

Abstract
As eukaryotes, fungi possess relatively few molecules sufficiently unique from mammalian cell components to be used as drug targets. Consequently, most current antifungals have significant host cell toxicity. Primary fungal pathogens (e.g., Histoplasma) are of particular concern, as few antifungals are effective in treating them. To identify additional antifungal candidates for the treatment of histoplasmosis, we developed a high-throughput platform for monitoring Histoplasma growth and employed it in a phenotypic screen of 3,600 commercially available compounds. Seven hit compounds that inhibited Histoplasma yeast growth were identified. Compound 41F5 has fungistatic activity against Histoplasma yeast at micromolar concentrations, with a 50% inhibitory concentration (IC50) of 0.87 μM, and has the greatest selectivity for yeast (at least 62-fold) relative to host cells. Structurally, 41F5 consists of an aminothiazole core with an alicyclic substituent at the 2-position and an aromatic substituent at the 5-position. 41F5 inhibits Histoplasma growth in liquid culture and similarly inhibits yeast cells within macrophages, the actual host environment of this fungal pathogen during infection. Importantly, 41F5 protects infected host cells from Histoplasma-induced macrophage death, making this aminothiazole hit compound an excellent candidate for development as an antifungal for Histoplasma infections.
INTRODUCTION
Systemic fungal infections represent an emerging threat to human health due to the increasing population of individuals with immune function deficiencies (e.g., persons with AIDS and organ transplant recipients). Furthermore, once established, fungal infections are notoriously difficult to clear, resulting in high mortality rates. At a cost of $15,000 to $54,000 per patient (1), fungal infections represent an annual nationwide health burden in the millions of dollars. In contrast to the opportunistic pathogens that cause disease only when host immunity is severely impaired, endemic dimorphic fungal pathogens, such as Histoplasma capsulatum, infect and cause disease even in immunocompetent individuals.
The antifungal drug challenge results from the shared eukaryotic nature of fungi and host cells and the difficulty of identifying factors sufficiently unique from their host that can be exploited as drug targets. Clinically, the main antifungal classes available for treatment of invasive fungal infections are the polyenes, the azoles, and the echinocandins. The polyene amphotericin B (AmB) interacts with the fungal sterol ergosterol, causing membrane disruption and leading to fungal cell death (2, 3). AmB also binds other sterols, including mammalian cholesterol, although with lower affinity (4). This cross-targeting of host sterols results in significant host toxicity; however, liposomal formulations of AmB have increased both the tolerability and efficacy of this drug for fungal disease (5). The azoles also affect fungal sterols but target the biosynthesis of ergosterol rather than the end product itself. The azoles (e.g., fluconazole [FLC], ketoconazole, itraconazole, and voriconazole) specifically inhibit the cytochrome P450 enzyme CYP51, which catalyzes the 14α-demethylation of lanosterol, an essential step in the production of sterols (6, 7). However, simultaneous targeting of host P450 enzymes by azoles poses a significant and demonstrated risk of detrimental side effects to the host (8, 9). The third, and most recently developed, family of antifungals for invasive mycoses is the echinocandin class, which inhibits production of fungal cell wall β-1,3-glucan. Although the echinocandins are better tolerated than the polyenes and the azoles due to the uniqueness of β-1,3-glucan for fungi, the echinocandins are ineffective for the treatment of the fungal pathogens Cryptococcus neoformans and Histoplasma capsulatum (10–12).
Histoplasma capsulatum, the etiologic agent of histoplasmosis, is endemic to the Ohio and Mississippi River valleys, where more than 80% of residents exhibit evidence of prior infection (13). As one of the dimorphic fungal pathogens, Histoplasma grows as a saprobic filamentous mold form in the environment and as a parasitic yeast form during mammalian infection. These two phases not only are morphologically quite distinct but also produce many phase-specific gene products (14–17). Lung infection can range from subclinical to severe respiratory disease depending upon the host's immune status and the inoculum (18). Disseminated histoplasmosis, the most deadly form of disease, is nearly universal among infections of immunocompromised individuals. However, the disease is not restricted to immunocompromised individuals, with the majority of hospitalizations due to histoplasmosis occurring in individuals without other comorbidities (19). Within the mammalian host, Histoplasma yeast cells infect and survive within phagocytes (e.g., alveolar macrophages). This intracellular localization of yeast cells during infection presents an additional permeability barrier to antifungal drug effectiveness. Replication of the yeast cells ultimately leads to destruction of macrophages and spread of the fungus to neighboring phagocytes.
The current clinical recommendation for management of histoplasmosis includes the use of the cytotoxic AmB and azole drugs (20), the combination of which depends on the severity of disease. Histoplasmosis requires prolonged antifungal therapy, typically 12 weeks to 24 months (20), significantly raising the cost of treatment and the potential negative side effects of current antifungals. The high rate of toxicity underscores the importance of developing new antifungal drugs, particularly those effective against intramacrophage yeast cells.
In the current study, we developed a high-throughput assay for efficiently monitoring Histoplasma yeast growth and utilized it to phenotypically screen a library of 3,600 commercially available compounds selected for having structural similarity to purines or purine analog scaffolds. From this purinome-focused chemical library, we identified a set of hit compounds with antifungal activity against Histoplasma yeasts and with minimal toxicity to host cells. The most potent and most selective compound, an aminothiazole, is fungistatic against Histoplasma yeast cells in culture and, importantly, also inhibits growth of intramacrophage yeast cells. Treatment of infected macrophages with the aminothiazole antifungal compound was protective against Histoplasma-induced macrophage death, demonstrating its potential utility as a therapeutic option for histoplasmosis.
MATERIALS AND METHODS
Fungal strains and culture.
Histoplasma capsulatum strains used in this study included wild-type strain G217B (ATCC 26032), G186A (ATCC 26027), and the uracil auxotroph derivative WU15 (21). Histoplasma yeast strains were grown in Histoplasma-macrophage medium (HMM) (22) at 37°C with constant agitation. For growth of WU15, HMM was supplemented with uracil (100 μg/ml). Histoplasma strain OSU76 was created by transformation of WU15 with the NotI-linearized pCR540 plasmid containing the tandem dimer tdTomato (Clontech) red fluorescent protein (RFP), driven by the constitutive Histoplasma histone 2B promoter. The growth rate of OSU76 compared to that of wild-type strain G217B was verified by turbidity measurements (absorbance at 595 nm). For single-cell fluorescence measurements, Histoplasma yeast cells were cultured for 48 h in HMM, HMM with 10% fetal bovine serum (FBS), 3M medium (a defined minimal medium supplemented with 0.7 mM l-cystine to support Histoplasma growth) (22), or RPMI 1640 medium supplemented with 0.7 mM l-cystine.
To determine the sensitivity of the microtiter-based growth assay, growth of replicate cultures of yeast at 4 days, either by turbidity or red fluorescent protein (RFP) readings, was used to derive the overall variation. Statistical power calculations were performed for 5%, 10%, 15%, 20%, and 25% reductions in yeast growth by using the SYSTAT (version 13) statistical calculation package (Systat Software, Inc.). The assay detection limit was defined as the minimum decrease in growth that provides 95% confidence (i.e., power level) in differences between two samples (100% growth compared to reduced growth).
For determination of the MIC for different fungal strains, microdilution assays were performed according to Clinical and Laboratory Standards Institute (CLSI) methods for yeasts and filamentous fungi (CLSI M27-A3 and M38-A2, respectively) (23, 52, 53). Aspergillus fumigatus conidia (strains Af293 and OSUAf1) were used to inoculate RPMI 1640 medium at 5 × 104 conidia/ml. After 72 h of growth at 37°C, wells were visually scored for the presence or absence of mold growth. Candida albicans (strain SC5314) and Cryptococcus neoformans (strain TDY271) yeast cells were inoculated into RPMI 1640 medium at 1 × 103 CFU/ml and incubated for 72 h. Yeast growth was measured by culture turbidity (absorbance at 595 nm). For MIC determination of Blastomyces dermatitidis (strains 26199 and T5319) and Histoplasma (strains G217B and G186A), the CLSI microdilution assay methodology (CLSI M27-A3) (52) was modified to accommodate yeast growth. Specifically, Blastomyces and Histoplasma yeast cells were enumerated by using a hemacytometer and used to inoculate HMM at 1 × 106 yeast cells/ml in 96-well microtiter plates, since consistent yeast growth requires higher inoculums and medium additives. Yeast growth was measured by the absorbance at 595 nm after 4 days. MICs were defined as the lowest concentration of drug that resulted in at least a 60% reduction in turbidity compared to the drug-free control.
Purinome-focused chemical library.
The purinome-focused library was constructed as described previously (24). Briefly, the increase in solved crystal structures for purine-utilizing proteins, their molecular folds, and the orientations of purines when bound enabled the use of a cheminformatic approach to develop a set of descriptors for compounds that would likely bind to proteins that utilize purine cofactors. Eighty-five parameters and filters (e.g., hydrogen bond donors and acceptors, number of rings, topological torsions, and characteristic purine-binding motifs) were optimized to allow faithful identification of 40 known inhibitors of purine-binding proteins. These parameters were then used to query approximately 2.4 million commercially available compounds from 19 vendors to select approximately 150,000 with potential interactions with purine-binding proteins. The descriptors were weighted and compounds were clustered by using Sorensen-type coefficients as similarity criteria between a reference and test molecule to select approximately 10,000 compounds. These final compounds were manually inspected for desired physiochemical properties to reduce this set to the final library screened.
High-throughput monitoring of Histoplasma growth.
Yeast cells were grown in 96-well microtiter plates by modification of protocols reported previously for Saccharomyces growth in microtiter plates (25). OSU76 yeast cells at the exponential phase of growth were enumerated by using a hemacytometer and added to microtiter plates at 2.5 × 105 yeast cells per well (for library screening) or 1 × 105 yeast cells per well (for follow-up screening) (2.5 × 106 yeast cells/ml and 1 × 106 yeast cells/ml, respectively) in HMM supplemented with 10 μg/ml tetracycline and 10 μg/ml gentamicin. Plates were incubated at 37°C in 5% CO2–95% air and were mixed daily on a microplate mixer to improve aeration. Growth was monitored by daily measurements of turbidity (absorbance at 595 nm) and RFP fluorescence (530-nm excitation and 590-nm emission wavelengths) on a Synergy2 plate reader (BioTek). For linear regression and computation of 50% inhibitory concentration (IC50) values, yeast growth at 4 days (93 to 102 h) was used. All tests were performed on three biological replicates, and differences were compared by using Student's t test. For determination of assay efficacy, the known antifungal drug AmB was added at concentrations from 0.003 μM to 1.7 μM. For library screening, compounds were added to yeast at a concentration of 10 μM. Compounds that inhibited yeast growth at this concentration were rescreened against yeast cells at concentrations from 0.0095 μM to 10 μM to establish an IC50 for yeast cells.
Evaluation of mammalian cell toxicity.
Mammalian cell lines used include P388D1 macrophage cells engineered to express the lacZ transgene (P388D1-lacZ cells) (26), J774A.1 macrophages (ATCC TIB-67), and the human hepatocyte cell line HepG2 (ATCC HB-8065). For mammalian cell culture, cells were maintained at 37°C in 5% CO2–95% air in basal medium supplemented with 10% FBS and 2 mM l-glutamine. Basal media were Ham's F-12 medium for P388D1-lacZ cells, Dulbecco's modified Eagle's medium (DMEM) for J774A.1 cells, and minimum essential medium (MEM) for HepG2 cells. Mammalian cells were incubated with antifungal inhibitors at a concentration range from 2.5 μM to 80 μM. After 48 h, cells were quantified either by measuring the macrophage-expressed lacZ activity (P388D1-lacZ cells [26]) or by measuring cell mass by crystal violet staining (J774A.1 and HepG2 cells) and quantification (27). For determination of lacZ activity, cells were lysed with 0.5% Triton X-100, and conversion of the LacZ substrate o-nitrophenyl-β-d-galactopyranoside (ONPG) (500 μg/ml) was measured spectrophotometrically. To quantify crystal violet staining, cells were fixed in 3% paraformaldehyde and stained with 0.1% crystal violet. Residual stain was removed by washing cells with water, and the retained crystal violet was solubilized in 10% acetic acid for 20 min with agitation. Crystal violet dye was quantified by measurement of the absorbance at 590 nm and normalized to values for untreated samples. Assays were performed on at least three replicate samples, and differences were tested for statistical significance by Student's t test.
Fungistatic/fungicidal testing.
Yeast suspensions of OSU76 were exposed to 41F5, AmB, or FLC at inhibitory concentrations or to dimethyl sulfoxide (DMSO) for 2, 8, or 24 h. The DMSO concentration used was 0.25%, which is equivalent to the maximum concentration of solvent present in drug-treated samples. This concentration is noninhibitory to Histoplasma yeast growth (data not shown). Following drug exposure, 10-fold serial dilutions of the yeast suspension were plated onto solid HMM to assess yeast viability. Plates were incubated at 37°C in 95% air–5% CO2 until colonies developed, and the colonies were imaged and CFU were enumerated by using an AlphaImager system (Cell Biosciences).
Intramacrophage yeast growth and viability.
P388D1-lacZ macrophages were infected with Histoplasma yeast cells, and intramacrophage yeast growth was measured by RFP fluorescence or CFU platings. Macrophages were seeded into wells of 96-well microtiter plates (for fluorescence measurements) at 2 × 104 cells per well or into wells of a 24-well plate (for CFU plating) at a concentration of 1 × 105 cells per well for 24 h prior to infection. For fluorescence measurements, OSU76 yeast cells were added to macrophages at a multiplicity of infection (MOI) of 2.5 yeast cells per macrophage in HMM buffered with 25 mM bicarbonate and supplemented with 10% FBS (cHMM-M). After 4 h, the medium was replaced with fresh cHMM-M to remove extracellular yeast cells, and the inhibitor 41F5 (or equivalent DMSO) was added. Simultaneously, yeast cells in the absence of macrophages were also incubated with drugs in cHMM-M under identical conditions. Yeast growth was monitored daily for 4 days by measuring the absorbance at 595 nm (for yeast alone) and RFP fluorescence (for intramacrophage yeast). At 4 days postinfection, wells were also assayed for remaining macrophages by using the LacZ assay, as described above. Intramacrophage yeast CFU counts were determined as described previously (28). Briefly, macrophages were infected with yeasts at a multiplicity of infection (MOI) of 0.5 yeast cells per macrophage in cHMM-M. At 2 h postinfection, extracellular yeast cells were removed by washing with phosphate-buffered saline (PBS) and replacing the medium with fresh cHMM-M. At the indicated time points postinfection, macrophages were lysed in Tris-buffered H2O, and lysate dilutions were plated onto HMM plates and incubated until individual colonies developed.
Fluorescence microscopy.
For visualization of intramacrophage yeast localization and growth, macrophages were plated onto 12-mm acid-washed coverslips and infected as described above. At the indicated time points, coverslips were washed with PBS and fixed in 3% paraformaldehyde for 20 min. Fixed samples were permeabilized and blocked in 0.2% saponin–3% bovine serum albumin (BSA). For determination of the intracellular localization of yeasts, infected macrophages were stained with antibody to mouse lysosome-associated membrane protein 1 (LAMP-1) (hybridoma 1D4B, developed by J. T. August, was obtained from the Developmental Studies Hybridoma Bank, developed under the auspices of the NICHD and maintained by The University of Iowa Department of Biology, Iowa City, IA), followed by anti-rat fluorescein isothiocyanate (FITC) (Thermo Scientific). To visualize intramacrophage replication of yeast cells in the presence of the inhibitor 41F5, macrophages were fixed at 2 h and 3 days of infection in the presence of drug. In all fixed samples, yeast cells were stained with Uvitex 3BSA in PBS (Ciba-Geigy). Samples were observed on a Nikon Eclipse Ti epifluorescence microscope equipped with a 40× oil immersion objective with differential interference contrast (DIC) optics. Images were acquired by using a CoolSnap HQ2 camera. z-stacks of Uvitex-fluorescent images were deconvolved by using NIS-Elements AR 4.0 imaging software (Nikon).
RESULTS
Development of a high-throughput method for monitoring Histoplasma growth.
To facilitate screening of a large number of compounds for antifungal activity against Histoplasma yeast, we developed a microtiter-based assay for yeast growth. We created a transgenic strain of Histoplasma (OSU76) that expresses RFP to provide fluorescence-based monitoring of yeast growth. RFP production in OSU76 is driven by the constitutive Histoplasma histone 2B promoter so that RFP is proportional to the cell number. Monitoring of growth by fluorescence provides two advantages over relying on culture turbidity alone. First, the greater dynamic range of RFP fluorescence increases the sensitivity of the assay. Fluorescence readings can detect as little as a 10% reduction in growth with 95% confidence, compared to the 25% reduction in growth needed for 95% confidence with 595 nm (OD595) readings in microtiter plates. Second, RFP measurement enables nondestructive monitoring of yeast growth within macrophages, thereby allowing yeast growth to be measured over time within a single monolayer of cells (see below). As Histoplasma yeast cells reside in macrophages during infection, intramacrophage growth of Histoplasma is the most physiologically relevant condition and the most accurate in vitro model for assessing the effectiveness of potential antifungal drugs.
We validated that OSU76 exhibits growth identical to wild-type growth in liquid medium and within macrophages. In flask-based liquid cultures, OSU76 showed normal growth kinetics compared to the wild-type G217B clinical isolate (Fig. 1A) and exhibited identical intramacrophage growth (see Fig. 6A and below). To determine if yeast population density and RFP fluorescence measurements of growth are correlated in the microtiter plate format, OSU76 or wild-type G217B yeast suspensions were added to wells at concentrations from 6.25 × 105 yeast cells/ml to 2.5 × 106 yeast cells/ml. Growth at 37°C was measured daily after inoculation either as culture turbidity (absorbance at 595 nm) or by RFP fluorescence. Consistent with flask-based Histoplasma yeast growth, culture turbidity in 96-well plates increased steadily with time, reflecting an increase in yeast cell numbers (Fig. 1B). RFP fluorescence similarly increased over time (Fig. 1C). Growth of OSU76 cells, as measured by absorbance and RFP fluorescence, was highly correlated (R2 = 0.96 to 0.97) over a range of yeast concentrations (Fig. 1D), indicating that both methods of measurement were suitable to monitor yeast growth in a microtiter plate. As plate readings reflect the overall fluorescence of the population, we also examined the distribution of RFP fluorescence of individual yeast cells by microscopy of cells grown under different conditions (Fig. 1E). Consistent with population-level fluorescence, the average RFP-yeast fluorescence of yeast cells grown in HMM was 24- ± 6-fold above that of RFP-negative yeast. Yeast fluorescence was tested in a medium reflecting growth in serum (HMM plus 10% FBS), a defined minimal medium (3 M) that may more closely reflect the composition of the phagosomal lumen, and RPMI medium, a medium commonly used in antimicrobial testing (23). The distributions of RFP fluorescence were highly similar, with RPMI medium showing a slightly higher mean fluorescence.
Fig 1.

Development of a fluorescence microtiter plate-based assay to monitor Histoplasma yeast growth. (A) Histoplasma G217B and OSU76 yeast growth in flasks. Growth was monitored over time by measurements of culture turbidity (absorbance at 595 nm). (B to D) Histoplasma yeast growth in microtiter plates. (B and C) Yeast cells were added to 96-well microtiter plates in a volume of 100 μl at 2.5 × 106, 1.25 × 106, or 6.25 × 105 yeast cells per ml, and growth was monitored daily by measuring the absorbance at 595 nm (B) or RFP fluorescence (C). (D) Correlation of Histoplasma yeast growth measured by RFP with growth determined by the absorbance at 595 nm. Data points represent 2.5 × 106, 1.25 × 106, or 6.25 × 105 yeast cells per ml, and a line of best fit shows correlation between the measurement methods (R2 = 0.96, 0.96, and 0.97, respectively). (E) Distribution of RFP fluorescence of individual OSU76 yeast cells. Fluorescence of yeasts was measured by microscopy following growth in HMM, HMM plus 10% serum, 3M minimal medium, or RPMI-based medium. Data represent the means (horizontal bars) ± standard deviations (boxes) and ranges (vertical lines; n = 100) of RFP-fluorescent yeast cells (OSU76) compared to RFP-negative yeast cells (G217B).
Fig 6.
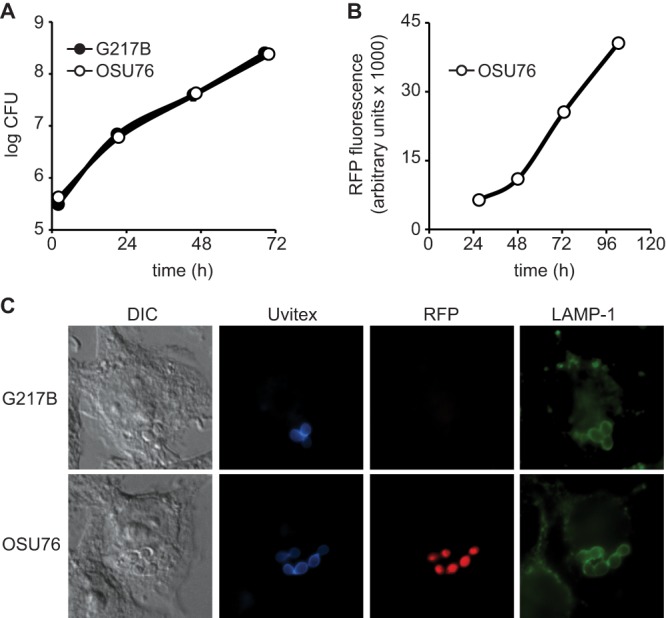
RFP-expressing Histoplasma yeast cells replicate and traffic in macrophages similarly to wild-type cells. (A and B) Intramacrophage growth of Histoplasma yeast cells. Yeast growth was measured by quantitative plating of macrophage lysates for viable yeast CFU (A) or measurement of RFP fluorescence (B). P388D1 macrophages were infected with Histoplasma G217B or OSU76 yeast cells at an MOI of 0.5:1 (A) or 2.5:1 (B) (yeast cells to macrophages). At 2, 21, 46, and 69 h postinfection, macrophages were lysed, and yeast cells were recovered and plated onto solid medium to determine CFU (A). At 27, 48, 73, and 103 h postinfection, RFP fluorescence from macrophages infected with OSU76 yeast cells was determined (B). Data points represent means ± standard deviations of yeast CFU counts or fluorescence (n = 3 replicates). (C) Characterization of the intracellular Histoplasma-containing compartment. At 2 h postinfection, macrophages were fixed and stained. Infected macrophages were visualized by differential interference contrast (DIC) microscopy and fluorescent markers for yeasts (Uvitex), RFP, and lysosome-associated membrane protein 1 (LAMP-1). Representative images are shown.
To validate that this high-throughput method could effectively detect yeast growth inhibition, we exposed yeast cells to increasing concentrations of the known antifungals amphotericin B (AmB) and fluconazole (FLC). From yeast growth based on both the absorbance at 595 nm and RFP fluorescence at 4 days, we computed IC50 levels for AmB and FLC. AmB demonstrated an IC50 of 0.09 μM by absorbance (595 nm) measurements and an IC50 of 0.08 μM by RFP measurements (Fig. 2A). The MIC for both methods of measurement was 0.21 μM, which is similar to what was reported previously for AmB against Histoplasma yeast in vitro (29). For FLC, IC50 values of 3.07 μM and 6.37 μM were calculated with OD595 and RFP fluorescence-based assays, respectively (data not shown). To determine if this high-throughput assay has sufficient power to discriminate growth inhibition, we calculated the Z′ factor (30) for AmB and FLC. For AmB concentrations of 0.22 μM and higher, the Z′ factor was ≥0.79. For FLC concentrations of 6.5 μM and higher, the Z′ factor was at least 0.73. These data, together with the assay sensitivity determinations described above, indicate that the microtiter plate-based growth assay is suitable for identifying compounds that inhibit Histoplasma growth.
Fig 2.
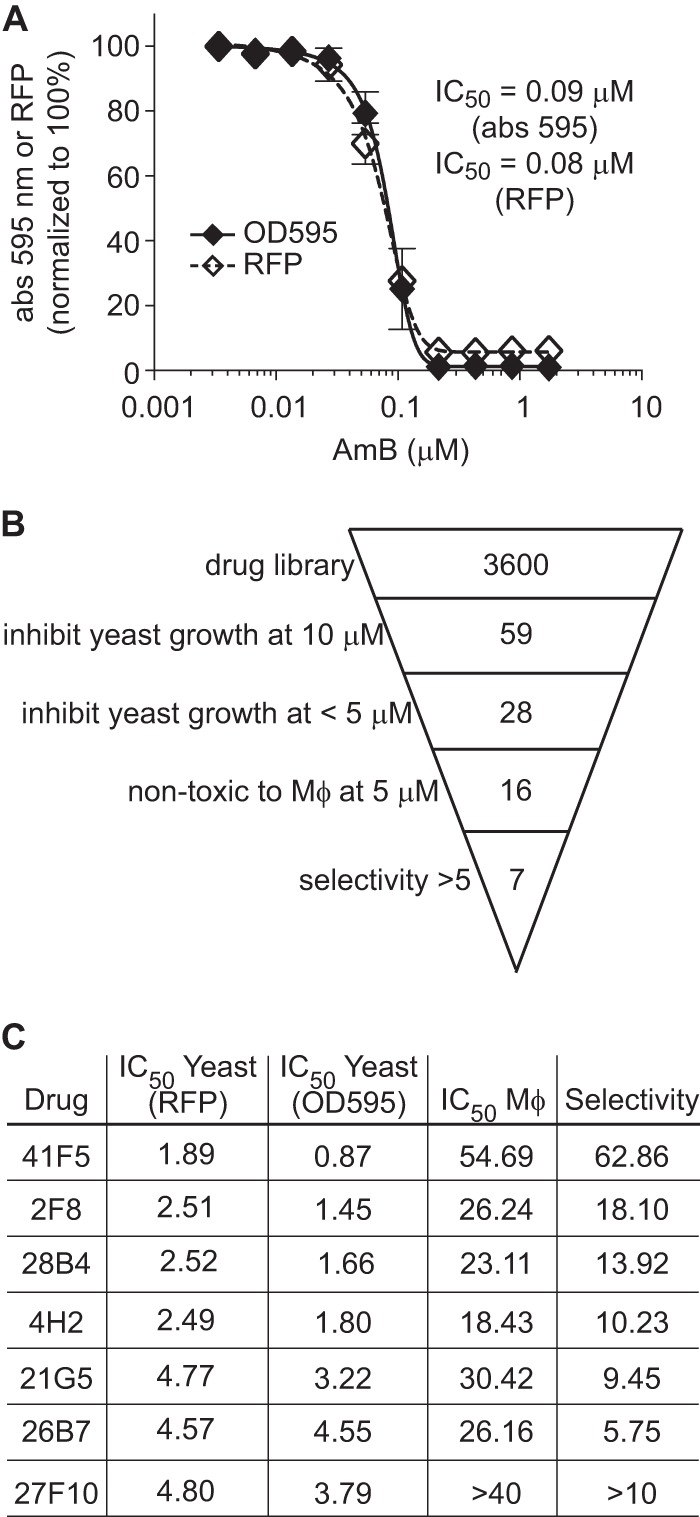
Identification of antifungal compounds from a purinome-focused chemical library. (A) Determination of the Z′ factor for microtiter plate-based assays of growth inhibition by measurements of culture turbidity (absorbance) and RFP fluorescence. OSU76 yeast cells were exposed to AmB at concentrations from 0.003 μM to 1.7 μM, and growth was measured at day 4 and normalized to growth of yeast in the absence of AmB. Values represent the means ± standard deviations of replicate cultures (n = 3). Data are representative of ≥3 independent experiments. (B) Overview of hit compound identification. A total of 3,600 compounds were screened for their ability to inhibit Histoplasma yeast growth by measurement of RFP fluorescence and absorbance at 595 nm. Compounds were initially screened at 10 μM. Dose-response tests further narrowed the 59 hits to 28, which were effective at concentrations of <5 μM. Macrophage (Mϕ) toxicity was determined by inhibition of P388D1-lacZ macrophage cells and identified 16 compounds that were nontoxic to host cells at 5 μM. Overall selectivity (yeast IC50 determined by the absorbance at 595 nm relative to the P388D1-lacZ IC50) was used to identify the top 7 hits with selectivity indices of >5. (C) Summary of IC50 and selectivity data for hit compounds. All concentrations are in μM.
Identification of antifungal hit compounds from a chemical library.
To identify new compounds that selectively inhibit Histoplasma growth, we used the high-throughput Histoplasma growth assay to screen a purinome-focused chemical library. An overview of the results is presented in Fig. 2B. The library of 3,600 compounds was selected from over 2.4 million commercially available compounds with structural similarity to purines or any known purine analog scaffold (library construction was defined in reference 24). Each compound in the library was added to Histoplasma yeast suspensions at a concentration of 10 μM (final DMSO concentration of 0.5%), and Histoplasma growth was tracked for 5 days by measurements of the absorbance at 595 nm and RFP fluorescence. Fifty-nine compounds that reduced yeast growth by at least 75% were identified (Fig. 2B). Dose-response growth curves were then performed with serial 2-fold dilutions of each of the 59 hit compounds, and the IC50 against Histoplasma yeast was calculated. From the IC50 curves, 28 compounds showed dose-dependent inhibition of yeast growth, with an IC50 of <5 μM. We used host cell toxicity as a secondary screen to identify compounds worth further investigation. To facilitate high-throughput determination of macrophage toxicity, we added hit compounds to cultures of LacZ-transgenic P388D1 macrophages, which produce β-galactosidase activity that is directly proportional to the number of macrophages present (26). Using macrophage replication as an indicator of nontoxicity, 16 of the 28 compounds were not toxic to macrophages at concentrations of 5 μM or lower, as determined by RFP fluorescence or by culture turbidity. Determination of the IC50 against macrophages for these 16 compounds allowed computation of overall yeast selectivity (ratio of the IC50 for yeasts compared to the IC50 for host cells). We identified 7 compounds that are at least 5-fold more toxic to yeast cells than to macrophages (Fig. 2C). Three of the seven compounds, 41F5, 2F8, and 4H2, contained a similar core structure, with an aminothiazole or aminothiophene with an aromatic ring substituent at the 5-position (Fig. 3). The structures of 41F5 and 2F8 both have an alicyclic substituent at the 2-position of the aminothiazole. Because 41F5 had the best selectivity for Histoplasma yeast cells compared to macrophages, this compound was identified as the top hit from our library screen and was prioritized for further characterization.
Fig 3.
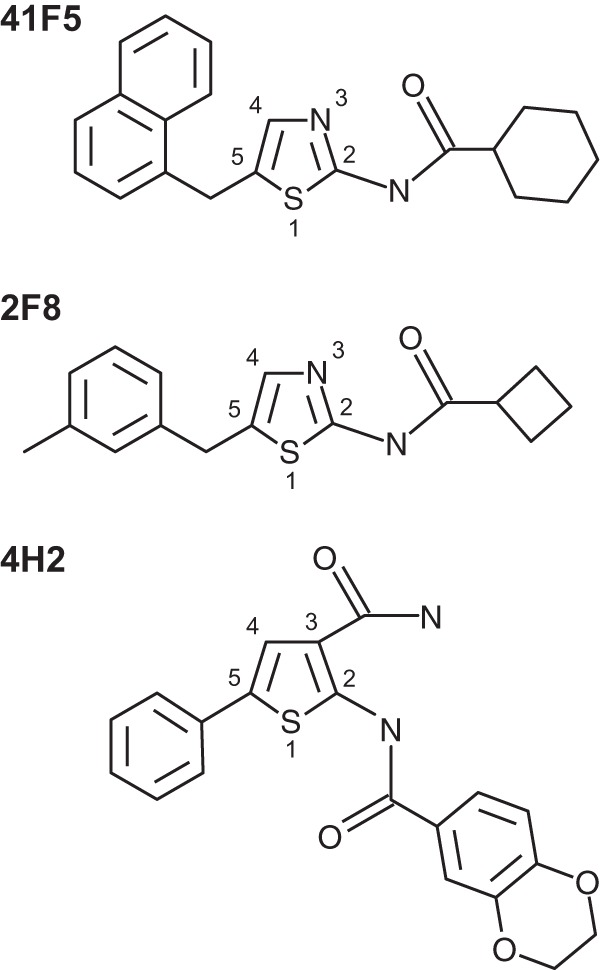
Hit compounds containing a similar core structure that inhibit Histoplasma yeasts. Compounds 41F5, 2F8, and 4H2 are shown with substituent numbering on the central thiazole (41F5 and 2F8) or thiophene (4H2).
Characterization of the antifungal properties of 41F5.
To verify the antifungal activity of 41F5, newly purchased and high-performance liquid chromatography (HPLC)-validated pure 41F5 was tested against Histoplasma yeast cells in culture. Similar to the primary library screen, 41F5 effectively inhibited Histoplasma yeast cells, as determined by culture turbidity (Fig. 4A) as well as RFP fluorescence (Fig. 4B). We sometimes observed elevated expression levels of RFP at subinhibitory concentrations. This phenomenon, whereby subinhibitory concentrations of cellular inhibitors increase transcription levels of genes compared to the levels under uninhibited conditions, was observed previously (31). This is independent of the promoter used for RFP expression, as RFP driven by the Histoplasma TEF1 promoter instead of the histone 2B promoter also exhibited this trend (data not shown). Since the elevated RFP expression level at subinhibitory concentrations decreases the accuracy of the linear regression (resulting in an underestimation of the compound's efficacy), we used absorbance measurements of culture turbidity for precise IC50 determinations and determination of the selectivity of the compounds (Fig. 2C). Multiple dose-response analyses with biological replicates determined the IC50 against Histoplasma OSU76 yeast cells to be 0.87 ± 0.16 μM (Fig. 4A). As further validation, the IC50 for 41F5 was also determined for Histoplasma wild-type strain G217B and was determined to be nearly identical to that for OSU76 (0.83 ± 0.08 μM) (Table 1).
Fig 4.

41F5 is fungistatic to Histoplasma yeast. (A and B) Determination of the IC50 of 41F5 against Histoplasma yeast cells using culture turbidity (absorbance at 595 nm) (A) or RFP fluorescence (B). Growth of OSU76 yeast cells was measured (absorbance or fluorescence) over 4 days of incubation with a 2-fold dilution series of compound 41F5. IC50 and MIC values for 41F5 were computed from linear regression of the data points using growth at 4 days. Data points represent the mean yeast growths ± standard deviations from three separate experiments. (C and D) Viability of Histoplasma yeast cells following treatment with antifungal inhibitors. OSU76 yeast suspensions were treated with 41F5 (3 μM), fluconazole (FLC) (6.86 μM), amphotericin B (AmB) (0.49 μM), or DMSO (vehicle control) (0.25%) for 2, 8, or 24 h, after which aliquots were removed and dilutions were plated onto solid medium to measure viable CFU. The amount of DMSO present in the vehicle control is equivalent to that present in the 41F5 treatments. Images in panel C are representative of three independent experiments. Panel D shows quantification of the viability of Histoplasma yeast cells after treatment with 41F5, FLC, and AmB for 2, 8, or 24 h. Data represent the mean viable CFU ± standard deviations. Statistically significant differences compared to the vehicle control (0.25% DMSO) are indicated by asterisks (ns, nonsignificant; ∗, P < 0.05; ∗∗, P < 0.01; ∗∗∗, P < 0.001).
Table 1.
MICs of 41F5 for different fungal pathogens
| Fungus (strain) | MIC (μM) |
|---|---|
| Candida albicans (SC531) | >40 |
| Aspergillus fumigatus (Af293) | >40 |
| Aspergillus fumigatus (OSUAf1) | >40 |
| Histoplasma capsulatum (G186A) | 1.25 |
| Histoplasma capsulatum (G217B) | 2 |
| Blastomyces dermatitidis (26199) | >40 |
| Blastomyces dermatitidis (T5319) | >40 |
| Cryptococcus neoformans (TDY271) | 1.25 |
To determine the type of antifungal activity of 41F5, we tested whether 41F5 inhibits yeast growth by direct killing of yeast cells or by inhibiting yeast growth and replication. Histoplasma yeast cells were treated with 41F5 for 2, 8, or 24 h, after which dilutions of yeast cells were plated onto solid medium to monitor yeast viability (Fig. 4C). As controls for fungicidal or fungistatic action, yeast cells were also treated with AmB and FLC at concentrations near their MIC (0.49 μM and 6.86 μM, respectively). DMSO was used at 0.25% for no-drug control treatments, which is equivalent to the amount of DMSO required to solubilize 41F5. Yeast cells treated with 41F5 showed no statistically significant loss of viability compared to the no-drug treatment at 2 and 8 h, similar to that for FLC treatment (Fig. 4C and D). Treatment with 41F5 was also not significantly different from FLC treatment. While not decreasing viability, treatment with 41F5 or with FLC prevented any increase in yeast CFU; at 24 h, yeast cells incubated without drug replicated about 8-fold, whereas 41F5 or FLC treatment resulted in CFU nearly identical to the starting numbers of yeast cells (107% and 97% of the initial CFU, respectively) (Fig. 4D). These results with 41F5 and FLC are in contrast with those for yeast cells treated with the fungicidal drug AmB, which reduced the number of viable yeast cells at all time points (Fig. 4C and D). Thus, 41F5 acts as a fungistatic inhibitor of Histoplasma yeast cells.
41F5 has inhibitory activity against a limited set of medically relevant fungi. To determine the spectrum of fungal pathogens susceptible to 41F5, we tested the compound for its ability to inhibit standard and clinical strains of Candida albicans, Aspergillus fumigatus, Cryptococcus neoformans, and Blastomyces dermatitidis as well as a second strain of Histoplasma (Table 1). 41F5 had no detectable activity against Candida albicans and Aspergillus fumigatus up to the maximal concentration tested (40 μM). 41F5 had antifungal activity against Cryptococcus neoformans with an MIC of 1 μM in RPMI medium. 41F5 was active (MIC of 1.25 μM) against a second strain of Histoplasma yeast (G186A) that is phylogenetically distinct from G217B. Surprisingly, 41F5 did not inhibit the growth of Blastomyces dermatitidis, a fungal species closely related to Histoplasma, even at the highest concentration tested (40 μM).
Determination of 41F5 toxicity to mammalian cells.
In the early phases of drug discovery, it is essential to assay compound toxicity to relevant host cells as well as general mammalian cell toxicity. Since macrophages are the host niche for Histoplasma during infection, we tested a graded series of 41F5 concentrations for toxicity to macrophage cells. Two macrophage lines were used: P388D1-lacZ cells (Fig. 5A) and J774A.1 cells (Fig. 5B). Concentrations of DMSO above 1% are inhibitory to P388D1-lacZ and J774A.1 macrophages, requiring comparison of drug-treated cells to cells treated with equivalent concentrations of the DMSO solvent. 41F5 cytotoxicities to both P388D1 (IC50 = 54.69 μM) and J774A.1 (IC50 = 74.65 μM) cells at 48 h were similar (Fig. 5A and B). In J774A.1 cells, 41F5 exhibited toxicity at 40 μM and 80 μM which was similar to that of the DMSO control, indicating that the actual IC50 values of 41F5 for these macrophages are higher than the computations from this assay. As an indicator of in vivo drug toxicity targets (i.e., the liver) (32), 41F5 was tested against the HepG2 liver epithelial cell line (Fig. 5C). 41F5 had no toxicity to HepG2 cells at 20 μM or lower. The 40 μM treatment caused some growth inhibition, and this toxicity was not due to DMSO, as the equivalent percentage of DMSO (1%) did not show a significant decrease in HepG2 cell numbers. The IC50 of 41F5 for HepG2 cells was 53.90 μM, closely resembling the IC50 for macrophage cells.
Fig 5.
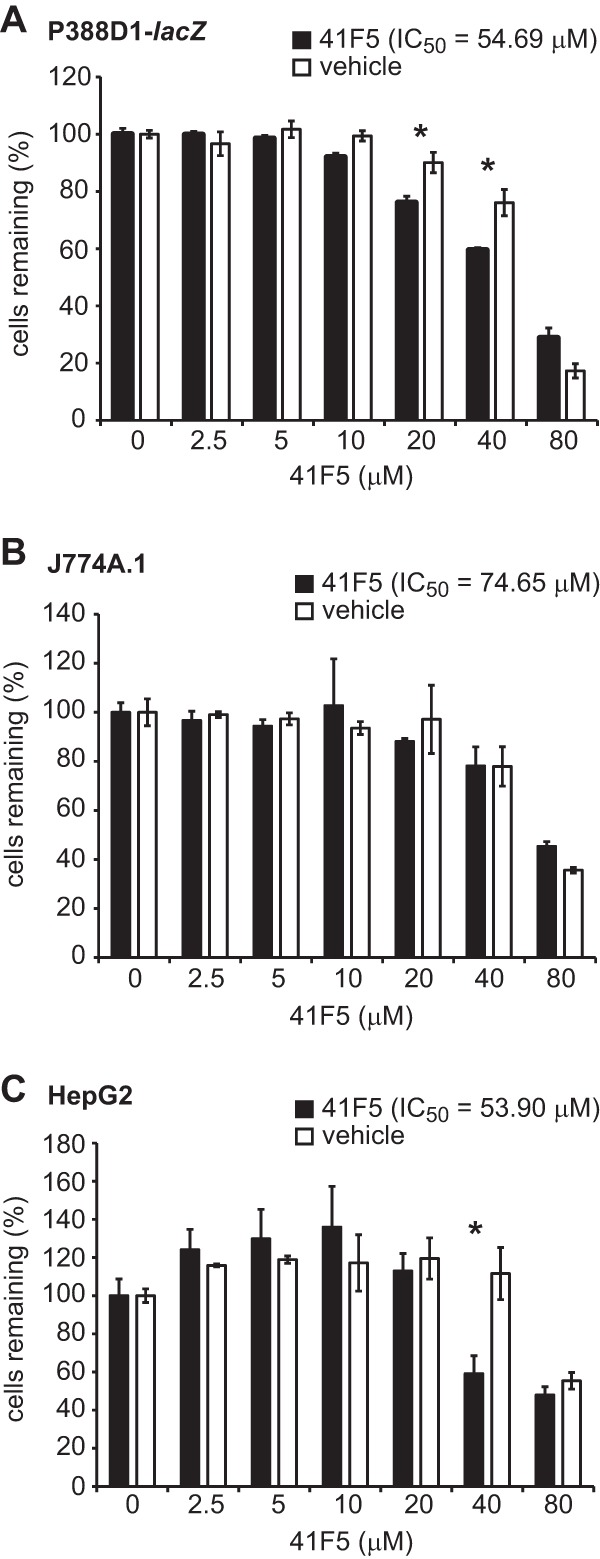
41F5 has low toxicity to mammalian cells at concentrations that inhibit Histoplasma yeast growth. Determination of the IC50 of 41F5 for P388D1-lacZ macrophages (A), J774A.1 macrophages (B), or HepG2 hepatocytes (C) was performed. Host cells were cultured with different concentrations of 41F5 or the vehicle-only control (DMSO). After 48 h, the remaining cells were quantified by using crystal violet and normalized to the value for cells cultured in the absence of an inhibitor. Data represent the means ± standard deviations (n = 3). Significant differences determined by Student's t test denote greater toxicity of 41F5 than of the DMSO control, indicated by asterisks (P < 0.05).
Effectiveness of 41F5 against intramacrophage yeast.
RFP-fluorescent yeast cells permit efficient and nondestructive monitoring of intramacrophage growth. To validate that RFP-fluorescent OSU76 yeast cells are identical to wild-type Histoplasma in their intramacrophage growth and infection characteristics, intracellular viability and trafficking of yeast cells were measured. Within macrophages, OSU76 and wild-type yeast cells had comparable survival and growth, as determined by quantitative plating of viable CFU over time (Fig. 6A). Due to the expression of RFP, OSU76 yeast cells permitted measurement of intramacrophage growth by RFP fluorescence, which closely parallels growth determined by CFU platings (Fig. 6B). Both intramacrophage OSU76 and wild-type yeast cells became localized to lysosome-associated membrane protein 1 (LAMP-1)-positive compartments at 2 h postinfection (Fig. 6C) and remained in these compartments throughout the duration of their intramacrophage life cycle (data not shown). In addition, we confirmed that OSU76 yeast cells produced the required Cbp1 (28, 33), Yps3 (34), and Sod3 (35) virulence factors (data not shown). Thus, OSU76 yeast cells accurately reflect wild-type Histoplasma yeast cells in their intracellular virulence characteristics and permit monitoring of intracellular growth by RFP fluorescence.
During infection, Histoplasma yeast cells enter and replicate within macrophages. Therefore, it is essential that potential drugs for the treatment of histoplasmosis can effectively inhibit intramacrophage yeast cells. To test whether 41F5 could target intramacrophage yeast cells, macrophages were infected with yeast for 4 h. This time is sufficient for >90% of the yeast cells to establish intracellular residence (data not shown), but it is not enough time for the yeast to replicate. After removal of noninternalized yeast cells, 41F5 or DMSO vehicle controls were added to the infected macrophages, and yeast growth was monitored over time. This procedure models the clinical situation whereby antifungals would be used to treat existing infections. Intramacrophage growth was monitored without destruction of host cells by measuring RFP fluorescence of Histoplasma yeast cells. A dose-response curve calculated for yeast growing within macrophages was compared to that of yeast growing in the absence of macrophages under identical growth conditions. The IC50 for yeast cells under these conditions is slightly elevated compared to that of yeast cells grown in liquid HMM culture due to the presence of serum in the medium required to maintain the macrophages; the addition of 10% serum increases the IC50 2-fold from 0.87 μM in the absence of serum to 1.69 μM in the presence of serum. Nonetheless, the IC50 of 41F5 is statistically indistinguishable for extracellular or intracellular yeast cells (Fig. 7A) under these growth conditions (1.69 μM and 1.77 μM, respectively). Thus, 41F5 effectively inhibits intramacrophage yeast cells, with no apparent reduction in potency.
Fig 7.

41F5 inhibits the growth of intramacrophage yeast. Macrophages were infected with OSU76 at an MOI of 2.5:1 (yeast cells to macrophages). After internalization (4 h postinfection), concentrations of 41F5 (from 0.07 μM to 20 μM) or equivalent solvent control (DMSO) (0.002% to 0.5%) were added. (A) 41F5 inhibition of growth of yeast cells in culture (absorbance at 595 nm) and intramacrophage yeast cells (RFP fluorescence) after 4 days. (B) Quantitation of 41F5-based protection of infected macrophages. Shown is the survival of P388D1-lacZ macrophages after 4 days of infection with treatment with 41F5 or equivalent DMSO. Macrophage survival (relative to that of uninfected macrophages) was quantified by measuring the remaining macrophage-expressed LacZ activity. Bars represent means ± standard deviations (n = 3). Data are representative of 3 independent experiments. Statistically significant macrophage protection determined by Student's t test is indicated by an asterisk (P < 0.05). (C) Visualization of intramacrophage yeast growth in the absence (DMSO only) or presence of 41F5 (2 μM or 5 μM) at 1 day or 3 days postinfection. Infected macrophages (DIC image) and Histoplasma yeast cells (blue) were visualized by microscopy after staining with the fluorescent fungal cell wall stain Uvitex 3BSA. Representative images are shown.
To determine whether 41F5-based inhibition of Histoplasma yeast can protect host macrophages, survival of infected macrophages was determined following treatment with 41F5. P388D1-lacZ macrophages were infected with Histoplasma yeast cells at an MOI of 2.5:1 (yeast cells to macrophages), and following infection, the macrophages were treated with a graded concentration series of 41F5. Infection of P388D1-lacZ monolayers with Histoplasma yeast at an MOI of as low as 0.25:1 (yeast cells to macrophages) results in significant loss of the host cell monolayer within 4 days of infection (26). Following 4 days of coincubation, surviving macrophages were quantified by measuring the remaining macrophage-produced β-galactosidase activity (Fig. 7B). At subinhibitory concentrations of 41F5 (less than 2 μM), Histoplasma yeast cells caused the death of at least 70% of macrophages. Treatment of macrophages with larger amounts of 41F5 had a protective effect on macrophage viability, with nearly 100% survival of macrophages at concentrations of 41F5 above 2.5 μM. To demonstrate that the protective effect on macrophage viability was due to 41F5-dependent inhibition of yeast growth, we visualized intracellular yeast populations by microscopy (Fig. 7C). On the first day of infection, intracellular yeast cells were present in all treatment groups. At 3 days postinfection, in the absence of 41F5, intracellular yeast cells had extensively proliferated, with the macrophage cytosol essentially filled with fungal cells. In contrast, treatment of infected macrophages with 2 μM or 5 μM 41F5 effectively prevented yeast replication. At 2 μM (less than the IC90 for intracellular yeast cells), some yeast replication was seen, but at 5 μM (above the MIC), there was virtually no yeast replication. Thus, 41F5 is a candidate antifungal that is active against intramacrophage Histoplasma cells and protects host cells from Histoplasma-induced death.
DISCUSSION
Despite regions of endemicity with a high prevalence of Histoplasma capsulatum and this pathogen's ability to successfully infect individuals irrespective of their immune status, it has been over 20 years since a new antifungal class has been developed that is effective against histoplasmosis. The currently recommended treatment options for histoplasmosis, AmB and itraconazole (20), are poorly tolerated, with a >10% risk that treatment with AmB and itraconazole must be discontinued due to drug complications (36). In the present study, we identified new hit compounds that can inhibit Histoplasma growth with limited toxicity to host cells. The development of a high-throughput microtiter plate-based growth assay, optionally using fluorescence of a transgenic RFP-expressing Histoplasma strain, facilitated efficient screening of a compound library.
Our efforts focused solely on the yeast form of Histoplasma, since this is the form present during mammalian infection and the form that causes disease. Investigation of the clinically appropriate form is essential, as highlighted by the lack of effectiveness of echinocandins against Histoplasma. Early studies indicated the echinocandins were potent inhibitors of Histoplasma, but those studies were performed by using the nonpathogenic mycelial form (37, 38). However, subsequent testing of the yeast form showed that the echinocandins were relatively ineffective against Histoplasma (10, 39–41). This developmental stage-specific resistance is not unique to Histoplasma, as different forms of Pneumocystis also exhibit different susceptibilities to β-glucan synthase inhibitors (42). In addition to the use of the pertinent form of the pathogen, we also employed a relevant host environment. Since Histoplasma yeast cells reside within macrophages during infection, our discovery criteria mandated that an effective antifungal candidate could inhibit intramacrophage yeast growth.
The identified aminothiazole 41F5 has good selectivity for Histoplasma and has potent antifungal activity against Histoplasma yeast cells both in vitro and within macrophages. The IC50 calculated for compound 41F5 is 0.87 μM, and the MIC is 2 μM. Even without compound optimization, these 41F5 concentrations are already comparable to the ones of other clinically established antifungals (43–46). Compound 41F5 also has a high degree of selectivity for Histoplasma yeast cells, an essential criterion for any treatment against a eukaryotic pathogen. Estimates of its toxicity to host cells range from 53.95 μM to 74.65 μM depending on the cell type. Due to the effects of the DMSO solvent used to solubilize 41F5, these concentrations are overestimates of the actual cytotoxicity of 41F5 to mammalian cells. Most importantly for potential clinical applicability, 41F5 can inhibit the growth and replication of intracellular Histoplasma yeast cells. The combined potency of 41F5 against intracellular yeast cells and low toxicity to macrophages translate into the ability of 41F5 to protect host macrophages from Histoplasma-dependent phagocyte killing. While compound 41F5 is not active against all fungal pathogens, it can inhibit Cryptococcus yeast cells in addition to Histoplasma. Phylogenetically, these two species are quite distantly related (a basidiomycete versus an ascomycete), but they do share the characteristic of significant residence within host macrophages.
The central thiazole of 41F5 is a structural core common to many compounds used to treat a range of diseases. The established molecular targets of these thiazoles are quite varied and include protein kinases, G proteins, and metabolic dehydrogenases (47–50). A feature shared by many of these molecular targets is their interaction with purine molecules. Consistent with this, the 41F5 compound was identified from a purinome-focused chemical library. Although the precise target of 41F5 is unknown, its origination from the purinome-focused library suggests that it inhibits a purine-binding or purine-utilizing factor in yeast cells. The current antifungal compound most similar in structure to 41F5 is the recently licensed topical antifungal abafungin, which is thought to permeabilize fungi by targeting ergosterol. Like 41F5, abafungin has ring systems extending from a central thiazole (51), and abafungin can inhibit Histoplasma growth in vitro. However, this was demonstrated only for the mycelial form and not for the clinically relevant yeast form (51). Unlike 41F5, abafungin is fungicidal. While both 41F5 and abafungin are thiazole-based compounds, there are important structural differences: 41F5 has substituents at the 2- and 5-positions, whereas abafungin has substituent groups at the 2- and 4-positions. Thus, 41F5 is distinct from abafungin in structure and in cidal versus static inhibition, suggesting that the cellular molecule inhibited by 41F5 represents a different antifungal drug target. Future structure-activity relationship studies should establish the target-specifying roles of the thiazole substituents. In addition, optimization of the physicochemical and pharmacokinetic properties of 41F5 may lead to development of an effective and less toxic therapeutic option for the treatment of histoplasmosis.
ACKNOWLEDGMENTS
We thank Timothy Haystead for generously providing the purinome-focused library. We thank Jesse Kwiek and Manjusha Kulkarni for technical assistance with screening. We thank Werner Tjarks and Ahmed Khalil at the OSU Division of Medicinal Chemistry and Pharmacognosy for discussion and critique of 41F5. We thank the OSU Clinical Microbiology Laboratory, Tamara Doering, Greg Gauthier, and Robert Cramer for providing fungal strains for testing.
This work was supported by pilot funds awarded from the Public Health Preparedness for Infectious Diseases Program at Ohio State University and from the Center for Clinical and Translational Science at Ohio State University, which is funded by NIH/NCATS grant UL1TR000090.
Footnotes
Published ahead of print 1 July 2013
REFERENCES
- 1.Wilson LS, Reyes CM, Stolpman M, Speckman J, Allen K, Beney J. 2002. The direct cost and incidence of systemic fungal infections. Value Health 5:26–34 [DOI] [PubMed] [Google Scholar]
- 2.Finkelstein A, Holz R. 1973. Aqueous pores created in thin lipid membranes by the polyene antibiotics nystatin and amphotericin B. Membranes 2:377–408 [PubMed] [Google Scholar]
- 3.Palacios DS, Dailey I, Siebert DM, Wilcock BC, Burke MD. 2011. Synthesis-enabled functional group deletions reveal key underpinnings of amphotericin B ion channel and antifungal activities. Proc. Natl. Acad. Sci. U. S. A. 108:6733–6738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hsuchen CC, Feingold DS. 1973. Selective membrane toxicity of the polyene antibiotics: studies on natural membranes. Antimicrob. Agents Chemother. 4:316–319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moen MD, Lyseng-Williamson KA, Scott LJ. 2009. Liposomal amphotericin B: a review of its use as empirical therapy in febrile neutropenia and in the treatment of invasive fungal infections. Drugs 69:361–392 [DOI] [PubMed] [Google Scholar]
- 6.Vanden Bossche H, Willemsens G, Marichal P. 1987. Anti-Candida drugs—the biochemical basis for their activity. Crit. Rev. Microbiol. 15:57–72 [DOI] [PubMed] [Google Scholar]
- 7.Georgopapadakou NH, Walsh TJ. 1996. Antifungal agents: chemotherapeutic targets and immunologic strategies. Antimicrob. Agents Chemother. 40:279–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Korashy HM, Shayeganpour A, Brocks DR, El-Kadi AO. 2007. Induction of cytochrome P450 1A1 by ketoconazole and itraconazole but not fluconazole in murine and human hepatoma cell lines. Toxicol. Sci. 97:32–43 [DOI] [PubMed] [Google Scholar]
- 9.Girois SB, Chapuis F, Decullier E, Revol BG. 2006. Adverse effects of antifungal therapies in invasive fungal infections: review and meta-analysis. Eur. J. Clin. Microbiol. Infect. Dis. 25:138–149 [DOI] [PubMed] [Google Scholar]
- 10.Hage CA, Connolly P, Horan D, Durkin M, Smedema M, Zarnowski R, Smith P, Wheat LJ. 2011. Investigation of the efficacy of micafungin in the treatment of histoplasmosis using two North American strains of Histoplasma capsulatum. Antimicrob. Agents Chemother. 55:4447–4450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Maligie MA, Selitrennikoff CP. 2005. Cryptococcus neoformans resistance to echinocandins: (1,3)beta-glucan synthase activity is sensitive to echinocandins. Antimicrob. Agents Chemother. 49:2851–2856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Messer SA, Jones RN, Moet GJ, Kirby JT, Castanheira M. 2010. Potency of anidulafungin compared to nine other antifungal agents tested against Candida spp., Cryptococcus spp., and Aspergillus spp.: results from the global SENTRY Antimicrobial Surveillance Program (2008). J. Clin. Microbiol. 48:2984–2987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Edwards LB, Acquaviva FA, Livesay VT, Cross FW, Palmer CE. 1969. An atlas of sensitivity to tuberculin, PPD-B, and histoplasmin in the United States. Am. Rev. Respir. Dis. 99(Suppl):1–132 [PubMed] [Google Scholar]
- 14.Holbrook ED, Edwards JA, Youseff BH, Rappleye CA. 2011. Definition of the extracellular proteome of pathogenic-phase Histoplasma capsulatum. J. Proteome Res. 10:1929–1943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hwang L, Hocking-Murray D, Bahrami AK, Andersson M, Rine J, Sil A. 2003. Identifying phase-specific genes in the fungal pathogen Histoplasma capsulatum using a genomic shotgun microarray. Mol. Biol. Cell 14:2314–2326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nguyen VQ, Sil A. 2008. Temperature-induced switch to the pathogenic yeast form of Histoplasma capsulatum requires Ryp1, a conserved transcriptional regulator. Proc. Natl. Acad. Sci. U. S. A. 105:4880–4885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Patel JB, Batanghari JW, Goldman WE. 1998. Probing the yeast phase-specific expression of the CBP1 gene in Histoplasma capsulatum. J. Bacteriol. 180:1786–1792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rippon JW. 1988. Medical mycology: the pathogenic fungi and the pathogenic actinomycetes, 3rd ed, p 381–423 WB Saunders Co, Philadelphia, PA [Google Scholar]
- 19.Chu JH, Feudtner C, Heydon K, Walsh TJ, Zaoutis TE. 2006. Hospitalizations for endemic mycoses: a population-based national study. Clin. Infect. Dis. 42:822–825 [DOI] [PubMed] [Google Scholar]
- 20.Wheat LJ, Freifeld AG, Kleiman MB, Baddley JW, McKinsey DS, Loyd JE, Kauffman CA. 2007. Clinical practice guidelines for the management of patients with histoplasmosis: 2007 update by the Infectious Diseases Society of America. Clin. Infect. Dis. 45:807–825 [DOI] [PubMed] [Google Scholar]
- 21.Marion CL, Rappleye CA, Engle JT, Goldman WE. 2006. An alpha-(1,4)-amylase is essential for alpha-(1,3)-glucan production and virulence in Histoplasma capsulatum. Mol. Microbiol. 62:970–983 [DOI] [PubMed] [Google Scholar]
- 22.Worsham PL, Goldman WE. 1988. Quantitative plating of Histoplasma capsulatum without addition of conditioned medium or siderophores. J. Med. Vet. Mycol. 26:137–143 [PubMed] [Google Scholar]
- 23.Fothergill AW. 2012. Antifungal susceptibility testing: Clinical Laboratory and Standards Institute (CLSI) methods, p 65–74 In Hall GS. (ed), Interactions of yeasts, moulds, and antifungal agents: how to detect resistance. Humana Press, Totowa, NJ [Google Scholar]
- 24.Fadden P, Huang KH, Veal JM, Steed PM, Barabasz AF, Foley B, Hu M, Partridge JM, Rice J, Scott A, Dubois LG, Freed TA, Silinski MA, Barta TE, Hughes PF, Ommen A, Ma W, Smith ED, Spangenberg AW, Eaves J, Hanson GJ, Hinkley L, Jenks M, Lewis M, Otto J, Pronk GJ, Verleysen K, Haystead TA, Hall SE. 2010. Application of chemoproteomics to drug discovery: identification of a clinical candidate targeting hsp90. Chem. Biol. 17:686–694 [DOI] [PubMed] [Google Scholar]
- 25.Marjanovic J, Chalupska D, Patenode C, Coster A, Arnold E, Ye A, Anesi G, Lu Y, Okun I, Tkachenko S, Haselkorn R, Gornicki P. 2010. Recombinant yeast screen for new inhibitors of human acetyl-CoA carboxylase 2 identifies potential drugs to treat obesity. Proc. Natl. Acad. Sci. U. S. A. 107:9093–9098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Edwards JA, Zemska O, Rappleye CA. 2011. Discovery of a role for Hsp82 in Histoplasma virulence through a quantitative screen for macrophage lethality. Infect. Immun. 79:3348–3357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kueng W, Silber E, Eppenberger U. 1989. Quantification of cells cultured on 96-well plates. Anal. Biochem. 182:16–19 [DOI] [PubMed] [Google Scholar]
- 28.Edwards JA, Alore EA, Rappleye CA. 2011. The yeast-phase virulence requirement for alpha-glucan synthase differs among Histoplasma capsulatum chemotypes. Eukaryot. Cell 10:87–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.LeMonte AM, Washum KE, Smedema ML, Schnizlein-Bick C, Kohler SM, Wheat LJ. 2000. Amphotericin B combined with itraconazole or fluconazole for treatment of histoplasmosis. J. Infect. Dis. 182:545–550 [DOI] [PubMed] [Google Scholar]
- 30.Zhang JH, Chung TD, Oldenburg KR. 1999. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J. Biomol. Screen. 4:67–73 [DOI] [PubMed] [Google Scholar]
- 31.Goh EB, Yim G, Tsui W, McClure J, Surette MG, Davies J. 2002. Transcriptional modulation of bacterial gene expression by subinhibitory concentrations of antibiotics. Proc. Natl. Acad. Sci. U. S. A. 99:17025–17030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hughes JP, Rees S, Kalindjian SB, Philpott KL. 2011. Principles of early drug discovery. Br. J. Pharmacol. 162:1239–1249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sebghati TS, Engle JT, Goldman WE. 2000. Intracellular parasitism by Histoplasma capsulatum: fungal virulence and calcium dependence. Science 290:1368–1372 [DOI] [PubMed] [Google Scholar]
- 34.Bohse ML, Woods JP. 2007. RNA interference-mediated silencing of the YPS3 gene of Histoplasma capsulatum reveals virulence defects. Infect. Immun. 75:2811–2817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Youseff BH, Holbrook ED, Smolnycki KA, Rappleye CA. 2012. Extracellular superoxide dismutase protects Histoplasma yeast cells from host-derived oxidative stress. PLoS Pathog. 8:e1002713. 10.1371/journal.ppat.1002713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang JL, Chang CH, Young-Xu Y, Chan KA. 2010. Systematic review and meta-analysis of the tolerability and hepatotoxicity of antifungals in empirical and definitive therapy for invasive fungal infection. Antimicrob. Agents Chemother. 54:2409–2419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Espinel-Ingroff A. 1998. Comparison of in vitro activities of the new triazole SCH56592 and the echinocandins MK-0991 (L-743,872) and LY303366 against opportunistic filamentous and dimorphic fungi and yeasts. J. Clin. Microbiol. 36:2950–2956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Graybill JR, Najvar LK, Montalbo EM, Barchiesi FJ, Luther MF, Rinaldi MG. 1998. Treatment of histoplasmosis with MK-991 (L-743,872). Antimicrob. Agents Chemother. 42:151–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kohler S, Wheat LJ, Connolly P, Schnizlein-Bick C, Durkin M, Smedema M, Goldberg J, Brizendine E. 2000. Comparison of the echinocandin caspofungin with amphotericin B for treatment of histoplasmosis following pulmonary challenge in a murine model. Antimicrob. Agents Chemother. 44:1850–1854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nakai T, Uno J, Ikeda F, Tawara S, Nishimura K, Miyaji M. 2003. In vitro antifungal activity of micafungin (FK463) against dimorphic fungi: comparison of yeast-like and mycelial forms. Antimicrob. Agents Chemother. 47:1376–1381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Finquelievich J, Landaburu MF, Pinoni V, Iovannitti CA. 2011. Determination of the therapeutic activity of caspofungin compared with the amphotericin B in an animal experimental model of histoplasmosis in hamster (Mesocrisetus auratus). Rev. Iberoam. Micol. 28:155–158 [DOI] [PubMed] [Google Scholar]
- 42.Cushion MT, Linke MJ, Ashbaugh A, Sesterhenn T, Collins MS, Lynch K, Brubaker R, Walzer PD. 2010. Echinocandin treatment of pneumocystis pneumonia in rodent models depletes cysts leaving trophic burdens that cannot transmit the infection. PLoS One 5:e8524. 10.1371/journal.pone.0008524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Espinel-Ingroff A, Fothergill A, Fuller J, Johnson E, Pelaez T, Turnidge J. 2011. Wild-type MIC distributions and epidemiological cutoff values for caspofungin and Aspergillus spp. for the CLSI broth microdilution method (M38-A2 document). Antimicrob. Agents Chemother. 55:2855–2859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pfaller MA, Castanheira M, Messer SA, Moet GJ, Jones RN. 2011. Echinocandin and triazole antifungal susceptibility profiles for Candida spp., Cryptococcus neoformans, and Aspergillus fumigatus: application of new CLSI clinical breakpoints and epidemiologic cutoff values to characterize resistance in the SENTRY Antimicrobial Surveillance Program (2009). Diagn. Microbiol. Infect. Dis. 69:45–50 [DOI] [PubMed] [Google Scholar]
- 45.Pfaller MA, Diekema DJ, Andes D, Arendrup MC, Brown SD, Lockhart SR, Motyl M, Perlin DS. 2011. Clinical breakpoints for the echinocandins and Candida revisited: integration of molecular, clinical, and microbiological data to arrive at species-specific interpretive criteria. Drug Resist. Updat. 14:164–176 [DOI] [PubMed] [Google Scholar]
- 46.Wheat J, Marichal P, Vanden Bossche H, Le Monte A, Connolly P. 1997. Hypothesis on the mechanism of resistance to fluconazole in Histoplasma capsulatum. Antimicrob. Agents Chemother. 41:410–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Das J, Chen P, Norris D, Padmanabha R, Lin J, Moquin RV, Shen Z, Cook LS, Doweyko AM, Pitt S, Pang S, Shen DR, Fang Q, de Fex HF, McIntyre KW, Shuster DJ, Gillooly KM, Behnia K, Schieven GL, Wityak J, Barrish JC. 2006. 2-Aminothiazole as a novel kinase inhibitor template. Structure-activity relationship studies toward the discovery of N-(2-chloro-6-methylphenyl)-2-[[6-[4-(2-hydroxyethyl)-1-piperazinyl)]-2-methyl-4-pyrimidinyl]amino)]-1,3-thiazole-5-carboxamide (dasatinib, BMS-354825) as a potent pan-Src kinase inhibitor. J. Med. Chem. 49:6819–6832 [DOI] [PubMed] [Google Scholar]
- 48.Hom RK, Bowers S, Sealy JM, Truong AP, Probst GD, Neitzel ML, Neitz RJ, Fang L, Brogley L, Wu J, Konradi AW, Sham HL, Toth G, Pan H, Yao N, Artis DR, Quinn K, Sauer JM, Powell K, Ren Z, Bard F, Yednock TA, Griswold-Prenner I. 2010. Design and synthesis of disubstituted thiophene and thiazole based inhibitors of JNK. Bioorg. Med. Chem. Lett. 20:7303–7307 [DOI] [PubMed] [Google Scholar]
- 49.Lu J, Shi M, Shoichet MS. 2009. Click chemistry functionalized polymeric nanoparticles target corneal epithelial cells through RGD-cell surface receptors. Bioconjug. Chem. 20:87–94 [DOI] [PubMed] [Google Scholar]
- 50.Sams AG, Mikkelsen GK, Larsen M, Langgard M, Howells ME, Schroder TJ, Brennum LT, Torup L, Jorgensen EB, Bundgaard C, Kreilgard M, Bang-Andersen B. 2011. Discovery of phosphoric acid mono-{2-[(E/Z)-4-(3,3-dimethyl-butyrylamino)-3,5-difluoro-benzoylimino]-thiazol-3-ylmethyl} ester (Lu AA47070): a phosphonooxymethylene prodrug of a potent and selective hA(2A) receptor antagonist. J. Med. Chem. 54:751–764 [DOI] [PubMed] [Google Scholar]
- 51.Borelli C, Schaller M, Niewerth M, Nocker K, Baasner B, Berg D, Tiemann R, Tietjen K, Fugmann B, Lang-Fugmann S, Korting HC. 2008. Modes of action of the new arylguanidine abafungin beyond interference with ergosterol biosynthesis and in vitro activity against medically important fungi. Chemotherapy 54:245–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Clinical and Laboratory Standards Institute 2008. Reference method for broth dilution antifungal susceptibility testing of yeasts; approved standard, 3rd ed, CLSI document M27-A3 Clinical and Laboratory Standards Institute, Wayne, PA [Google Scholar]
- 53.Clinical and Laboratory Standards Institute 2008. Reference method for broth dilution antifungal susceptibility testing of filamentous fungi; approved standard, 2nd ed, CLSI document M38-A2 Clinical and Laboratory Standards Institute, Wayne, PA [Google Scholar]