMRA (final concentration, 200 nM; TAMRA is 6-carboxytetramethylrhodamine), distilled H2O, and template (10 μl). Amplification was performed on a 7500 real-time PCR system (Applied Biosystems, Foster City, CA). PCR conditions were 50°C for 2 min, 95°C for 10 min, and 40 cycles of 95°C for 15 s and 60°C for 60 s. Quantification standards were prepared by cloning the US17 amplicon in the pCR2.1-TOPO plasmid vector (Invitrogen, Carlsbad, CA). Serial 10-fold dilutions of plasmid from 7.0 to 1.0 log10 copies/reaction mixture were included with each assay and used to establish a standard curve. The slope and R2 value of the standard curve were −3.3 ± 0.1 and >0.990, respectively. Assay controls included quantified CMV AD169 DNA (10 copies/reaction; Advanced Biotechnologies Inc.) and quantified Towne CMV at 3.0 and 5.0 log10 copies/ml. Quantitative CMV data are expressed as the number of viral DNA copies per milliliter.
Toxicity in cancer cells and primary HFFs.
HeLa (human cervical adenocarcinoma) and HCT116 (human colorectal carcinoma) cells were maintained in Dulbecco modified Eagle medium (DMEM) containing 10% fetal bovine serum. At 12 to 16 h prior to drug treatment, 2 × 103cells were seeded on each well of 96-well tissue culture plates. Drugs were diluted in DMEM containing 10% fetal bovine serum and applied to the cells. At 72 h after drug treatment, 20 μl of 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolim bromide (MTT) solution (5 mg/ml in PBS) was added into each well, followed by shaking of the plates at 150 rpm for 5 min and incubation for 4 h (37°C, 5% CO2). Medium was removed, plates were dried, and the MTT formazan in each well was dissolved in 100 μl DMSO. After shaking of the plates at 150 rpm for 10 min, the absorbance of each well was recorded at 560 nm on a GloMax-Multi+ detection system (Promega). The CC50 was calculated for each compound. HFF viability was also determined using the MTT assay, which was performed at the same time points and cell densities as the antiviral assay.
Time-of-addition and time-of-removal studies.
HFFs were infected with Towne CMV as previously described. At 0, 6, 12, 24, 36, and 48 hpi, the medium was replaced with a medium containing AS, 558, 606, 838, TPP, and GCV.
For time-of-removal studies, the medium containing the compounds was removed at 0, 6, 12, 24, 36, and 48 hpi, cells were washed three times with PBS, and drug-free medium was added. Cells were incubated at 37°C, and luciferase assays were performed at 72 hpi. In the add-on assay, 0 hpi served as the positive control (the compound was added just after infection), and in the removal assay, 0 hpi served as the negative control (no compound was added to the wells).
CMV reversibility assay.
HFFs were infected and treated with the compounds AS, 558, 606, 838, TPP, and GCV. At 24, 48, and 72 hpi, the media were removed and replaced with fresh media without compounds. Cells were incubated for 6 days postinfection, and supernatants were collected for viral DNA quantification by real-time PCR (12).
Pretreatment assay.
HFFs were treated with the compounds AS, 558, 606, 838, TPP, and GCV for 24 h prior to CMV infection. Compounds were removed, and cells were washed with phosphate-buffered saline (PBS) three times before infection. After infection, cells were incubated in medium containing no compounds. The following assays were performed: DNA replication (by real-time PCR of US17 in cell lysates at 48 hpi), late gene expression (by luciferase assay at 72 hpi), and virus DNA yield (by real-time PCR of US17 in supernatants at 96 hpi).
Western blot analysis.
HFFs were infected with Towne CMV and treated with AS, 558, 606, 838, TPP, and GCV. At the indicated times, cells were harvested in sample buffer (radioimmunoprecipitation assay buffer; Tris, 50 mM; NaCl, 150 mM; SDS, 0.1%; sodium deoxycholate, 0.5%; NP-40, 1%; protease inhibitor cocktail) and boiled. Proteins were separated by electrophoresis and transferred to a polyvinylidene difluoride (PVDF) membrane (Bio-Rad Laboratories, Hercules, CA). After blocking, blots were probed with the following primary antibodies: mouse anti-human CMV immediate early 1 (IE1) and IE2 (MAB810; Millipore, Billerica, MA), mouse anti-human CMV UL83, pp65 (Vector Laboratories Inc., Burlingame, CA), mouse anti-human CMV UL44 (Santa Cruz, Santa Cruz, CA), and mouse anti-human β-actin (Sigma). After washing five times, blots were probed with horseradish peroxidase-conjugated anti-mouse antibody (Sigma) for 1 h at room temperature. Following washing with PBS-Tween 20, protein bands were visualized by chemiluminescence using SuperSignal West Dura and Pico reagents (Pierce Chemical, Rockford, IL) according to the manufacturer's protocol.
Drug combination.
The combined inhibitory effect of 606 and TPP on HCMV replication was determined in infected HFFs. Compounds were added at different dilutions starting with concentrations that resulted in full inhibition. Analysis was performed using the Bliss equation, in which the activity of the drug combination represents the product of two probabilistically independent events (16):
where D is the drug concentration, m is the slope, and EC50 is the effective concentration resulting in 50% virus inhibition. The combined effect of two inhibitors (fraction unaffected [fu]) is computed as the product of the individual effects of the two inhibitors, fu1 and fu2.
If the ratio of the observed fold inhibition divided by the expected fold inhibition is greater than 1, the combination of compounds is considered synergistic. If the ratio is less than 1, the combination is considered antagonistic, and if it equals 1, the combination is additive.
RESULTS
We previously reported that the oxygen bridge within the artemisinins' backbones was necessary for their anti-CMV activities (11). While a deoxy dimer (dimer 574) of dimer primary alcohol, compound 606, lost its anti-CMV activity even when it was used at a high concentration of 100 μM, a deoxy dimer of 838 (dimer 806) retained modest anti-CMV activity (Fig. 2), suggesting that the diphenyl phosphate (DPP) moiety of dimer 838 could contribute to its anti-CMV activity.
Fig 2.

(A) Chemical structures of deoxy dimers 574 (deoxy dimer of compound 606) and 806 (deoxy dimer of compound 838). (B) Dose-response curves of deoxy dimers 574 and 806. The dose-dependent anti-CMV activity was measured for compounds 806 and 574 by determination of luciferase activity in CMV-infected HFFs at 72 hpi. The EC50s and CC50s of the four compounds appear in the inset. Mean values ± SDs of triplicate determinations from three independent experiments are shown.
The contribution of DPP to CMV inhibition by artemisinins was tested by determining the anti-CMV activities of a monomeric DPP compound as well as monophenyl phosphate (MPP) derivatives of artemisinin monomer and dimer (Fig. 3). The DPP moiety improved the anti-CMV activity of both monomeric and dimeric artemisinins, but the MPP moiety decreased their anti-CMV activity (Fig. 3).
Fig 3.

The anti-CMV activity of MPP and DPP derivatives of dimer primary alcohol, compound 606 (A), and artemisinin monomer, AS (B). HFFs were infected and treated with monomeric or dimeric artemisinins at the indicated drug concentrations. Luciferase activity was measured at 72 hpi. Mean values ± SDs of triplicate determinations from at least three independent experiments are shown.
Because the dimer and monomer forms of DPP were found to have improved anti-CMV activities compared to their parent compounds, mono-, di-, and triphenyl phosphates were tested for their independent anti-CMV activity (Fig. 4). Treatment with TPP resulted in decreased pp28-luciferase activity, although its selectivity index was low: the EC50 was 57 ± 4.1 μM, the CC50 was 133 ± 23 μM, and the SI (CC50/EC50) was 2.33 ± 0.26. DPP and MPP did not inhibit CMV replication. Although treatment with DPP resulted in a moderate increase of luciferase activity, virus yield revealed no inhibition or enhancement of CMV replication.
Fig 4.

Anti-CMV activity of MPP, DPP, and TPP. CMV-infected HFFs were treated with MPP, DPP, and TPP, and luciferase activity was measured at 72 hpi. Mean values ± SDs of triplicate determinations from three independent experiments are shown.
Since artemisinins were reported in previous studies to inhibit cancer cell growth (17–19) and we found that artemisinin-derived dimers inhibit cancer cell growth at concentrations similar to those at which they show anti-CMV activities but are nontoxic to primary HFFs (11), the effects of artemisinin-MPP and artemisinin-DPP on cancer cell growth and their toxicity profile in primary HFFs were tested (Table 1). The anticancer activity of dimers 838, 762, and 606 correlated with their anti-CMV activities, but the linkage of DPP to a monomeric artemisinin, while improving its anti-CMV activity, was not associated with significant changes in inhibiting cancer cell growth. The addition of DPP to monomeric artemisinins increased their toxicity in HFFs, an effect that was not observed for dimeric artemisinins.
Table 1.
Anti-CMV activity, cellular toxicity, and anticancer activity of artemisinin-derived phosphates, their parent compounds, and TPPa
| Compound (MW) | EC50 (μM) for CMV | CC50 (μM) for HFFs | SI (CMV) | CC50 (μM) for HeLa cells | CC50 (μM) for HCT116 cells |
|---|---|---|---|---|---|
| Dimer primary alcohol (606) | 0.16 ± 0.01 | 48.1 ± 2.6 | 292 ± 21 | 1.06 ± 0.2 | 0.1 ± 0.00 |
| Dimer DPP (838) | 0.039 ± 0.00 | 56.69 ± 4.69 | 1,430.95 ± 122 | 0.19 ± 0.08 | 0.04 ± 0.00 |
| Dimer MPP (762) | 1.57 ± 0.08 | 56.49 ± 3.24 | 35.93 ± 2.77 | 18.56 ± 1.2 | 6.67 ± 0.7 |
| Artesunate (406) | 6.6 ± 0.4 | 71.7± 4 | 11 ± 0.9 | 7.1 ± 0.4 | 11.3 ± 0.6 |
| Monomer DPP (558) | 0.24 ± 0.01 | 4.88 ± 0.4 | 20.15 ± 2.03 | 8.11 ± 0.6 | 3.82 ± 0.3 |
| Monomer MPP (482) | 38.3 ± 2.3 | 55.04 ± 2.8 | 1.43 ± 011 | 68.4 ± 4.7 | 43.5 ± 2.1 |
| TPP (326) | 57 ± 4.1 | 133 ± 23 | 2.33 ± 0.26 | 51.9 ± 7.2 | 70.7 ± 9.8 |
The EC50s, CC50s, and SIs of artemisinin derivatives and TPP against CMV and their cytotoxicity in HeLa and HCT 116 are shown. Data represent mean values ± SDs of triplicate determinations from at least three independent experiments.
We tested the contribution of the chemical components of 838 (dimer and DPP) to its unique and highly selective anti-CMV activities: the irreversibility of CMV replication upon withdrawal of 838 and significant CMV inhibition when HFFs were treated with the compound prior to infection. We reported that artemisinins are early inhibitors of CMV replication. When dimer 838 was removed at 12 hpi, it had already achieved nearly complete CMV inhibition, while AS required longer than 40 h to achieve the same effect on CMV replication (12). To address whether the pattern of dimer 838 removal was a result of its dimer backbone or DPP, an add-on and removal assays were performed (Fig. 5). In the add-on assay (Fig. 5A), DPP derivatives 558 and 838 had similar activity patterns as their backbone structures, AS and 606. However, in the removal assay (Fig. 5B), DPP derivatives displayed an earlier effect than their backbone dimers. Dimer and monomer DPP grouped together at the 24-h time point, while AS and dimer 606 grouped together and separately from the compounds containing DPP. Removal at 6 hpi showed a similar pattern for dimers 606 and 838. Dimer 838 showed an activity pattern similar to that of dimer 606, in that early removal was already effective, but it was also similar to compound 558 by achieving complete inhibition when removed at 24 hpi. The dimer structure and DPP group together appeared to contribute to the unique removal curve pattern of dimer 838. TPP exhibited a different pattern from the artemisinins in both the add-on assay and removal curve study. Although still highly effective when added at 36 and 48 hpi, TPP was slower than GCV in achieving full virus inhibition at different time points of removal.
Fig 5.

Add-on and removal assays of artemisinin monomer (AS), monomer DPP (compound 558), dimer primary alcohol (compound 606), dimer DPP (compound 838), TPP, and GCV. Compounds were added (A) or removed (B) at 0, 6, 12, 24, 36, and 48 hpi. CMV inhibition was measured by determination of luciferase expression at 72 hpi. Mean values ± SDs of triplicate determinations from two independent experiments are shown.
To determine whether the irreversible activity of dimer 838 was a result of its dimer unit or DPP, compounds were added and removed at the indicated times (Fig. 6). At day 6 postinfection, virus DNA in supernatants was quantified by real-time PCR. DPP derivatives were irreversible inhibitors of CMV replication, while their backbones were reversible inhibitors. Dimer 838 was completely irreversible when removed after 24 h, while monomer DPP (compound 558) was irreversible only when removed after 3 days. Unlike 838 and 558, TPP was a reversible CMV inhibitor.
Fig 6.

Reversibility of CMV replication after treatment with artemisinin monomer (AS), monomer DPP (compound 558), artemisinin dimer primary alcohol (compound 606), dimer DPP (compound 838), TPP, and GCV. Compounds were present in CMV-infected cells over the indicated intervals (in days). Virus DNA in supernatants at 6 days postinfection was quantified by real-time PCR. Mean values ± SDs of triplicate determinations from two independent experiments are shown.
The effect of treatment with DPP and non-DPP derivatives prior to CMV infection was tested. HFFs were treated with the compounds for 24 h, and then cells were washed, compounds were removed, and cells were infected with CMV (MOI = 1). The virus DNA yield in supernatants collected at 96 hpi was measured by real-time PCR. pp28-luciferase activity was measured in cell lysates collected at 72 hpi. DNA replication was determined by real-time PCR from cell DNA collected at 48 hpi. Dimer 838 was the only compound that was highly effective in CMV inhibition when added to HFFs before infection (Fig. 7A). Treatment with compound 558 or 606 prior to CMV infection was not effective against CMV replication.
Fig 7.

(A) Effect of pretreatment with artemisinin monomer (AS), monomer DPP (compound 558), dimer primary alcohol (compound 606), dimer DPP (compound 838), TPP, and GCV on CMV replication. (B) Inhibition of DNA replication, pp28 expression, and virus DNA yield by artemisinin monomer (AS), monomer DPP (compound 558), dimer primary alcohol (compound 606), dimer DPP (compound 838), TPP, and GCV. HFFs were treated with the indicated compounds for 24 h prior to infection. Compounds were removed just before infection. After infection, cells were grown in medium free of compounds. DNA replication in cell lysates collected at 48 hpi was measured by real-time PCR. pp28-luciferase activity was measured in cell lysates collected at 72 hpi. Virus DNA yield in supernatants collected at 96 hpi was measured by real-time PCR. Mean values ± SDs of triplicate determinations from three independent experiments are shown.
The activity of DPP and non-DPP compounds was tested at different stages of virus replication. Following infection and treatment with the indicated compounds, virus DNA yield was measured at 96 hpi, pp28-luciferase activity was measured at 72 hpi, and DNA replication was measured at 48 hpi. For both artemisinins and TPP, the inhibition of virus DNA yield was stronger than the inhibition of DNA replication and pp28 expression (Fig. 7B).
The effect of the compounds on the expression of selected CMV genes was determined. Compounds were added after virus infection, and cell lysates were collected for Western blotting at 24, 48, and 72 hpi. The four artemisinins had similar effects on the inhibition of CMV gene expression. All had minimal inhibition of immediate early 1 (IE1) protein expression but significant inhibition of IE2, the early UL44 protein, and the late tegument protein pp65 (Fig. 8). Despite the differences in their anti-CMV activities, all artemisinins showed a similar pattern of inhibition of CMV gene expression. Thus, despite differences in the add-on, removal, and reversibility studies, the effects on CMV gene expression were similar between non-DPP- and DPP-containing dimers. Compounds 838 and TPP had different effects on gene expression (Fig. 8). TPP showed strong inhibition of viral gene expression. It inhibited IE1 expression, in addition to expression of IE2 and all other assayed proteins, more effectively than 838.
Fig 8.
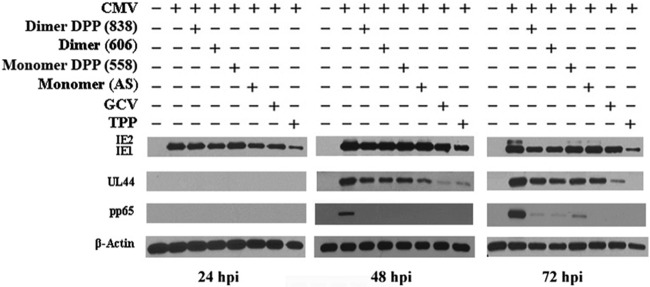
Effects of AS, 558, 606, 838, TPP, and GCV on CMV gene expression. The expression of CMV proteins was measured in infected HFFs. Compounds were added after virus infection, and cell lysates were collected for Western blotting at 24, 48, and 72 hpi.
Finally, to mimic a noncovalently linked form of 838, the combination of dimer primary alcohol (compound 606)-GCV or 606-TPP was tested against CMV (Table 2). While 606 and TPP were synergistic for CMV inhibition, the combination of GCV and TPP was only additive.
Table 2.
Combination effect of 606 or GCV with TPPa
| Compound 1 only |
Compound 2 (TPP) only |
Fold inhibition for the combination |
||||
|---|---|---|---|---|---|---|
| Compound | Concn (μM) | Fold inhibition | Concn (μM) | Fold inhibition | Theoretical | Actual |
| 606 | 0.09 | 1.71 ± 0.01 | 50 | 1.30 ± 0.01 | 2.21 ± 0.00 | 8.15 ± 0.25 |
| 606 | 0.09 | 1.71 ± 0.01 | 60 | 1.96 ± 0.17 | 3.34 ± 0.01 | 10.68 ± 1.05 |
| 606 | 0.12 | 2.70 ± 0.04 | 50 | 1.30 ± 0.01 | 3.50 ± 0.01 | 9.01 ± 0.76 |
| 606 | 0.12 | 2.70 ± 0.04 | 60 | 1.96 ± 0.17 | 5.29 ± 0.02 | 12.18 ± 2.82 |
| GCV | 1.7 | 1.71 ± 0.06 | 50 | 1.30 ± 0.01 | 2.21 ± 0.01 | 1.88 ± 0.12 |
| GCV | 1.7 | 1.71 ± 0.06 | 60 | 1.96 ± 0.17 | 3.35 ± 0.02 | 3.48 ± 0.48 |
Mean values ± SDs of triplicate determinations from three independent experiments are shown.
DISCUSSION
Our work reveals that the linkage of diphenyl phosphate (DPP) to artemisinin-derived dimer contributes to its unique and selective anti-CMV activities. Understanding the anti-CMV effects of dimer 838 resulted from our previous findings of the unique anti-CMV activities of this compound compared to other artemisinins. Removal of the peroxide unit in the dimer backbone of 838 resulted in a deoxy compound (compound 806) that retained modest anti-CMV activity, but compound 574 (a deoxy version of dimer primary alcohol 606) completely lost anti-CMV activity (Fig. 2). Thus, we hypothesized that the linkage of the DPP group to artemisinin-derived dimer could enhance its anti-CMV activity. Further investigation of the contribution of DPP to the activities of artemisinin-derived monomer and dimer (MWs, 558 and 838, respectively) revealed that compound 558 also had enhanced activity against CMV replication compared to AS (Fig. 3). These results, along with previous data showing that dimer DPP (dimer 838) is a stronger CMV inhibitor than dimer primary alcohol (compound 606), suggest that DPP could contribute to the enhanced anti-CMV activity of artemisinin-derived monomers and dimers. The detailed anti-CMV assays also suggest that DPP may provide artemisinins with improved affinity to their potential targets in CMV-infected cells. In the removal assay (Fig. 5), compounds 558 and 838 acted similarly, while their parent compounds (AS and 606, respectively) grouped together. In addition, 558 and 838 were irreversible inhibitors, although 838 showed stronger irreversibility than 558 (Fig. 6). CMV irreversibility, a feature of the DPP group and not of the dimer backbone, was achieved when DPP was chemically linked to artemisinins. Thus, DPP may play a role in the affinity of binding of artemisinins to their targets.
The contribution of the DPP group to CMV inhibition by artemisinins was determined by testing the anti-CMV activity of monophenyl-, diphenyl-, and triphenyl phosphates. Among the three compounds, only triphenyl phosphate showed some effect on CMV replication, and its selectivity index was low and similar to that of deoxy dimer 806. These results may have been expected on the basis of the chemical structures of these compounds; TPP is similar structurally to monomer DPP (compound 558) or dimer DPP (compound 838). The anti-CMV activities of TPP were evaluated using several virological assays (Fig. 5 to 8) and were found to be distinct from the anti-CMV activities of 838. Its removal curve showed a slower effect, and it was a reversible anti-CMV agent. The inhibition of CMV gene expression by TPP was different from that of artemisinins. Taken together, the activities of TPP are different from those of artemisinins.
Although DPP contributed to the improved anti-CMV activity of compounds 838 and 558, its activity cannot be considered a result of simple chemical linkage to artemisinins or an independent agent. It appears to be the interaction of DPP with the artemisinin backbone structure that results in the overall anti-CMV effect. Although DPP contributed to the irreversibility of 838 and 558, this effect likely represents that of the combination of artemisinins and DPP and not the individual effect of each group. Similarly, the anti-CMV activity of 838 before virus infection may also be due to the combination of dimer and DPP. Taken together, the DPP group attached to artemisinins is not an independent CMV inhibitor, but it converts its structure to a TPP analog. Dimer 838 therefore represents a unique and complex chemical structure that leads to highly selective CMV inhibition.
Would compound 558 or 838 be a candidate for CMV therapy? While the addition of DPP to artemisinin monomer improved its anti-CMV activity and SI compared to those of AS, it also increased its toxicity in HFFs. Although additional cell types should be tested, 558 may or may not be a candidate for in vivo studies. In the case of dimers, 838 was superior to 606 in CMV inhibition without additional toxicity in HFFs. We believe that compound 838 is a lead compound for in vivo studies because of its unique properties and its very high SI. Compared to monomeric artemisinins, 838 has two separate chemical components, dimer and DPP, both of which contribute to the anti-CMV activity of 838. As an irreversible inhibitor of CMV, 838 should also be evaluated in vivo for its pharmacokinetic properties.
ACKNOWLEDGMENTS
This study was supported by NIH grant 1R01AI093701 and March of Dimes grant 6-FY11-268 (to R.A.-B.) and NIH grant AI 34885 (to G.H.P.).
Footnotes
Published ahead of print 17 June 2013
REFERENCES
- 1.Adjuik M, Babiker A, Garner P, Olliaro P, Taylor W, White N. 2004. Artesunate combinations for treatment of malaria: meta-analysis. Lancet 363:9–17 [DOI] [PubMed] [Google Scholar]
- 2.Efferth T, Romero MR, Wolf DG, Stamminger T, Marin JJ, Marschall M. 2008. The antiviral activities of artemisinin and artesunate. Clin. Infect. Dis. 47:804–811 [DOI] [PubMed] [Google Scholar]
- 3.Efferth T, Marschall M, Wang X, Huong SM, Hauber I, Olbrich A, Kronschnabl M, Stamminger T, Huang ES. 2002. Antiviral activity of artesunate towards wild-type, recombinant, and ganciclovir-resistant human cytomegaloviruses. J. Mol. Med. 80:233–242 [DOI] [PubMed] [Google Scholar]
- 4.Kaptein SJ, Efferth T, Leis M, Rechter S, Auerochs S, Kalmer M, Bruggeman CA, Vink C, Stamminger T, Marschall M. 2006. The anti-malaria drug artesunate inhibits replication of cytomegalovirus in vitro and in vivo. Antiviral Res. 69:60–69 [DOI] [PubMed] [Google Scholar]
- 5.Shapira MY, Resnick IB, Chou S, Neumann AU, Lurain NS, Stamminger T, Caplan O, Saleh N, Efferth T, Marschall M, Wolf DG. 2008. Artesunate as a potent antiviral agent in a patient with late drug-resistant cytomegalovirus infection after hematopoietic stem cell transplantation. Clin. Infect. Dis. 46:1455–1457 [DOI] [PubMed] [Google Scholar]
- 6.Wolf DG, Shimoni A, Resnick IB, Stamminger T, Neumann AU, Chou S, Efferth T, Caplan O, Rose J, Nagler A, Marschall M. 2011. Human cytomegalovirus kinetics following institution of artesunate after hematopoietic stem cell transplantation. Antiviral Res. 90:183–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chou S, Marousek G, Auerochs S, Stamminger T, Milbradt J, Marschall M. 2011. The unique antiviral activity of artesunate is broadly effective against human cytomegaloviruses including therapy-resistant mutants. Antiviral Res. 92:364–368 [DOI] [PubMed] [Google Scholar]
- 8.Lau PK, Woods ML, Ratanjee SK, John GT. 2011. Artesunate is ineffective in controlling valganciclovir-resistant cytomegalovirus infection. Clin. Infect. Dis. 52:279. [DOI] [PubMed] [Google Scholar]
- 9.Ribeiro IR, Olliaro P. 1998. Safety of artemisinin and its derivatives. A review of published and unpublished clinical trials. Med. Trop. (Mars.) 58(3 Suppl):50–53 [PubMed] [Google Scholar]
- 10.Arav-Boger R, He R, Chiou CJ, Liu J, Woodard L, Rosenthal A, Jones-Brando L, Forman M, Posner GH. 2010. Artemisinin-derived dimers have greatly improved anti-cytomegalovirus activity compared to artemisinin monomers. PLoS One 5:e10370. 10.1371/journal.pone.0010370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.He R, Mott BT, Rosenthal AS, Genna DT, Posner GH, Arav-Boger R. 2011. An artemisinin-derived dimer has highly potent anti-cytomegalovirus (CMV) and anti-cancer activities. PLoS One 6:e24334. 10.1371/journal.pone.0024334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.He R, Park K, Cai H, Kapoor A, Forman M, Mott B, Posner GH, Arav-Boger R. 2012. Artemisinin-derived dimer diphenyl phosphate is an irreversible inhibitor of human cytomegalovirus replication. Antimicrob. Agents Chemother. 56:3508–3515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Alagbala AA, McRiner AJ, Borstnik K, Labonte T, Chang W, D'Angelo JG, Posner GH, Foster BA. 2006. Biological mechanisms of action of novel C-10 non-acetal trioxane dimers in prostate cancer cell lines. J. Med. Chem. 49:7836–7842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.He R, Sandford G, Hayward GS, Burns WH, Posner GH, Forman M, Arav-Boger R. 2011. Recombinant luciferase-expressing human cytomegalovirus (CMV) for evaluation of CMV inhibitors. Virol. J. 8:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tanaka Y, Kanda Y, Kami M, Mori S, Hamaki T, Kusumi E, Miyakoshi S, Nannya Y, Chiba S, Arai Y, Mitani K, Hirai H, Mutou Y. 2002. Monitoring cytomegalovirus infection by antigenemia assay and two distinct plasma real-time PCR methods after hematopoietic stem cell transplantation. Bone Marrow Transplant. 30:315–319 [DOI] [PubMed] [Google Scholar]
- 16.Jilek BL, Zarr M, Sampah ME, Rabi SA, Bullen CK, Lai J, Shen L, Siliciano RF. 2012. A quantitative basis for antiretroviral therapy for HIV-1 infection. Nat. Med. 18:446–451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Efferth T, Dunstan H, Sauerbrey A, Miyachi H, Chitambar CR. 2001. The anti-malarial artesunate is also active against cancer. Int. J. Oncol. 18:767–773 [DOI] [PubMed] [Google Scholar]
- 18.Posner GH, Ploypradith P, Parker MH, O'Dowd H, Woo SH, Northrop J, Krasavin M, Dolan P, Kensler TW, Xie S, Shapiro TA. 1999. Antimalarial, antiproliferative, and antitumor activities of artemisinin-derived, chemically robust, trioxane dimers. J. Med. Chem. 42:4275–4280 [DOI] [PubMed] [Google Scholar]
- 19.Efferth T. 2007. Willmar Schwabe Award 2006: antiplasmodial and antitumor activity of artemisinin—from bench to bedside. Planta Med. 73:299–309 [DOI] [PubMed] [Google Scholar]