

Abstract
It has been reported that autophagy is involved in the replication of many viruses. In this study, we screened 89 medicinal plants, using an assay based on the inhibition of the formation of the Atg12-Atg5/Atg16 heterotrimer, an important regulator of autophagy, and selected Silybum marianum L. for further study. An antiviral assay indicated that silybin (S0), the major active compound of S. marianum L., can inhibit influenza A virus (IAV) infection. We later synthesized 5 silybin derivatives (S1 through S5) and found that 23-(S)-2-amino-3-phenylpropanoyl-silybin (S3) had the best activity. When we compared the polarities of the substituent groups, we found that the hydrophobicity of the substituent groups was positively correlated with their activities. We further studied the mechanisms of action of these compounds and determined that S0 and S3 also inhibited both the formation of the Atg12-Atg5/Atg16 heterotrimer and the elevated autophagy induced by IAV infection. In addition, we found that S0 and S3 could inhibit several components induced by IAV infection, including oxidative stress, the activation of extracellular signal-regulated kinase (ERK)/p38 mitogen-activated protein kinase (MAPK) and IκB kinase (IKK) pathways, and the expression of autophagic genes, especially Atg7 and Atg3. All of these components have been reported to be related to the formation of the Atg12-Atg5/Atg16 heterotrimer, which might validate our screening strategy. Finally, we demonstrated that S3 can significantly reduce influenza virus replication and the associated mortality in infected mice. In conclusion, we identified 23-(S)-2-amino-3-phenylpropanoyl-silybin as a promising inhibitor of IAV infection.
INTRODUCTION
Although autophagy is thought to be a component of the host innate immunity mechanism, many viruses, such as hepatitis C virus (HCV), human immunodeficiency virus (HIV), coxsackievirus B3 (CVB3), and influenza A virus (IAV), have evolved mechanisms to evade, subvert, or exploit this machinery for their own replication. It has been reported that inhibition of autophagy can efficiently inhibit IAV replication and IAV-induced autophagic cell death; this autophagic cell death ultimately leads to acute lung injury and high mortality (1–3). Inhibition of autophagy has been proposed as a good target for novel antiviral drugs (1, 4, 5).
Macroautophagy (here referred to as autophagy) is strictly regulated. During the elongation of the autophagosome, Atg12 is covalently conjugated to Atg5 via its C-terminal glycine, and Atg16L1 is noncovalently conjugated to Atg5 via its N terminus. These associations ultimately form the Atg12-Atg5/Atg16 heterotrimer, which is crucial for the conversion of LC3I to LC3II, which in turn is necessary for the formation of autophagosomes (6). In this study, we chose to target the Atg12-Atg5/Atg16 heterotrimer for drug screening, because the formation of this heterotrimer can be regulated at several levels. First, the formation of the Atg12-Atg5/Atg16 heterotrimer is directly regulated by Atg7, Atg10, Atg3, oxidative signals, and ATP. It is well known that the conjugation of Atg12 to Atg5 requires ubiquitin-like reactions involving Atg7 and Atg10 (6). The formation of the Atg12-Atg5/Atg16 heterotrimer can also be regulated by Atg3; Atg3 deficiency causes a reduction in the conjugation of Atg12 to Atg5 (7). Reactive oxygen species (ROS) can colocalize with the Atg12-Atg5-positive phagophores, and oxidative stress can induce the deacetylation of FoxO1, which binds to Atg7 and activates Atg12; this process is required for the formation of the Atg12-Atg5 heterodimer (8). In addition, ATP is essential for the de novo reconstitution of the Atg12-Atg5 heterodimer (9). Second, the degradation of the Atg12-Atg5 complex can be directly regulated by intracellular Ca2+ and calpain 1, which promote the cleavage of Atg5; inhibiting calpain 1 is sufficient to increase the amount of the Atg12-Atg5 conjugate (10). Third, the expression of autophagic genes can be regulated by different signal pathways. IκB kinase (IKK) is required for the starvation-induced expression of Atg5 and Beclin 1 (11). The depletion of any IKK subunits limits autophagy, while constitutively active IKK subunits are sufficient to stimulate autophagy via the TAB2/TAB3-Beclin 1-TAK1 pathway (12). ROS-mediated JNK activation can upregulate the expression of Atg5, Atg7, and Beclin 1 (13). Hypoxia can increase the transcription of LC3B and Atg5 (14). HMGB1 can promote the formation and accumulation of the Atg12-Atg5/Atg16 complex through the phosphatidylinositol 3-kinase C3 (PI3KC3)-MEK-extracellular signal-regulated kinase (ERK) pathway (15). In summary, the formation of the Atg12-Atg5/Atg16 heterotrimer is regulated by many factors and is an excellent target for drug screening.
The bimolecular fluorescence complementation-fluorescence resonance energy transfer (BiFC-FRET) assay is a recently described technology that combines two techniques, BiFC and FRET. BiFC is based on the principle that two nonfluorescent fragments of a fluorescent protein, when brought together by the interaction of proteins fused to each nonfluorescent fragment, can reconstitute an intact fluorescent protein. FRET takes advantage of two fluorescent molecules; in FRET, an excited fluorescent molecule (donor) transfers its energy to a nearby light-absorbing molecule (acceptor) and excites it. This transfer is dependent on the proximity of the two molecules, and the technique is used to analyze molecular interactions quantitatively with spatial and temporal resolution (16–19). In our study, we constructed three plasmids (pMC-atg5, pMN-atg12, and pEGFP-atg16) to form a BiFC-FRET system in which Atg5 and Atg12 are fused with the N and C fragments of a red fluorescent protein, respectively. After the BiFC reaction, the intact fluorescent proteins are reconstituted through the interaction between Atg5 and Atg12 and are used as acceptors; Atg16 is fused to enhanced green fluorescent protein (EGFP) and used as a donor, The BiFC-FRET reaction takes place when the Atg12-Atg5/Atg16 heterotrimer is fully formed.
Using this BiFC-FRET screening model, we screened 89 medicinal plants and selected Silybum marianum L. for further study. We further synthesized 5 types of amino acid derivatives (S1 through S5) of silybin (S0) and found 23-(S)-2-amino-3-phenylpropanoyl-silybin (S3) had the best activity. Next, we determined the influences of S0 and S3 on oxidative stress, the activation of the ERK/p38 and IKK/NF-κB pathways, and the expression of autophagic genes, all of which are reported to be able to regulate the formation of the Atg12-Atg5/Atg16 complex, and this approach may validate the design of our screening strategy. In conclusion, we identified S3 as a promising inhibitor of IAV infection.
MATERIALS AND METHODS
Medicinal plants and compounds.
Eighty-nine medicinal plants were collected from the Yulin medicine market (Guangxi, China). Each specimen was deposited in our laboratory. Crude extracts were prepared as described in the 2005 edition of the Chinese Pharmacopoeia (53). Silybin (purity, >98%) was purchased from the National Institutes for Food and Drug Control (Beijing, China). The amino acid derivatives of silybin were synthesized at Jiangsu University, China. The process of chemical synthesis and 1H-nuclear magnetic resonance (NMR) analysis is shown in Fig. S1 and S2 in the supplemental material.
Cytotoxicity assays.
The cytotoxicity of the tested drugs in MDCK, A549, and Vero cells was measured using the 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) method (20). Briefly, The cells were seeded in 96-well microplates (1 × 104 cells/well) for 24 h. After treatment with the different concentrations of the tested drugs for 48 h, the cell viability was measured using the MTT method. The 50% cytotoxic concentration (CC50) was defined as the compound concentration required to reduce by 50% the cell viability, compared with that of the mock treatment group (0.5% dimethyl sulfoxide [DMSO]).
Plasmid construction.
The N and C segments of mCherry, a red fluorescent protein (GenBank accession number HQ423140.1), corresponding to amino acids 1 to 159 and 160 to 262, respectively, were inserted into the pcDNA 3.0 plasmid; these constructs were named pMC and pMN, respectively. The plasmids were designed as previously reported (21). Human atg5 (NM_004849.2) and mutant Atg5, in which Lys130 of Atg5 was changed to Arg, were inserted into the pMC plasmid; these plasmids were named pMC-atg5 and pMC-mut-atg5, respectively. The human atg12 gene (NM_004707.3) was inserted into pMN, and the construct was named pMN-atg12. The human atg16L1 (NM_030803.6) and mutant Atg16L1, in which the Atg5-binding domain in the N terminus of Atg16L1 was deleted, were inserted into pEGFP-C1, and these plasmids were named pEGFP-atg16 and pEGFP-mut-atg16, respectively. The human LC3B gene (NM_022818.4) was inserted into the pEGFP-C1 plasmid, and this construct was named pEGFP-LC3. A linker sequence (RPACKIPNDLKQKVMNH) was inserted into each fusion protein (21). The primers are listed in Table S1 in the supplemental material. The results of double enzyme digestions of all constructs are shown in Fig. S3 in the supplemental material, and the plasmids were verified by DNA sequencing.
Drug-screening assay.
A549 cells were seeded in 96-well microplates for 24 h and then cotransfected with pMC-atg5, pMN-atg12, and pEGFP-Atg16 plasmids using Lipofectamine 2000 (Invitrogen). After 6 h, the cells were infected with IAV (multiplicity of infection [MOI] = 2.0) and treated or not with test drugs for 8 h. After 1 h at 4°C, the fluorescence intensities (FI) were determined using a microplate reader (Tecan Infinite M1000). In the BiFC assay, the FI was measured at 610 nm (FI610) after excitation at 587 nm. In the BiFC-FRET assay, the FI was measured at 610 and 509 nm after excitation at 488 nm, and the BiFC-FRET efficiency (BiFC-FRETe) was expressed as the ratio of acceptor (610-nm) to donor (509-nm) emission intensities and calculated as follows:
| (3) |
where RFUsmp is the FI of the sample group and RFUback is the FI of the background. The Z′ factor is a statistical parameter to quantify the suitability of a particular assay for use in a high-throughput screen; σc+ and σc− are the standard deviations of the negative and blank groups (BG), respectively; and μc+ and μc− are the average values of the negative and blank groups, respectively (22–24). To visualize this assay, A549 cells were plated on coverslips and incubated for 1 h at 4°C. After incubation, the cells were fixed using 3% formaldehyde and then visualized using an upright fluorescence microscope (Nikon Eclipse 90i).
TCID50 and antiviral assay using the sulforhodamine B (SRB) method.
A stock solution of IAV subtype A/ShanTou/169/06 (H1N1) was diluted with Dulbecco's modified Eagle's medium (DMEM) E0 containing 2.5 μg/ml trypsin and 3.2% bovine serum albumin (BSA) in serial dilutions and then incubated with MDCK cells for 48 h; the 50% tissue culture infective dose (TCID50) was calculated according to the method of Reed and Muench.
Antiviral activities were evaluated by the SRB method using cytopathic effect (CPE) reduction (25). Briefly, MDCK cells (1 × 104 cells/well) were seeded in 96-well plates for 24 h. Exactly 0.09 ml of virus suspension (50 TCID50) and 0.01 ml medium containing various concentrations of test compounds were added to the wells. At 48 h, the cells were washed and 100 μl −20°C 70% acetone was added. After removing the acetone, the plates were dried and 100 μl 0.4% (wt/vol) SRB was added. After another wash, the plates were dried and 100 μl 10 mM Tris base solution was added. The optical density (OD) was read at 562 nm. Three wells were included as negative (virus-infected non-drug-treated) and mock (noninfected non-drug-treated) controls. DMSO (0.5%) was used as the solvent in each group. The percent protection of a test compound (cell viability) was calculated as follows:
The concentration providing 50% protection was defined as the 50% effective concentration (EC50). The selective index (SI) was defined as the CC50 divided by the EC50.
Plaque formation, plaque inhibition, and time course assays.
The viral titers of various supernatants were also determined by plaque formation assays, as described in our previous report (26), and the plaques (diameter > 1 mm) were counted. Plaque inhibition assays of test compounds were also performed as previously reported (26). Briefly, MDCK cells were incubated with IAV (MOI = 0.001) with or without test compounds for 1 h; after washing, E0 medium with or without test compounds was added. After 48 h, the supernatant was collected and the viral titer was determined by a plaque formation assay. The time course assay comprised four tests: (i) before infection, the virus was incubated with a medium containing test compounds for 3 h; (ii) before infection, MDCK cells were incubated with a medium containing test compounds for 3 h; (iii) test compounds were added during viral adsorption; and (iv) test compounds were added at different postinfection (p.i.) time points. The MOI for the time course assay was 2.0. After 8 h, the supernatants were collected and the viral titer was determined by a plaque formation assay. Ribavirin and 0.5% DMSO were used as the positive control (PC) and negative control (NC), respectively.
Indirect immunofluorescence assay.
MDCK cells were infected (MOI = 5.0) and treated or not with test compounds. At 4 h p.i., the cells were fixed and blocked and then reacted with anti-NP primary antibody. After washing, the cells were incubated with a Cy3-conjugated anti-rabbit antibody (Santa Cruz), stained with DAPI (4′,6-diamidino-2-phenylindole), and observed using an upright fluorescence microscope (Nikon Eclipse 90i).
RT-PCR and qRT-PCR.
Real-time (RT)-PCR was performed using A549 cells, an MOI of 0.001, and an incubation time of 24 h. Quantitative RT-PCR (qRT-PCR) was performed using MDCK cells. After IAV infection (MOI = 5.0), the MDCK cells were treated with S0, S3, or ribavirin at −1 to 0 (adsorption), 0 to 2, 2 to 4, and 4 to 6 h p.i. qRT-PCR was performed in a 20-μl reaction mixture containing forward and reverse primers (50 nM each), 1× SYBR green master mix (Invitrogen), and various templates. The NP and GAPDH primers are shown in Table S1 in the supplemental material.
Western blotting and co-IP assays.
Anti-LC3B, anti-Atg3, anti-Atg5, anti-Atg7, anti-Atg12, anti-Atg16, anti-IKKβ, anti-p65, anti-p-JNK, anti-JNK, anti-p-ERK, anti-ERK, anti-p-p38, anti-p38, anti-NP, and anti-β-actin antibodies were purchased from Cell Signaling Technology Inc. To detect NF-κB p65, the nuclear protein was extracted. Western blotting was performed as previously reported (26). The formation of the Atg12-Atg5/Atg16 complex was detected by a coimmunoprecipitation (co-IP) assay, following the protocol of the Co-Immunoprecipitation Kit (Thermo Scientific 23600). Normal rabbit IgG was used as a control.
Analysis of EGFP-LC3 puncta.
A549 cells were transfected with the pEGFP-LC3 plasmid using Lipofectamine 2000 (Invitrogen). After 6 h, the cells were infected (MOI = 2.0) and treated or not with test compounds; after 8 h, the cells were visualized using an upright fluorescence microscope (1,000×). The ratio of cells containing EGFP-LC3 dots to cells expressing EGFP was calculated in 10 fields chosen at random from three independent experiments.
Antioxidant assay.
Levels of reduced glutathione (GSH), malondialdehyde (MDA), NO, OH−, total superoxide dismutase (T-SOD), glutathione reductase (GR), catalase (CAT), and glutathione peroxidase (GSH-Px) were determined using commercially available kits (Jiancheng Bioengineering Institute, Nanjing, China).
RNA interference.
Atg7 small interfering RNA (siRNA) (sc-41447), Atg3 siRNA (sc-72582), and control siRNA were purchased from Santa Cruz Biotechnology and used according to the manufacturer's siRNA transfection protocol.
In vivo efficacy study.
BALB/c mice (20 ± 2 g) were housed for 5 days for acclimation and maintained on standard rodent chow with water ad libitum. The mice were anesthetized by intraperitoneal injection of ketamine (100 mg/kg of body weight) and then infected intranasally with five 50% mouse lethal doses (MLD50) of A/PR8 virus in a 50-μl volume to achieve 100% lethality. DMSO-PBS (10% [vol/vol]), S0 (25 mg/kg/day), S3 (25 mg/kg/day), and ribavirin (75 mg/kg/day) were given twice a day (at 12-hour intervals) for 10 days to the infected animals by oral gavage, starting 24 h before virus exposure (27). The mice were weighed daily for 21 days. Three mice per group were euthanized at day 6, when the virus titers in the lungs reached the maximum (28), and the viral titers in their lungs were analyzed. Briefly, each lung was removed, weighed, and homogenized in minimal essential medium (MEM), and the viral titer was determined by the TCID50 assay with MDCK cells. The number of wells showing a positive cytopathic effect was scored, and the titer (TCID50) per milligram of lung tissue was calculated by standard procedures (28–30). All experimental procedures were approved by the Institutional Animal Care and Use Committee (IACUC) of Shantou University.
Statistical analysis.
The statistical significance of the comparisons between treated groups was assessed by one-way analysis of variance (ANOVA) with least significant difference (LSD) and Waller-Duncan multiple-comparison methods using SPSS13.0 software. All values are expressed as means ± standard deviations (SD). Each experiment was repeated at least three times. P values below 0.05 were considered statistically significant. Statistical analysis of the survival curve by a log rank (Mantel-Cox) χ2 test was conducted using GraphPad Prism 5 software.
RESULTS
Rationale and design of our screening model and results of a drug-screening assay.
Our drug-screening model consisted of three plasmids, pMC-atg5, pMN-atg12, and pEGFP-atg16. The human Atg5 and Atg12 genes were fused with the N and C segments of a red fluorescent protein (RFP), mCherry, corresponding to amino acids 1 to 159 and 160 to 262, respectively. The splitting of the red fluorescent protein mCherry was done as previously reported (21). Atg16 was fused to EGFP. After cotransfection, red fluorescent proteins would first reconstitute by the BiFC reaction, due to the interaction of Atg5 and Atg12; to measure the BiFC reaction, the FI was determined at 610 nm after excitation at 587 nm. Subsequently, the FRET reaction would take place, due to the interaction between the Atg12-Atg5 heterodimer and the Atg16 protein; to evaluate this FRET reaction, the FI was determined at 509 and 610 nm after excitation at 488 nm, and the BiFC-FRETe was expressed as the ratio of ΔFI610 to ΔFI509 (Fig. 1A).
Fig 1.

Design of our drug-screening model. (A) Construction of plasmids and schematic representation of the BiFC-FRET technique. The human Atg5 and Atg12 genes were fused to the N and C fragments, respectively, of a red fluorescent protein (mCherry). Atg16 was fused to EGFP. After cotransfection, the BiFC reaction took place between the Atg5 and Atg12 fusion proteins, and the FI was measured at 610 nm after excitation at 587 nm; subsequently, the FRET reaction took place between the Atg5-Atg12 heterodimer and the Atg16 fusion protein, and the FI was measured at 509 and 610 nm after excitation at 488 nm, and BiFC-FRETe was expressed as the ratio of ΔFI610 to ΔFI509. (B) Influences of 0.5% DMSO, mut-Atg5, mut-Atg16, and IAV infection (MOI = 2.0; infection time, 8 h) on the BiFC reaction between the Atg5 and Atg12 fusion proteins. (C) Influences of DMSO, mut-Atg5, mut-Atg16, and IAV infection on the BiFC-FRET reaction between the Atg5-Atg12 heterodimer and the Atg16 fusion protein. (D) Influences of DMSO, mut-Atg5, mut-Atg16, and IAV infection on cell viability, as determined by the MTT method. (E) Influences of DMSO, mut-Atg5, mut-Atg16, and IAV infection on the transcriptional activity of the plasmids determined by Western blotting using anti-Flag antibody. After cotransfection for 6 h, only the IAV-plus-DMSO group was infected with IAV, and each group was incubated for another 8 h. The zones and average gray values of each band were detected and quantified with BandScan 5.0 software. The multiplication product of the zone area and the average gray value was calculated, and the results are expressed as the ratio of Flag to β-actin. The data are expressed as the means ± SD of two independent experiments with three replicates each. *, P < 0.05, and **, P < 0.01 versus the untreated group; ##, P < 0.01 versus the IAV-plus-DMSO group.
To detect the stability and suitability of this model, we first determined the influence of DMSO (0.5%) on the model, because all drugs were dissolved in 0.5% DMSO. This experiment showed that the BiFC signal and BiFC-FRETe in DMSO-treated samples were not significantly different from those in the untreated group. Second, we determined the effects of the mutations in Atg5 and Atg16 on the model. The mutation in Atg5 (mut-Atg5) changed Lys130 to Arg, which disrupted the interaction between Atg5 and Atg12. The mutation in Atg16 (mut-Atg16) deleted the Atg5-binding domain in the N terminus of Atg16, which disrupted the interaction between Atg5 and Atg16. The results showed that, after cotransfection with pMC-mut-atg5, pMN-atg12, and pEGFP-atg16, the BiFC signal and BiFC-FRETe were significantly decreased compared to the untreated group; after cotransfection with pMC-atg5, pMN-atg12, and pEGFP-mut-atg16, the BiFC signal showed no significant change, but BiFC-FRETe was significantly decreased. These results matched our predictions. Finally, we determined the influence of IAV infection (MOI = 2.0; 8 h; plus 0.5% DMSO) on the model. After IAV infection, the BiFC signal and BiFC-FRETe were significantly increased compared to the DMSO group, which meant that IAV infection could promote the formation of the Atg12-Atg5 and Atg12-Atg5/Atg16 complexes (Fig. 1B and C). In addition, we determined the cell viability of each group; these results showed there was no significant change between the groups (Fig. 1D). We also determined the transcriptional activity of the plasmids, in which all fusion proteins were under the cytomegalovirus (CMV) promoter, by Western blotting using anti-Flag antibody, and no significant differences could be observed between the constructs (Fig. 1E).
Using this model, we analyzed 89 medicinal plants, and we observed that S. marianum L. possessed excellent activity in the assay. Recently, Song and Choi have also reported that silymarin, the crude extract of S. marianum L., has excellent anti-IAV activity that is even stronger than that of oseltamivir (31). In our study, we purchased silybin (purity, >98%), the major active component of S. marianum L., and chemically synthesized 5 types of amino acid derivatives (see Fig. S1 and S2 in the supplemental material). In our study, the Z′ factor was 0.5699. The Z′ factor is a statistical parameter that is used to quantify the suitability of a particular assay for use in a high-throughput screen. According to a previous report (24), our screening model was valid (Z′ > 0.5).
Anti-IAV activities of silybin and its derivatives.
The cytotoxicities of S0 and its derivatives (S1 to S5) are shown in Fig. 2A; S0 and its derivatives did not show any significant cytotoxicity in MDCK cells at concentrations up to 400 μM. Rather, all of them significantly elevated the viability of cells at concentrations ranging from 6.25 to 100 μM. The estimated CC50 was greater than 400 μM. These results were similar to those in previous reports (32–34).
Fig 2.

Anti-IAV activities of silybin and its derivatives. (A) Results of the cytotoxicity assay of MDCK cells as determined by the MTT method. (B) Results of the antiviral assay determined using the SRB method. The CC50, EC50, and SI (CC50/EC50) values are shown below the graphs; The data are expressed as means ± SD of two independent experiments with three replicates each.
The anti-IAV activities of S0 and its derivatives were determined using the SRB method (Fig. 2B). The EC50s of S0, S1, S2, S3, S4, S5, and ribavirin were 70.73, 13.83, 5.32, 2.36, 3.81, 25.71, and 43.44 μM, respectively. The order of anti-IAV efficacy was S3 > S4 > S2 > S1 > S5 > ribavirin > S0. We compared the polarities of the substituent groups and found that the hydrophobicities of the substituent groups were positively correlated with their activities. Among these derivatives, S3 showed the best activity. We also estimated the anti-IAV activities of S0 and S3 by the classical plaque inhibition assay; S3 showed stronger activity than S0, and at a concentration of 80 μM, S3 showed stronger activity even than ribavirin (200 μM) (see Fig. S4 in the supplemental material). The anti-IAV efficacy of S0 was similar to the anti-HCV and anti-HIV efficacies reported by researchers. Ahmed-Belkacem et al. showed that silybin could inhibit the replication of HCV with a 50% inhibitory concentration of ∼100 μM in cell culture (35). McClure et al. showed that silybin could suppress HIV-1 infection in TZM-bl cells at concentrations ranging from 40 to 324 μM (36).
To determine which stage(s) of the IAV replication cycle was affected by S0 and S3, we performed a time course assay. Pretreatment of virus suspensions or cells with S0 or S3 before infection had no significant influence on the virus yield (Fig. 3A and B), and S0 and S3 did not significantly influence the adsorption of virus (Fig. 3C). The anti-IAV activities of S0 and S3 occurred at 1 to 4 h p.i. (Fig. 3D and E). We then investigated whether S0 and S3 inhibited viral protein and mRNA synthesis at this stage (1 to 4 h p.i.) by Western blotting and qRT-PCR analysis. As Fig. S5A and B in the supplemental material shows, S0 and S3 could significantly inhibit NP production at 0 to 2 and 2 to 4 h p.i., whereas at −1 to 0 (adsorption) and 4 to 6 h p.i., there was no significant difference between the groups. We also used indirect immunofluorescence assays to confirm this result. As shown in the supplemental material, S0 and S3 significantly reduced the expression of IAV NP. These experiments showed that S0 and S3 inhibited IAV replication at 1 to 4 h p.i.
Fig 3.
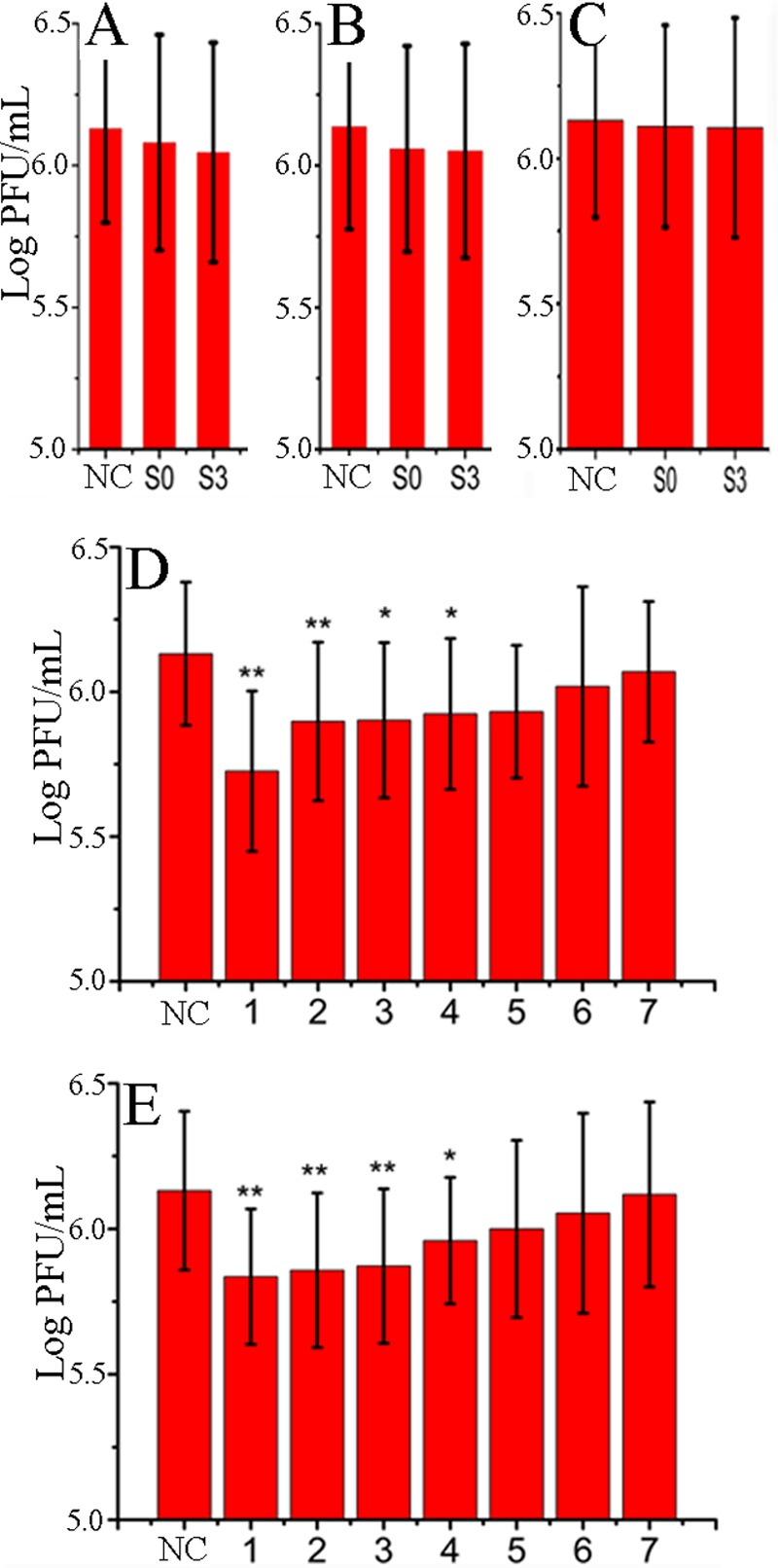
S0 and S3 inhibited IAV proliferation at 1 to 4 h p.i., as determined by a time course assay. (A) Before infection, virus was incubated with medium containing S0 or S3 for 3 h. (B) Before infection, MDCK cells were incubated with medium containing S0 or S3 for 3 h. (C) S0 or S3 was added during virus adsorption. (D and E) S0 or S3 was added at different time points after virus challenge, and the titer was determined at 8 h p.i. The concentrations of S0 and S3 were 100 and 80 μM, respectively. DMSO (0.5%) was used as an NC (virus-only group). The data are expressed as means ± SD of 3 experiments, each performed in duplicate. *, P < 0.05, and **, P < 0.01 versus the NC.
S0 and S3 can inhibit the formation of the Atg12-Atg5/Atg16 heterotrimer induced by IAV infection.
Because our screening assay was based on inhibition of the formation of the Atg12-Atg5/Atg16 heterotrimer, we determined the influence of S0 and S3 on the formation of the heterotrimer. The results showed that after infection, the BiFC signal and BiFC-FRETe (Fig. 4B, E, and F) were significantly enhanced compared to the control group (Fig. 4A). S0 and S3 significantly decreased this elevated BiFC signal and BiFC-FRETe induced by IAV infection (Fig. 4C, D, E and F). A co-IP assay also showed the same result (Fig. 4G). These experiments showed that S0 and S3 can inhibit the formation of the Atg12-Atg5/Atg16 heterotrimer after IAV infection.
Fig 4.

S0 and S3 inhibited the formation of the Atg12-Atg5/Atg16 heterotrimer after IAV infection. (A to D) A549 cells were cotransfected with the pMC-atg5, pMN-atg12, and pEGFP-atg16 plasmids for 6 h, and then A549 cells were treated with 0.5% DMSO only (A), infected with IAV (MOI = 2.0; 8 h) (B), and infected with IAV (MOI = 2.0) and treated with S0 (100 μM) (C) and S3 (80 μM) (D). After incubation for 8 h, each group was visualized using an upright fluorescence microscope. Ex, excitation. (E) Results of the BiFC assay. The fluorescence intensity was measured at 610 nm after excitation at 587 nm. RLU, relative light units. (F) Results of the BiFC-FRET assay. The fluorescence intensity was measured at 509 and 610 nm after excitation at 488 nm, and BiFC-FRETe was expressed as the ratio of ΔFI610 to ΔFI509. (G) Coimmunoprecipitation assay to detect the influence of S0 and S3 on the formation of the Atg5-Atg12/Atg16 heterotrimer. After IAV infection, cells were collected and subjected to a coimmunoprecipitation assay. The data are expressed as means ± SD of two independent experiments with three replicates each. *, P < 0.05, and **, P < 0.01 versus the virus-only group.
S0 and S3 can inhibit the accumulation of autophagosomes induced by IAV infection.
As discussed previously, S0 and S3 can inhibit the formation of the Atg12-Atg5/Atg16 heterotrimer and can inhibit IAV replication at 1 to 4 h p.i., which is the period when IAV-induced autophagy took place (1). In this study, we determined the effects of S0 and S3 on IAV-induced autophagy. We investigated the levels of LC3II using Western blot analysis. At 8, 16, and 24 h p.i., the ratio of LC3II to β-actin in the NC (virus-only group) was significantly increased compared to that of the BG (no-IAV-infection group); ribavirin (PC), S0, and S3 significantly decreased these ratios compared to the NC group (Fig. 5A). Additionally, LC3II could accumulate on autophagosomes, and we constructed a plasmid that expressed the fusion protein EGFP-LC3. As shown in Fig. 5B, the ratio of cells containing EGFP-LC3 dots to cells expressing EGFP in the NC group was significantly higher than that of the BG group, whereas the ratios in the ribavirin-, S0-, and S3-treated groups were significantly decreased. These findings indicated that S0 and S3 could inhibit the accumulation of autophagosomes induced by IAV infection.
Fig 5.

S0 and S3 inhibit the elevation of autophagy induced by IAV. (A) Effects of S0 and S3 on the conversion of LC3I to LC3II determined by Western blotting. A549 cells were infected or not with IAV (MOI = 0.001) and treated or not with ribavirin, S0, or S3. At 8, 16, or 24 h p.i., the cells were collected and subjected to Western blotting using a human LC3 antibody. The zones and average gray values of each band were detected and quantified with BandScan 5.0 software, the multiplication product of the zone area and the average gray value was calculated, and the results are expressed as the ratio of LC3II to β-actin. (B) S0 and S3 inhibited the dot-like aggregation of EGFP-LC3II. A549 cells were transfected with pEGFP-LC3 plasmid and infected with IAV (MOI = 2.0), and at 8 h p.i., the percentages of cells containing EGFP-LC3 dots compared to cells expressing EGFP were calculated. (f) The concentrations of S0, S3, and ribavirin were 100, 80, and 200 μM, respectively. The data are expressed as means ± SD of three independent experiments. *, P < 0.05, and **, P < 0.01 versus the NC group (virus-only group).
S0 and S3 can inhibit oxidative stress, and the activation of the ERK/p38 mitogen-activated protein kinase (MAPK) and IKK pathways induced by IAV infection.
As discussed above, oxidative stress can regulate the formation of the Atg12-Atg5/Atg16 heterotrimer; we speculated that the ability of S0 and S3 to inhibit the formation of the Atg12-Atg5/Atg16 heterotrimer was related to their antioxidant activities, because S0 had been reported to possess antioxidant activity (37, 38). In our study, we investigated the effects of S0 and S3 on IAV-induced oxidative stress and found that after IAV infection, MDA, NO, and OH− were significantly increased, whereas GSH, T-SOD, GR, and CAT were significantly decreased, but S0 and S3 could significantly inhibit the oxidative stress induced by IAV infection (Table 1).
Table 1.
S0 and S3 inhibit the oxidative stress induced by IAVa
| Groupb | GSH (nmol/g tissue) | MDA (nmol/mg protein) | NO (nmol/mg protein) | OH− (U/mg protein) | T-SOD (U/mg protein) | GR (nmol NADPH oxidized) | CAT (nmol H2O2 consumed/min/mg protein) | GSH-Px (103 U/mg protein) |
|---|---|---|---|---|---|---|---|---|
| BG | 29.33 ± 2.53c | 0.073 ± 0.022c | 32.67 ± 2.78c | 90.41 ± 8.76c | 7.06 ± 0.96c | 97.33 ± 8.74c | 59.67 ± 6.74c | 2.03 ± 0.33 |
| NC | 13.33 ± 2.73 | 0.277 ± 0.032 | 78.77 ± 8.83 | 268.62 ± 21.83 | 2.57 ± 0.36 | 36.67 ± 6.84 | 27.77 ± 7.31 | 2.13 ± 0.29 |
| PC | 21.67 ± 2.28d | 0.157 ± 0.036c | 62.67 ± 5.79d | 209.47 ± 26.65c | 4.35 ± 0.78 | 79.83 ± 3.45c | 35.33 ± 4.02d | 2.57 ± 0.33d |
| S0 | 22.67 ± 6.01c | 0.091 ± 0.021c | 52.67 ± 7.01c | 120.33 ± 14.06c | 5.76 ± 0.92d | 87.67 ± 8.36c | 47.33 ± 3.86c | 3.04 ± 0.43c |
| S3 | 26.67 ± 6.96c | 0.087 ± 0.026c | 48.33 ± 8.83c | 106.67 ± 13.08c | 6.73 ± 0.93d | 90.33 ± 9.59c | 50.67 ± 4.73c | 3.21 ± 0.73c |
Data are expressed as the means ± SD of two independent experiments with three replicates each.
In noninfected or infected A549 cells, 0.5% DMSO was used as a BG or NC, respectively, and ribavirin was used as a PC. The concentrations of S0, S3, and ribavirin were 100, 80, and 200 μM, respectively. MOI = 0.001; incubation time, 24 h.
P < 0.01 versus the NC group.
P < 0.05 versus the NC group.
Additionally, the IKK/NF-κB and ERK/JNK/p38 MAPK pathways are oxidant-sensitive signal pathways and are involved in the regulation of autophagy. IKK is required for the expression of Atg5 and Beclin 1 (11). ROS-mediated JNK activation can upregulate the expression of Atg5, Atg7, and Beclin 1 (39). The PI3KC3-MEK-ERK pathway can enhance the accumulation of the Atg12-Atg5/Atg16 heterotrimer (15). p38 MAPK is reported to link oxidative stress to autophagic gene expression (40). As shown in Fig. 6A, IAV infection activated the IKK/NF-κB and ERK/JNK/p38 MAPK signal pathways, and S0 and S3 significantly inhibited the activation of the IKK/NF-κB and ERK/p38 MAPK pathways induced by IAV infection but had little effect on the activation of the JNK pathway. Correspondingly, an oxidant (H2O2; 100 μM), an IKK activator (lipopolysaccharide [LPS]; 10 μM), an ERK activator (epidermal growth factor [EGF]; 100 ng/ml) and a p38 activator (anisomycin; 10 μM) all significantly antagonized the anti-IAV activities of S0 and S3 (Fig. 6B).
Fig 6.
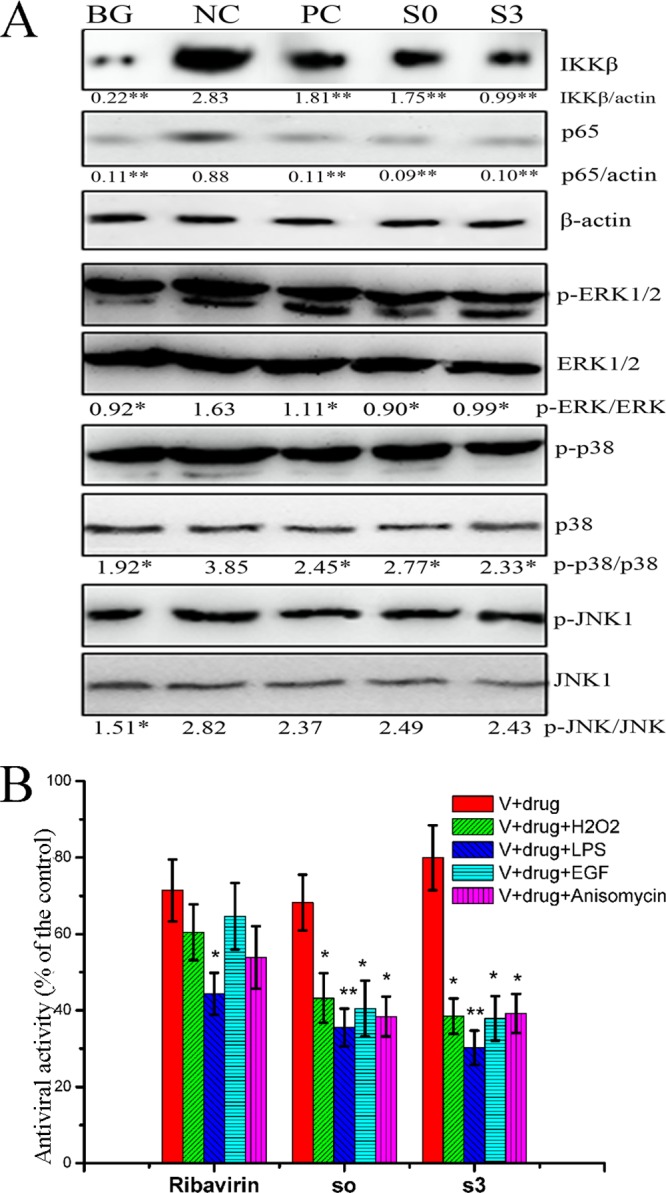
S0 and S3 inhibited the activation of the IKK and ERK/p38 MAPK pathways, and an oxidant and IKK, ERK, and p38 activators antagonized the anti-IAV activities of S0 and S3. (A) S0 and S3 inhibited the activation of the IKK and ERK/p38 MAPK pathways after IAV infection, as determined by Western blotting. A549 cells were infected and treated or not with ribavirin, S0, and S3. After 24 h, the cells were collected and subjected to Western blotting. In noninfected or infected A549 cells, 0.5% DMSO was used as a BG or NC, respectively, and ribavirin was used as a PC (MOI = 0.001; incubation time, 24 h). The zones and average gray values of each band were detected and quantified with BandScan 5.0 software, the multiplication product of the zone area and the average gray value was calculated, and the results were expressed as the ratios of the target genes to β-actin. (B) An oxidant and IKK, ERK, and p38 activators antagonized the anti-IAV activities of S0 and S3. A549 cells were infected (MOI = 0.001) and treated or not with ribavirin, S0, or S3; the cells were simultaneously treated or not with H2O2 (100 μM), LPS (10 μM), EGF (100 ng/ml), and anisomycin (10 μM). After 24 h, the antiviral activity (cell viability) was determined by the SRB method. The concentrations of S0, S3, and ribavirin were 100, 80, and 200 μM, respectively. The data are expressed as the means ± SD of three independent experiments. *, P < 0.05, and **, P < 0.01 versus the NC (A) or virus (V)-plus-drug control (B).
S0 and S3 can inhibit the expression of autophagic genes induced by IAV infection.
It has been reported that IAV infection can upregulate the levels of Beclin 1, Atg16, and the Atg5-Atg12 heterodimer (41). In this study, as shown in Fig. 7A and B, the expression levels of Atg7, Beclin 1, Atg5, Atg3, and Atg12 in the NC group were significantly increased compared to those in the BG group. S0 and S3 significantly inhibited the expression of Atg7, Beclin 1, Atg5, and Atg3 at both the mRNA and protein levels compared to the NC group, but the effect on the expression of Atg12 was not significant (P > 0.05).
Fig 7.

S0 and S3 inhibit the expression of autophagic genes induced by IAV infection and the effects of Atg7 and Atg3 depletion on the formation of the Atg12-Atg5/Atg16 complex and viral yield. (A and B) Effects of S0 and S3 on the expression of autophagic genes after IAV infection as determined by RT-PCR (A) and Western blotting (B). In noninfected or infected A549 cells, 0.5% DMSO was used as a BG or NC, respectively, and ribavirin was used as a PC (MOI = 0.001; incubation time, 24 h). The zones and average gray values of each band were detected and quantified with BandScan 5.0 software, the multiplication product of the zone area and the average gray value was calculated, and the results are expressed as the ratio of the target genes to β-actin. *, P < 0.05, and **, P < 0.01 versus the NC group. (C) Depletion of Atg7 and Atg3 by siRNA inhibited the formation of the Atg12-Atg5/Atg16 complex and reduced viral yield. (a) Abundances of Atg7 and Atg3 after siRNA treatment, as determined by Western blotting. (b) After siRNA treatment, A549 cells were infected with IAV (MOI = 0.001), and the expression level of the Atg5-Atg12/Atg16 complex was determined by co-IP assay at 24 h p.i. (c) The effects of Atg7 and Atg3 depletion on the viral yield were measured by the SRB method at 24 and 48 h p.i. The data are expressed as the means ± SD of three independent experiments. *, P < 0.05, and **, P < 0.01 versus the nondepleted control.
In addition, as mentioned above, the formation of the Atg12-Atg5/Atg16 complex is regulated by Atg7 and Atg3. Here, we also determined the effects of Atg7 and Atg3 depletion by siRNA on the level of the Atg12-Atg5/Atg16 heterotrimer and viral yield. As shown in Fig. 7C, the depletion of Atg7 and Atg3 by siRNA decreased the formation of the Atg12-Atg5/Atg16 complex and reduced the viral yield. Therefore, we speculated that the decreased expression of Atg7 and Atg3 induced by S0 and S3 might inhibit the formation of the Atg12-Atg5/Atg16 complex and reduce the viral yield.
Antiviral activity of S0 and S3 in vivo.
To evaluate whether S0 and S3 could also be effective in controlling influenza A virus infection in vivo, we performed an experiment using BALB/c mice as recipients for the PR8 virus. The mice were infected with a lethal dose of influenza virus A/PR8 and then treated with DMSO-PBS, S0, S3, or ribavirin for 10 days. As expected, the mice treated with the DMSO-PBS vehicle control all succumbed to infection within 8 days, with 100% mortality. In contrast, 40% and 60% of the mice in the S0 and S3 treatment groups, respectively, survived (Fig. 8A); this survival rate is significantly higher than that of the control group. To further verify the antiviral activity of S0 and S3 in vivo, we quantified viral titers in the lungs at day 6. The mice treated with the 25-mg/kg/day dose of S3 had at least a 100-fold decrease in virus titers in their lungs (Fig. 8B). In summary, we have shown that S3 can significantly reduce influenza virus replication and its associated mortality in infected animals.
Fig 8.

In vivo anti-influenza virus activities of S0 and S3 in the lethal mouse model. Mice were infected with a lethal dose of A/PR8 virus (5× MLD50) and treated with DMSO-PBS (16% [vol/vol]), ribavirin (75 mg/kg/day), S0 (25 mg/kg/day), or S3 (25 mg/kg/day) for 10 days from day 0. (A) Survival curves of infected mice (n = 10). The results are expressed as the percent survival evaluated daily for 21 days. (B) Influenza virus titers in the lungs of mice at day 6 postinfection. Virus titers in lung homogenates were determined as described in Materials and Methods. The data are expressed as the means ± SD of three independent experiments. **, P < 0.01 versus the untreated group.
DISCUSSION
Autophagy is an evolutionarily conserved process, and its dysfunction leads to many diseases. It has become a novel therapeutic target for treatment of neurodegenerative diseases, cancers, cardiovascular disease, kidney diseases, and osteoarthritis (42). Autophagy is also involved in the replication of many viruses, such as IAV, HCV, herpes simplex virus (HSV), and CVB3/B4 (1, 4, 5). Therefore, a drug-screening model targeting autophagy is very useful for the treatment of these diseases. In addition, there are two anti-IAV drug-screening strategies: virus-based and cell-based drug-screening models. The virus-based drug-screening model targets IAV proteins, such as neuraminidase and the M2 ion channel. Due to the fact that the RNA-dependent RNA polymerase of IAV lacks a proofreading function, IAV possesses high genetic variability; thus, virus-based drug-screening models often lead to the inevitable selection of drug-resistant viral mutants, and drug-resistant variants can rapidly generate. However, human genetic variability is very low, so cell-based drug-screening models targeting cell components that are essential for virus replication often decrease the emergence of drug-resistant strains (43). We believe that our screening model is very useful for the development of novel drugs that can be used to treat autophagy-related diseases and different viral infections.
There are many crucial regulatory mechanisms in the autophagy signaling pathway, such as the mTOR and Beclin 1 complexes. The regulation of mTOR is achieved through phosphorylation and dephosphorylation reactions (44). Western blotting is the usual method used to detect the activation of mTOR, but this technique is not amenable to large-scale, high-throughput drug screening. Though the Beclin 1 complex is also a crucial autophagic regulatory mechanism, there are too many proteins that interact with Beclin 1, such as Atg14L, UVRAG, Bcl2, MyD88, TRIF, HMGB1, Bif-1, Ambra1, nPIST, VMP1, SLAM, PINK1, and Survivin (45), for the Beclin 1 complex to be used as a target for high-throughput drug screening. In addition, there are two other ubiquitin-like systems, the Atg8 (LC3) and Atg12-Atg5/Atg16 conjugates (46). LC3II can accumulate on the autophagosomes, which seems to make the fusion protein EGFP-LC3 a good drug-screening target. However, the ratio of cells containing EGFP-LC3 dots to cells expressing EGFP must be calculated manually, as in Fig. 5B, so it is also inappropriate for large-scale, high-throughput drug screening. Ultimately, we chose the Atg12-Atg5/Atg16 heterotrimer as our drug-screening target.
The BiFC-FRET assay is a newly developed technique. In this study, we explain that the BiFC-FRETe should be different from the FRET efficiency (FRETe), though both of them can be expressed as the ratio of acceptor to donor emission intensities. FRETe is dependent on the distance between the donor and the acceptor (a sixth-power relationship) (47). BiFC-FRETe is dependent on the distance between the donor and the acceptor and on the quantity of the fluorophore reconstructed by the BiFC reaction. If the BiFC reaction is completely disrupted, the BiFC-FRET system will lack an acceptor (or a donor) fluorophore, and the BiFC-FRETe will be zero.
Using this model, we screened 89 medicinal plants and picked S. marianum L. The anti-IAV activity of the crude extract of S. marianum L. has been reported previously (31). Here, we synthesized 5 amino acid derivatives of silybin and found that 23-(S)-2-amino-3-phenyl-propanoyl-silybin has the best activity. Comparing the polarities of the substituent groups, we found that the hydrophobicities of the substituent groups are positively correlated with their activities. Based on this finding, we will try to synthesize many other silybin derivatives and compare their activities.
To date, two major models have been used to explain the mechanism of IAV-induced acute lung injury, the oxidative-stress and cytokine storm hypotheses. For highly pathogenic IAV, oxidative stress may be the major contributor to IAV-induced acute lung injury (48). Oxidative stress can activate the JNK/ERK/p38 MAPK and IKK pathways, and these signaling pathways can directly or indirectly regulate the formation of the Atg5-Atg12/Atg16 complex and the rate of autophagy (11, 13–15, 49, 50), so we measured the effects of S0 and S3 on oxidative stress and the activation of these signal pathways. Based on our results, we speculate that S0 and S3 may inhibit oxidative stress or the activation of the ERK/p38 MAPK and IKK pathways, subsequently decreasing the expression of autophagic genes (especially the Atg7 and Atg3 genes, which directly regulate the formation of the Atg12-Atg5/Atg16 complex) and ultimately inhibiting autophagy and virus replication. In fact, it has been reported that silybin (25 to 50 μM) can inhibit the activation of the JNK/AP-1, ERK/MEK/Raf, and NHE1/IκBα signal pathways (51, 52). In addition, we determined that S0 and S3 can also inhibit infection by CVB3, the replication of which also involves autophagy (see Fig. S6 in the supplemental material). Similarly, silybin can also inhibit the replication of HCV and HIV-1 (35, 36), two additional viruses with replication processes that involve autophagy.
In conclusion, using a BiFC-FRET drug-screening method that is based on the formation of the Atg5-Atg12/Atg16 complex, we identified 23-(S)-2-amino-3-phenyl-propanoyl-silybin as a promising inhibitor of IAV infection.
Supplementary Material
ACKNOWLEDGMENT
This work has been funded by the National Natural Science Foundation of China (no. 31170852, 81001322, and 81172795).
Footnotes
Published ahead of print 8 July 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.00759-13.
REFERENCES
- 1.Zhou Z, Jiang X, Liu D, Fan Z, Hu X, Yan J, Wang M, Gao GF. 2009. Autophagy is involved in influenza A virus replication. Autophagy 5:321–328 [DOI] [PubMed] [Google Scholar]
- 2.Sun Y, Li C, Shu Y, Ju X, Zou Z, Wang H, Rao S, Guo F, Liu H, Nan W, Zhao Y, Yan Y, Tang J, Zhao C, Yang P, Liu K, Wang S, Lu H, Li X, Tan L, Gao R, Song J, Gao X, Tian X, Qin Y, Xu KF, Li D, Jin N, Jiang C. 2012. Inhibition of autophagy ameliorates acute lung injury caused by avian influenza A H5N1 infection. Sci. Signal. 5:ra16. 10.1126/scisignal.2001931 [DOI] [PubMed] [Google Scholar]
- 3.Ma J, Sun Q, Mi R, Zhang H. 2011. Avian influenza A virus H5N1 causes autophagy-mediated cell death through suppression of mTOR signaling. J. Genet. Genomics 38:533–537 [DOI] [PubMed] [Google Scholar]
- 4.Yoon SY, Ha YE, Choi JE, Ahn J, Lee H, Kweon HS, Lee JY, Kim DH. 2008. Coxsackievirus B4 uses autophagy for replication after calpain activation in rat primary neurons. J. Virol. 82:11976–11978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dreux M, Chisari FV. 2009. Autophagy proteins promote hepatitis C virus replication. Autophagy 5:1224–1225 [DOI] [PubMed] [Google Scholar]
- 6.Mizushima N, Yoshimori T, Ohsumi Y. 2003. Role of the Apg12 conjugation system in mammalian autophagy. Int. J. Biochem. Cell Biol. 35:553–561 [DOI] [PubMed] [Google Scholar]
- 7.Sou YS, Waguri S, Iwata J, Ueno T, Fujimura T, Hara T, Sawada N, Yamada A, Mizushima N, Uchiyama Y, Kominami E, Tanaka K, Komatsu M. 2008. The Atg8 conjugation system is indispensable for proper development of autophagic isolation membranes in mice. Mol. Biol. Cell 19:4762–4775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhao Y, Yang J, Liao W, Liu X, Zhang H, Wang S, Wang D, Feng J, Yu L, Zhu WG. 2010. Cytosolic FoxO1 is essential for the induction of autophagy and tumour suppressor activity. Nat. Cell Biol. 12:665–675 [DOI] [PubMed] [Google Scholar]
- 9.Shao Y, Gao Z, Feldman T, Jiang X. 2007. Stimulation of ATG12-ATG5 conjugation by ribonucleic acid. Autophagy 3:10–16 [DOI] [PubMed] [Google Scholar]
- 10.Xia HG, Zhang L, Chen G, Zhang T, Liu J, Jin M, Ma X, Ma D, Yuan J. 2010. Control of basal autophagy by calpain1 mediated cleavage of ATG5. Autophagy 6:61–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Comb WC, Cogswell P, Sitcheran R, Baldwin AS. 2011. IKK-dependent, NF-kappaB-independent control of autophagic gene expression. Oncogene 30:1727–1732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Niso-Santano M, Criollo A, Malik SA, Michaud M, Morselli E, Marino G, Lachkar S, Galluzzi L, Maiuri MC, Kroemer G. 2012. Direct molecular interactions between Beclin 1 and the canonical NFkappaB activation pathway. Autophagy 8:268–270 [DOI] [PubMed] [Google Scholar]
- 13.Byun JY, Yoon CH, An S, Park IC, Kang CM, Kim MJ, Lee SJ. 2009. The Rac1/MKK7/JNK pathway signals upregulation of Atg5 and subsequent autophagic cell death in response to oncogenic Ras. Carcinogenesis 30:1880–1888 [DOI] [PubMed] [Google Scholar]
- 14.Rouschop KM, van den Beucken T, Dubois L, Niessen H, Bussink J, Savelkouls K, Keulers T, Mujcic H, Landuyt W, Voncken JW, Lambin P, van der Kogel AJ, Koritzinsky M, Wouters BG. 2010. The unfolded protein response protects human tumor cells during hypoxia through regulation of the autophagy genes MAP1LC3B and ATG5. J. Clin. Invest. 120:127–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu L, Yang M, Kang R, Wang Z, Zhao Y, Yu Y, Xie M, Yin X, Livesey KM, Lotze MT, Tang D, Cao L. 2011. HMGB1-induced autophagy promotes chemotherapy resistance in leukemia cells. Leukemia 25:23–31 [DOI] [PubMed] [Google Scholar]
- 16.Sabariegos R, Picazo F, Domingo B, Franco S, Martinez MA, Llopis J. 2009. Fluorescence resonance energy transfer-based assay for characterization of hepatitis C virus NS3-4A protease activity in live cells. Antimicrob. Agents Chemother. 53:728–734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tsai MT, Cheng YH, Liu YN, Liao NC, Lu WW, Kung SH. 2009. Real-time monitoring of human enterovirus (HEV)-infected cells and anti-HEV 3C protease potency by fluorescence resonance energy transfer. Antimicrob. Agents Chemother. 53:748–755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim YS, Cha HJ. 2006. High-throughput and facile assay of antimicrobial peptides using pH-controlled fluorescence resonance energy transfer. Antimicrob. Agents Chemother. 50:3330–3335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yu X, Sainz B, Jr, Uprichard SL. 2009. Development of a cell-based hepatitis C virus infection fluorescent resonance energy transfer assay for high-throughput antiviral compound screening. Antimicrob. Agents Chemother. 53:4311–4319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dai JP, Chen J, Bei YF, Han BX, Wang S. 2009. Influence of borneol on primary mice oral fibroblasts: a penetration enhancer may be used in oral submucous fibrosis. J. Oral Pathol. Med. 38:276–281 [DOI] [PubMed] [Google Scholar]
- 21.Fan JY, Cui ZQ, Wei HP, Zhang ZP, Zhou YF, Wang YP, Zhang XE. 2008. Split mCherry as a new red bimolecular fluorescence complementation system for visualizing protein-protein interactions in living cells. Biochem. Biophys. Res. Commun. 367:47–53 [DOI] [PubMed] [Google Scholar]
- 22.Mirza H, Teo JD, Upcroft J, Tan KS. 2011. A rapid, high-throughput viability assay for Blastocystis spp. reveals metronidazole resistance and extensive subtype-dependent variations in drug susceptibilities. Antimicrob. Agents Chemother. 55:637–648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen CZ, Kulakova L, Southall N, Marugan JJ, Galkin A, Austin CP, Herzberg O, Zheng W. 2011. High-throughput Giardia lamblia viability assay using bioluminescent ATP content measurements. Antimicrob. Agents Chemother. 55:667–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sui Y, Wu Z. 2007. Alternative statistical parameter for high-throughput screening assay quality assessment. J. Biomol. Screen. 12:229–234 [DOI] [PubMed] [Google Scholar]
- 25.Choi HJ, Lim CH, Song JH, Baek SH, Kwon DH. 2009. Antiviral activity of raoulic acid from Raoulia australis against Picornaviruses. Phytomedicine 16:35–39 [DOI] [PubMed] [Google Scholar]
- 26.Dai JP, Wang GF, Li WZ, Zhang L, Yang JC, Zhao XF, Chen XX, Xu YX, Li KS. 2012. High-throughput screening for anti-influenza A virus drugs and study of the mechanism of procyanidin on influenza A virus-induced autophagy. J. Biomol. Screen. 17:605–617 [DOI] [PubMed] [Google Scholar]
- 27.Sidwell RW, Huffman JH, Bailey KW, Wong MH, Nimrod A, Panet A. 1996. Inhibitory effects of recombinant manganese superoxide dismutase on influenza virus infections in mice. Antimicrob. Agents Chemother. 40:2626–2631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Conti G, Magliani W, Conti S, Nencioni L, Sgarbanti R, Palamara AT, Polonelli L. 2008. Therapeutic activity of an anti-idiotypic antibody-derived killer peptide against influenza A virus experimental infection. Antimicrob. Agents Chemother. 52:4331–4337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kumar N, Sharma NR, Ly H, Parslow TG, Liang Y. 2011. Receptor tyrosine kinase inhibitors that block replication of influenza A and other viruses. Antimicrob. Agents Chemother. 55:5553–5559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Smee DF, Hurst BL, Wong MH, Bailey KW, Tarbet EB, Morrey JD, Furuta Y. 2010. Effects of the combination of favipiravir (T-705) and oseltamivir on influenza A virus infections in mice. Antimicrob. Agents Chemother. 54:126–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Song JH, Choi HJ. 2011. Silymarin efficacy against influenza A virus replication. Phytomedicine 18:832–835 [DOI] [PubMed] [Google Scholar]
- 32.Angeli JP, Barcelos GR, Serpeloni JM, Barbosa F, Jr, Nersesyan A, Mantovani MS. 2010. Evaluation of the genotoxic and anti-genotoxic activities of silybin in human hepatoma cells (HepG2). Mutagenesis 25:223–229 [DOI] [PubMed] [Google Scholar]
- 33.Fu H, Lin M, Katsumura Y, Yokoya A, Hata K, Muroya Y, Fujii K, Shikazono N. 2010. Protective effects of silybin and analogues against X-ray radiation-induced damage. Acta Biochim. Biophys. Sin. (Shanghai) 42:489–495 [DOI] [PubMed] [Google Scholar]
- 34.Wang F, Huang K, Yang L, Gong J, Tao Q, Li H, Zhao Y, Zeng S, Wu X, Stockigt J, Li X, Qu J. 2009. Preparation of C-23 esterified silybin derivatives and evaluation of their lipid peroxidation inhibitory and DNA protective properties. Bioorg. Med. Chem. 17:6380–6389 [DOI] [PubMed] [Google Scholar]
- 35.Ahmed-Belkacem A, Ahnou N, Barbotte L, Wychowski C, Pallier C, Brillet R, Pohl RT, Pawlotsky JM. 2010. Silibinin and related compounds are direct inhibitors of hepatitis C virus RNA-dependent RNA polymerase. Gastroenterology 138:1112–1122 [DOI] [PubMed] [Google Scholar]
- 36.McClure J, Lovelace ES, Elahi S, Maurice NJ, Wagoner J, Dragavon J, Mittler JE, Kraft Z, Stamatatos L, Horton H, De Rosa SC, Coombs RW, Polyak SJ. 2012. Silibinin inhibits HIV-1 infection by reducing cellular activation and proliferation. PLoS One 7:e41832. 10.1371/journal.pone.0041832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yang LX, Huang KX, Li HB, Gong JX, Wang F, Feng YB, Tao QF, Wu YH, Li XK, Wu XM, Zeng S, Spencer S, Zhao Y, Qu J. 2009. Design, synthesis, and examination of neuron protective properties of alkenylated and amidated dehydro-silybin derivatives. J. Med. Chem. 52:7732–7752 [DOI] [PubMed] [Google Scholar]
- 38.Yao J, Zhi M, Minhu C. 2011. Effect of silybin on high-fat-induced fatty liver in rats. Braz. J. Med. Biol. Res. 44:652–659 [DOI] [PubMed] [Google Scholar]
- 39.Kim MJ, Woo SJ, Yoon CH, Lee JS, An S, Choi YH, Hwang SG, Yoon G, Lee SJ. 2011. Involvement of autophagy in oncogenic K-Ras-induced malignant cell transformation. J. Biol. Chem. 286:12924–12932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McClung JM, Judge AR, Powers SK, Yan Z. 2010. p38 MAPK links oxidative stress to autophagy-related gene expression in cachectic muscle wasting. Am. J. Physiol. Cell Physiol. 298:C542–C549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Matarrese P, Nencioni L, Checconi P, Ciarlo L, Gambardella L, Ascione B, Sgarbanti R, Garaci E, Malorni W, Palamara AT. 2011. Pepstatin A alters host cell autophagic machinery and leads to a decrease in influenza A virus production. J. Cell. Physiol. 226:3368–3377 [DOI] [PubMed] [Google Scholar]
- 42.Lotz M, Carames B. 2012. Autophagy: a new therapeutic target in cartilage injury and osteoarthritis. J. Am. Acad. Orthop. Surg. 20:261–262 [DOI] [PubMed] [Google Scholar]
- 43.Muller B, Krausslich HG. 2009. Antiviral strategies. Handb. Exp. Pharmacol. 2009:1–24. 10.1007/978-3-540-79086-0_1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Laplante M, Sabatini DM. 2009. mTOR signaling at a glance. J. Cell Sci. 122:3589–3594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Itakura E, Mizushima N. 2009. Atg14 and UVRAG: mutually exclusive subunits of mammalian Beclin 1-PI3K complexes. Autophagy 5:534–536 [DOI] [PubMed] [Google Scholar]
- 46.Geng J, Klionsky DJ. 2008. The Atg8 and Atg12 ubiquitin-like conjugation systems in macroautophagy. Protein modifications: beyond the usual suspects review series. EMBO Rep. 9:859–864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Adedeji AO, Singh K, Calcaterra NE, DeDiego ML, Enjuanes L, Weiss S, Sarafianos SG. 2012. Severe acute respiratory syndrome coronavirus replication inhibitor that interferes with the nucleic acid unwinding of the viral helicase. Antimicrob. Agents Chemother. 56:4718–4728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vlahos R, Stambas J, Selemidis S. 2012. Suppressing production of reactive oxygen species (ROS) for influenza A virus therapy. Trends Pharmacol. Sci. 33:3–8 [DOI] [PubMed] [Google Scholar]
- 49.Geiler J, Michaelis M, Naczk P, Leutz A, Langer K, Doerr HW, Cinatl J., Jr 2010. N-acetyl-L-cysteine (NAC) inhibits virus replication and expression of pro-inflammatory molecules in A549 cells infected with highly pathogenic H5N1 influenza A virus. Biochem. Pharmacol. 79:413–420 [DOI] [PubMed] [Google Scholar]
- 50.Bellot GL, Liu D, Pervaiz S. 2013. ROS, autophagy, mitochondria and cancer: Ras, the hidden master? Mitochondrion 13:155–162 [DOI] [PubMed] [Google Scholar]
- 51.Lin CM, Chen YH, Ma HP, Wang BW, Chiu JH, Chua SK, Ong JR, Shyu KG. 2012. Silibinin inhibits the invasion of IL-6-stimulated colon cancer cells via selective JNK/AP-1/MMP-2 modulation in vitro. J. Agric. Food Chem. 60:12451–12457 [DOI] [PubMed] [Google Scholar]
- 52.Trappoliere M, Caligiuri A, Schmid M, Bertolani C, Failli P, Vizzutti F, Novo E, di Manzano C, Marra F, Loguercio C, Pinzani M. 2009. Silybin, a component of sylimarin, exerts anti-inflammatory and anti-fibrogenic effects on human hepatic stellate cells. J. Hepatol. 50:1102–1111 [DOI] [PubMed] [Google Scholar]
- 53.Ministry of Health of the People's Republic of China 2005. Pharmacopoeia of the People's Republic of China. People's Medical Publishing House, Beijing, China [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.