

Abstract
In an era of rapidly emerging antimicrobial-resistant bacteria, it is critical to understand the importance of the relationships among drug exposure, duration of therapy, and selection of drug resistance. Herein we describe the results of studies designed to determine the ceftolozane-tazobactam exposure necessary to prevent the amplification of drug-resistant bacterial subpopulations in a hollow-fiber infection model. The challenge isolate was a CTX-M-15-producing Escherichia coli isolate genetically engineered to transcribe a moderate level of blaCTX-M-15. This organism's blaCTX-M-15 transcription level was confirmed by relative quantitative reverse transcription-PCR (qRT-PCR), β-lactamase hydrolytic assays, and a ceftolozane MIC value of 16 mg/liter. In these studies, the experimental duration (10 days), ceftolozane-tazobactam dose ratio (2:1), and dosing interval (every 8 h) were selected to approximate those expected to be used clinically. The ceftolozane-tazobactam doses studied ranged from 125-62.5 to 1,500-750 mg. Negative- and positive-control arms included no treatment and piperacillin-tazobactam at 4.5 g every 6 h, respectively. An inverted-U-shaped function best described the relationship between bacterial drug resistance amplification and drug exposure. The least- and most-intensive ceftolozane-tazobactam dosing regimens, i.e., 125-62.5, 750-375, 1,000-500, and 1,500-750 mg, did not amplify drug resistance, while drug resistance amplification was observed with intermediate-intensity dosing regimens (250-125 and 500-250 mg). For the intermediate-intensity ceftolozane-tazobactam dosing regimens, the drug-resistant subpopulation became the dominant population by days 4 to 6. The more-intensive ceftolozane-tazobactam dosing regimens (750-375, 1,000-500, and 1,500-750 mg) not only prevented drug resistance amplification but also virtually sterilized the model system. These data support the selection of ceftolozane-tazobactam dosing regimens that minimize the potential for on-therapy drug resistance amplification.
INTRODUCTION
The pharmacokinetic-pharmacodynamic (PK-PD) challenges to understanding the joint actions of β-lactam–β-lactamase inhibitor combinations are multidimensional. Some levels of complexity are unique to β-lactam–β-lactamase inhibitor combinations, while others are identical to those of single-drug antibacterial regimens. An example of the former is that the magnitude of exposure necessary for efficacy of either the β-lactam or β-lactamase inhibitor across clinically relevant exposures is inversely related to the exposure of the partner agent. Therefore, any PK-PD relationships or target thresholds identified are conditional.
One challenge that is common to both β-lactam–β-lactamase inhibitor combinations and single-drug regimens is identifying the exposure necessary to prevent the amplification of drug-resistant bacterial subpopulations. Tam et al. previously demonstrated that the relationship between drug exposure and drug resistance amplification takes the functional form of an inverted U (1). That is, at low and high drug exposures, drug resistance amplification is lower than that with intermediate exposures. Moreover, Tam et al. demonstrated that the drug exposure necessary to prevent drug resistance amplification increases with the duration of therapy.
Ceftolozane is a novel cephalosporin with broad-spectrum Gram-negative activity whose mechanism of action is to interfere with cell wall production. The combination of ceftolozane and tazobactam, a β-lactamase inhibitor known to prevent the most common β-lactamase enzymes (including CTX-M-15) from hydrolyzing β-lactam antibiotics, targets the organism and its most common resistance mechanism.
We recently investigated the PK-PD of tazobactam in combination with ceftolozane against an isogenic CTX-M-15-producing Escherichia coli triplet set genetically engineered to transcribe different levels of blaCTX-M-15 (2). The percentage of the dosing interval that tazobactam concentrations remained above a threshold (%Time>threshold) was identified as the PK-PD exposure measure that was most closely associated with efficacy. The %Time>threshold values for tazobactam associated with net bacterial stasis and with 1- and 2-log10 CFU reductions in bacteria at 24 h were approximately 35, 50, and 70, respectively, regardless of enzyme transcription level. The threshold tazobactam concentrations associated with these targets ranged from 0.05 to 0.25 mg/liter. However, one limitation of this study was that the experimental duration was 24 h, so the study was not designed to examine the impact of therapy duration on the amplification of drug-resistant bacterial subpopulations.
The objective of the studies described herein was to identify the ceftolozane-tazobactam exposure necessary to prevent drug resistance amplification. In these studies, the experimental duration, ceftolozane-tazobactam dose ratio, and dosing interval were selected to approximate those expected to be used clinically.
MATERIALS AND METHODS
Bacterium, antimicrobials, and β-lactamase inhibitor.
Ceftolozane and tazobactam were provided by Cubist Pharmaceuticals (Lexington, MA), while piperacillin was obtained from Sigma (St. Louis, MO). The challenge organism utilized in these studies was a recombinant E. coli strain (GenBank accession number KC355192) provided by JMI Laboratories (North Liberty, IA). The amount of CTX-M-15 expression was determined previously by quantitative reverse transcription-PCR (qRT-PCR) and measurement of the hydrolytic activity of the enzyme produced (2).
Media and in vitro susceptibility studies.
Susceptibility testing studies were performed according to Clinical and Laboratory Standards Institute guidelines (3) for broth microdilution and agar dilution methods, utilizing cation-adjusted Mueller-Hinton (MH) broth and MH agar (BD Laboratories, Franklin Lakes, NJ). Strain susceptibilities to ceftolozane and piperacillin were determined alone and in combination with tazobactam at a fixed concentration (4 mg/liter). All susceptibility studies were performed in triplicate over a 2-day period, and the MIC results presented represent the modes from these studies. E. coli ATCC 25922 was utilized as an internal control for all susceptibility testing.
Mutation frequency studies.
The frequency of mutation to drug resistance was estimated by plating 4 ml of log-phase growth suspension onto agar containing ceftolozane at 3 or 5 times the baseline MIC and tazobactam at a fixed concentration (4 mg/liter). The bacterial concentration within the suspension was determined by quantitative culture, and the ratio of growth found on the drug-containing plates to that of the starting inoculum provided an estimate of the drug resistance frequency within a total population. This assay was performed in duplicate, and for each trial, a subset of isolates was taken from the drug-containing plates and tested for a change in the MIC from the baseline to confirm decreased susceptibility.
Hollow-fiber infection model.
The hollow-fiber infection model has been described previously (4). In brief, this pharmacodynamic system allows pathogens to grow in the peripheral chamber of a hollow-fiber cartridge. The peripheral chamber is separated from the central compartment by semipermeable membranes with pore sizes that are large enough to allow nutrients, drugs, and bacterial metabolites to transverse freely into and out of the peripheral compartment but too small for bacteria to leave the peripheral compartment. Fresh medium is circulated through the hollow-fiber cartridge from the central compartment by use of peristaltic pumps. Drug is pumped into the central compartment under computer control, using multiple infusion pumps to simulate different half-lives, and is continually diluted in the central compartment without diluting the pathogen in the peripheral compartment. Due to the high surface-area-to-volume ratio, drug concentrations equilibrate rapidly in the periphery. Specimens for quantitative culture and drug concentration assay can be removed from the peripheral compartment through sampling ports.
Resistance amplification prevention studies.
Each study consisted of a ceftolozane-tazobactam combination regimen and three control regimens. In total, six ceftolozane-tazobactam dosing regimens simulating human free serum pharmacokinetics were evaluated using a fixed 2:1 ratio infused over 1 h every 8 h. The ceftolozane-tazobactam dosing regimens evaluated were 125-62.5, 250-125, 500-250, 750-375, 1,000-500, and 1,500-750 mg. In each study, control regimens included a no-treatment growth control, ceftolozane infused at 375 mg over 1 h every 8 h (demonstrating limited bactericidal activity), and piperacillin-tazobactam infused at 4.5 g over 0.5 h every 6 h. A tazobactam control arm was omitted because this compound has been shown to have no therapeutic effect on the study isolate (2). All studies were conducted in duplicate over 10 days.
In these studies, the initial challenge isolate inoculum was prepared from an overnight culture grown on Trypticase soy agar (TSA) plus 5% sheep blood (BD Laboratories) at 35°C. Colonies from the culture were grown to mid-log phase and then suspended in an Erlenmeyer flask containing cation-adjusted MH medium set in a water-shaker bath at 35°C and 125 rotations per minute. The bacterial concentration within the flask of MH broth was determined by measuring the optical density and using a previously confirmed growth curve for the challenge isolate. Subsequently, 15 ml of the bacterial suspension was inoculated into the extracapillary space of a hollow-fiber cartridge (FiberCell Systems, Frederick, MD) at a concentration of 1.0 × 108 CFU/ml.
Within the hollow-fiber cartridge, bacteria were exposed to fluctuating free drug concentrations that simulated the half-lives in humans of 2.5 h for ceftolozane and 1 h for tazobactam. Protein binding was assumed to be 20% for ceftolozane (Cubist, data on file) and 30% for tazobactam (Zosyn package insert). Over the course of the 10-day experiment, 1-ml samples were taken from the extracapillary space, washed twice with sterile normal saline, serially diluted, and quantitatively cultured on drug-free TSA plates with 5% sheep blood to determine the effect of treatment on the total bacterial population. Portions of each sample were plated on MH agar plates containing ceftolozane at 3 times the baseline MIC plus tazobactam at a fixed concentration (4 mg/liter) for enumeration of the resistant subpopulation. MIC values were determined for a subset of isolates found growing on the drug-containing plates on days 1, 3, 6, and 10 of each study.
Pharmacokinetic validation studies.
Over the first 48 h of each study, 1-ml specimens for drug assay were collected from the peripheral compartment at 1, 3, 5, 7, 9, 23, 25, 29, 31, 33, and 48 h and then immediately frozen at −80°C until assayed for drug concentration. Ceftolozane, piperacillin, and tazobactam concentrations were measured by liquid chromatography-tandem mass spectrometry (LC-MS/MS), which was completed by Microconstants, San Diego, CA.
Analytical method.
All samples were assayed by LC-MS/MS (Waters, Milford, MA). The standard curves for ceftolozane and piperacillin were linear over ranges of 0.1 to 500 mg/liter and 0.5 to 500 mg/liter, respectively, and that for tazobactam was quadratic and ranged from 0.1 to 100 mg/liter. The lower quantification limit was 0.1 mg/liter for ceftolozane and tazobactam and was slightly higher for piperacillin, at 0.5 mg/liter. The intraday coefficients of variation (CV) for ceftolozane were ≤5.8% and ≤0.95% at concentrations of 0.3 mg/liter and 400 mg/liter, respectively; for tazobactam, the intraday CV were ≤5.3% and ≤6.5% at concentrations of 0.3 and 80 mg/liter, respectively. The interday CV for piperacillin were ≤4.8% and ≤5.4% at concentrations of 1.5 mg/liter and 400 mg/liter, respectively.
Pharmacokinetic-pharmacodynamic analysis.
The pharmacokinetic parameters and PK-PD measures for each study are presented in Table 1. A quadratic spline was applied to the data from the resistance amplification dose-ranging studies.
Table 1.
Ceftolozane-tazobactam pharmacokinetic parameters and PK-PD measures attained for each study
| Regimen (dose [mg]) | Parameter | Value |
|
|---|---|---|---|
| Ceftolozane | Tazobactam | ||
| Ceftolozane (375) | Cmax (mg/liter) | 19.2 | |
| AUC0–24 (mg · h/liter) | 178 | ||
| %Time>thresholda | 12.5 | ||
| Ceftolozane-tazobactam | Cmax (mg/liter) | 8.1 | 1.5 |
| (125-62.5) | AUC0–24 (mg · h/liter) | 75.1 | 6.6 |
| %Time>thresholdb | 100.0 | 75.0 | |
| Ceftolozane-tazobactam | Cmax (mg/liter) | 13 | 3.1 |
| (250-125) | AUC0–24 (mg · h/liter) | 120.3 | 13.1 |
| %Time>thresholdb | 100.0 | 87.5 | |
| Ceftolozane-tazobactam | Cmax (mg/liter) | 27.2 | 6.1 |
| (500-250) | AUC0–24 (mg · h/liter) | 252.2 | 26.3 |
| %Time>thresholdb | 100.0 | 98.0 | |
| Ceftolozane-tazobactam | Cmax (mg/liter) | 39.7 | 9.1 |
| (750-375) | AUC0–24 (mg · h/liter) | 368.3 | 39.4 |
| %Time>thresholdb | 100.0 | 100.0 | |
| Ceftolozane-tazobactam | Cmax (mg/liter) | 49.2 | 12.2 |
| (1,000-500) | AUC0–24 (mg · h/liter) | 456.2 | 52.5 |
| %Time>thresholdb | 100.0 | 100.0 | |
| Ceftolozane-tazobactam | Cmax (mg/liter) | 78.6 | 18.3 |
| (1,500-750) | AUC0–24 (mg · h/liter) | 728.4 | 78.8 |
| %Time>thresholdb | 100.0 | 100.0 | |
The ceftolozane threshold was 16 mg/liter, which was the ceftolozane MIC in the absence of tazobactam.
The ceftolozane threshold was 0.25 mg/liter, which was the ceftolozane MIC in the presence of 4 mg/liter tazobactam. The tazobactam threshold was 0.05 mg/liter, as identified previously (2).
RESULTS
In vitro susceptibility studies.
The MIC values for ceftolozane and piperacillin alone were determined to be 16 and 256 mg/liter, respectively. The MIC values for ceftolozane and piperacillin in the presence of 4 mg/liter tazobactam were 0.25 and 2 mg/liter, respectively. MIC values for piperacillin-tazobactam were within CLSI-recommended values for all studies (data not shown).
Mutation frequency studies.
The average density of the drug-resistant subpopulation at 3 times the baseline ceftolozane MIC was 1 CFU for every 7.26 log bacteria. Similarly, at 5 times the baseline ceftolozane-tazobactam MIC, the average density of the resistant subpopulation was 1 CFU for every 7.51 log bacteria. The average frequency of mutation to piperacillin-tazobactam resistance was 1 CFU for every 6.75 and 6.98 log bacteria at 3 and 5 times the baseline MIC value, respectively. The isolates taken from the mutation frequency study drug-containing plates had MIC values ranging from only 1 to 2 mg/liter, regardless of the concentration of drug within the drug-containing agar plate.
Pharmacokinetic validation studies.
For all dosing regimens, the targeted ceftolozane, piperacillin, and tazobactam pharmacokinetic profiles were well simulated in the hollow-fiber infection model, as evidenced by the agreement between observed and targeted concentration-time profiles for each drug. Figure 1 shows relationships between the observed and targeted concentrations for all dosing regimens of ceftolozane, piperacillin, and tazobactam studied. The r2 values for these relationships were 0.9827 for ceftolozane, 0.9932 for piperacillin, and 0.9668 for tazobactam.
Fig 1.
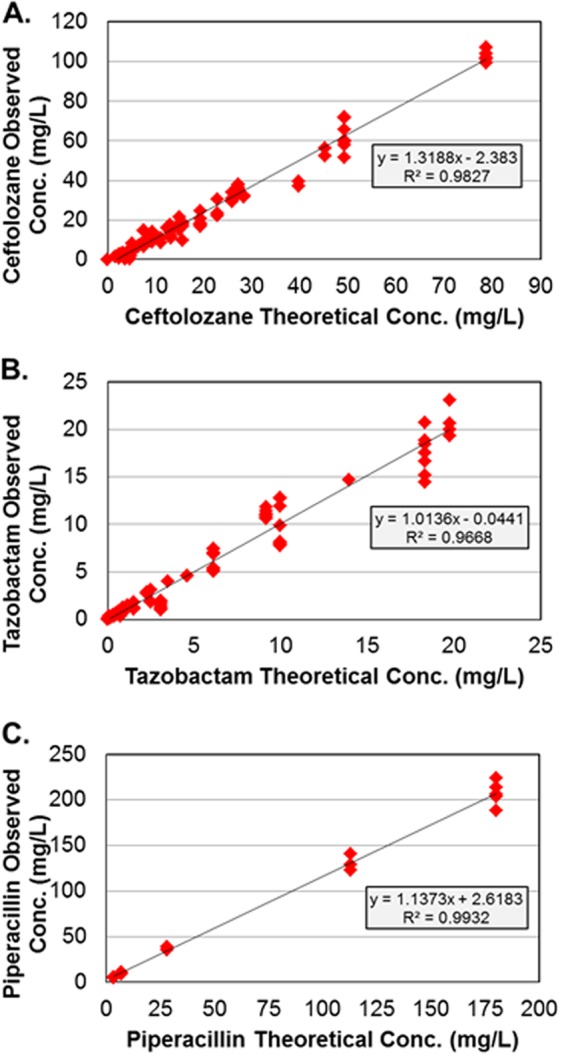
Relationships between observed and targeted ceftolozane (A), tazobactam (B), and piperacillin (C) concentrations.
Resistance amplification prevention studies.
Figure 2 shows the change in total population bacterial density over 10 days for each study. In the no-treatment growth control arms (Fig. 2A), the bacteria grew well, with the bacterial density increasing from 1.0 × 107.6 to 1.0 × 1010.2 CFU/ml by 2 days. Similarly, the ceftolozane control arm had growth similar to that of the no-treatment growth control arms, reaching an average bacterial density of 1.0 × 1010.4 CFU/ml by 2 days. The piperacillin-tazobactam control arm produced a 4-log10 CFU/ml reduction by 2 days and prevented drug resistance amplification over the entirety of the 10-day study period (Fig. 2A).
Fig 2.

Emergence of resistance during drug administration. (A) Negative control (no treatment) and active controls (ceftolozane at 375 mg every 8 h [Q8h] and piperacillin [PIP]-tazobactam at 4.5 g every 6 h). (B) Ceftolozane-tazobactam (TOL/TAZ) dosing regimens ranging from 125-62.5 to 1,500-750 mg infused every 8 h.
The range of ceftolozane-tazobactam dosing regimens evaluated resulted in a full spectrum of drug effects (Fig. 2B). The least intensive of the ceftolozane-tazobactam dosing regimens, 125-62.5 mg, resulted in no killing and, by day 1, selected a stable ratio of resistant to wild-type populations that remained throughout the 10 days. The next two least-intensive ceftolozane-tazobactam dosing regimens, 250-125 and 500-250 mg, resulted in close to immediate drug resistance amplification, and the drug-resistant subpopulation exceeded that of the no-treatment control by day 2 and, in the case of the 500-250-mg dosing regimen, nearly replaced the total population by day 3. Each of the three more-intensive ceftolozane-tazobactam dosing regimens, 750-375, 1,000-500, and 1,500-750 mg, resulted in a >3-log10 CFU/ml reduction by day 1 and prevented drug resistance amplification over the entirety of the 10-day study period. It is noteworthy that these three dosing regimens virtually sterilized the model system, as evidenced by total bacterial burdens of less than the quantification limit by day 4 and for the remaining 6 study days. Figure 3 shows the relationship between the change in log10 CFU of the drug-resistant subpopulation at 240 h and dose, which is represented by a quadratic spline. Doses of 125-62.6 mg and ≥750-375 mg did not amplify resistance, with the latter resulting in almost a 4-log kill. Resistance was amplified only with the 250-125- and 500-250-mg dosing regimens.
Fig 3.
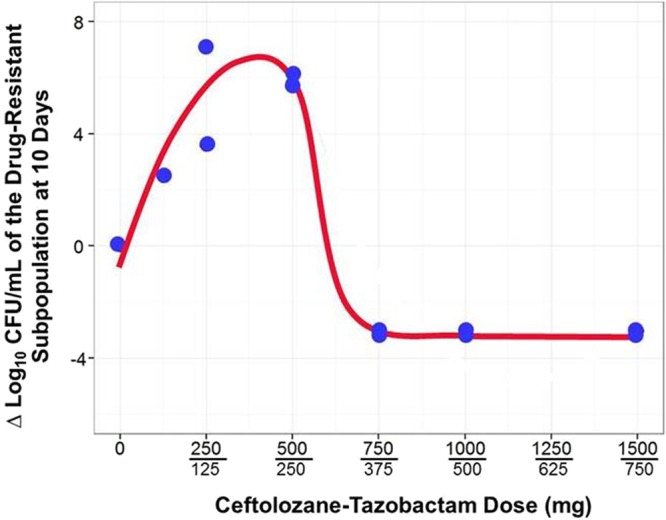
Relationship between ceftolozane-tazobactam exposure and change in bacterial density of the drug-resistant subpopulation.
Finally, across the intermediate ceftolozane-tazobactam regimens studied, the MIC values for isolates cultured on drug-containing plates were 8 to 32 mg/liter for ceftolozane-tazobactam. As would be expected, there was a tendency for the MIC values to increase with the duration of therapy, starting with 8 mg/liter initially and culminating with 32 mg/liter toward the end of therapy.
DISCUSSION
The objective of these studies was to identify the ceftolozane-tazobactam exposure necessary to prevent drug resistance amplification. In these studies, the experimental duration, ceftolozane-tazobactam dose ratio, and dosing interval were selected to approximate those expected to be used clinically for urinary tract infections and intra-abdominal infections (1,000-500 mg ceftolozane-tazobactam given every 8 h).
We successfully discriminated among ceftolozane-tazobactam dosing regimens that resulted in drug resistance amplification and those that did not. While the ceftolozane-tazobactam dosing regimen of 125-62.5 mg failed, it minimally amplified drug resistance. Ceftolozane-tazobactam dosing regimens of 250-125 and 500-250 mg also failed, and they greatly amplified drug resistance. For these dosing regimens, the drug-resistant population became the dominant population after 4 to 6 days of therapy. Moreover, the MICs for isolates collected from drug-containing plates increased with time. For instance, the ceftolozane-tazobactam MIC value for the 250-125-mg dosing regimen was 2 mg/liter on day 1 and 16 mg/liter on day 10. The ceftolozane-tazobactam dosing regimen of 750-375 mg and those with even greater doses prevented drug resistance amplification. The more-intensive ceftolozane-tazobactam dosing regimens not only drove the drug-resistant subpopulation below the quantification limit of the model system but also, by day 4, drove the entire bacterial population toward extinction. Cultures of the entire volume of medium contained in the hollow-fiber cartridges were sterile for the 1,000-500- and 1,500-750-mg dosing regimens.
As expected, the relationship between drug resistance amplification and drug exposure took a hormetic, or inverted-U, form (Fig. 3). Hormetic exposure-response relationships have long been recognized. In 1888, Hugo Paul Friedrich Schulz, a German pharmacologist, observed this phenomenon while studying the effects of formic acid on yeast cultures. He noted that low doses of formic acid stimulated the growth of yeast cells, while higher doses were lethal (5). In general, the biologic reason for such a relationship is the presence of two receptors with differing binding characteristics that move a drug or chemical response in opposite directions.
More recently, Tam and colleagues identified a hormetic function that described the relationship between Pseudomonas aeruginosa density and garenoxacin exposure in a hollow-fiber infection model (1). The hormetic relationship was due to the presence of two bacterial subpopulations, one of which was drug susceptible and the other of which was drug resistant. At low drug exposures, the growth of the garenoxacin-susceptible subpopulation was suppressed, while that of the garenoxacin-resistant subpopulation was amplified. Eventually, at high garenoxacin exposures, both the drug-susceptible and -resistant subpopulations were suppressed. In the study described herein, a large starting inoculum (1.0 × 108 CFU/ml) was intentionally selected to enrich the probability of the presence of a drug-resistant subpopulation(s). Moreover, the bacterial burden studied is clinically relevant to circumstances of pneumonia or closed-space infections such as meningitis and intra-abdominal abscesses.
Piperacillin-tazobactam was selected as a positive control in these experiments for two reasons. First, piperacillin-tazobactam has long been used successfully clinically for indications similar to those for which ceftolozane-tazobactam is being developed, namely, intra-abdominal infections and pneumonia. Second, the exposure of tazobactam coadministered with ceftolozane is 42% (6) of that with piperacillin due to piperacillin inhibiting the tubular secretion of tazobactam (7), a drug-drug interaction absent with ceftolozane (8). In the studies described herein, it is important that the simulated concentrations of tazobactam reflect the difference seen due to the tubular secretion of tazobactam and that there was essentially no difference in the time course of antibacterial effect between the piperacillin-tazobactam dosing of 4.5 g every 6 h and the ceftolozane-tazobactam dosing regimen of 750-375 mg every 8 h or the regimens with greater doses.
It should also be recognized that the work described herein involved a single strain that was a genetic construct. Additional work using clinical isolates expressing a full range of drug resistance determinants, including the expression of multiple β-lactamase enzymes, drug efflux, and porin mutations, may be warranted. In addition, similar studies with other target pathogens, such as P. aeruginosa and Klebsiella pneumoniae, appear to be warranted.
In conclusion, we successfully discriminated ceftolozane-tazobactam dosing regimens that resulted in drug resistance amplification from those that did not. Ceftolozane-tazobactam dosing regimens of 750-375 mg and greater prevented resistance amplification and, by day 4, drove the entire bacterial population toward extinction. These results were similar to those for piperacillin-tazobactam infused at 4.5 g every 6 h, which was used as a positive control. These data will aid in the selection of optimal ceftolozane-tazobactam dosing regimens to minimize the potential for on-therapy drug resistance amplification.
ACKNOWLEDGMENTS
We thank Kim A. Charpentier from ICPD (Latham, NY) and Lalitagauri M. Deshpande from JMI Laboratories (North Liberty, IA) for manuscript assistance and technical support.
This study was sponsored by Cubist Pharmaceuticals Inc., Lexington, MA. The Institute for Clinical Pharmacodynamics (B.V., S.M.B., A.F., and P.G.A.) has received research support from Achaogen, Astellas, AstraZeneca, Basilea Pharmaceutica, Bayer HealthCare, Bristol-Meyers Squibb, Cempra Pharmaceuticals, Cerexa, Cubist Pharmaceuticals, Durata Pharmaceuticals, Fedora Pharmaceuticals, Forest Research Institute, Furiex Pharmaceuticals, GlaxoSmithKline, Meiji Seika Pharma, Nabriva Therapeutics, Nimbus, Pfizer, PolyMedix, Rib-X, Roche Bioscience, Rempex Pharmaceuticals, Rib-X Pharmaceuticals, Rock Therapeutics, Tetraphase Pharmaceuticals, and The Medicines Company. JMI Laboratories, Inc. (R.E.M., M.C., and R.N.J.), received research and educational grants from 2009 to 2012 from the following entities: American Proficiency Institute (API), Anacor, Astellas, AstraZeneca, Bayer, Basilea Pharmaceuticals, Cempra/Forest, Cerexa, Contrafect, Cubist, Daiichi, Dipexium, Durata, Enanta, Fedora, Furiex, GlaxoSmithKline, Johnson & Johnson (Ortho McNeil), LegoChem Biosciences Inc., Meiji Seika Kaisha, Merck, Nabriva, Novartis, Pfizer (Wyeth), Rempex, Rib-X Pharmaceuticals, Seachaid, Shionogi, The Medicines Company, Theravance, ThermoFisher, and some other corporations. Some JMI employees are advisors/consultants for Astellas, Cubist, Pfizer, Cempra, Cerexa/Forest, J&J, and Theravance. In regard to speakers' bureaus and stock options, there are none to declare. L.F. and J.S. are both employees and stockholders of Cubist Pharmaceuticals.
Footnotes
Published ahead of print 17 June 2013
REFERENCES
- 1.Tam VH, Louie A, Deziel MR, Weiguo L, Leary R, Drusano GL. 2005. Bacterial-population responses to drug-selective pressure: examination of garenoxacin's effect on Pseudomonas aeruginosa. J. Infect. Dis. 192:420–428 [DOI] [PubMed] [Google Scholar]
- 2.VanScoy B, Mendes RE, Nicasio AM, Castanheira M, Bulik CC, Okusanya OO, Bhavnani SM, Forrest A, Jones RN, Friedrich LV, Steenbergen JN, Ambrose PG. 2013. Pharmacokinetics-pharmacodynamics of tazobactam in combination with ceftolozane in an in vitro infection model. Antimicrob. Agents Chemother. 57:2809–2814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Clinical and Laboratory Standards Institute 2012. Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically. Approved standard—. 9th ed. CLSI document M07-A9 Clinical and Laboratory Standards Institute, Wayne, PA [Google Scholar]
- 4.Louie A, Deziel MR, Liu WL, Drusano GL. 2007. Impact of resistance selection and mutant growth fitness on the relative efficacies of streptomycin and levofloxacin for plague therapy. Antimicrob. Agents Chemother. 51:2661–2667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schulz H. 2003. Contemporary medicine as presented by its practitioners themselves, Leipzig, 1923:217–250 Nonlinearity Biol. Toxicol. Med. 1:295–318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chandorkar G, Huntington J, Parsons T, Gotfried M, Rodvold KA, Umeh O. 2012. Intrapulmonary penetration of ceftolozane-tazobactam and piperacillin-tazobactam in healthy adult subjects. J. Antimicrob. Chemother. 67:2463–2469 [DOI] [PubMed] [Google Scholar]
- 7.Komuro M, Maeda T, Kakuo H, Matsushita H, Shimada J. 1994. Inhibition of the renal excretion of tazobactam by piperacillin. J. Antimicrob. Chemother. 34:555–564 [DOI] [PubMed] [Google Scholar]
- 8.Miller B, Hershberger E, Benzinger D, Trinh M, Friedland I. 2012. Pharmacokinetics and safety of intravenous ceftolozane-tazobactam in healthy adult subjects following single and multiple ascending doses. Antimicrob. Agents Chemother. 56:3086–3091 [DOI] [PMC free article] [PubMed] [Google Scholar]