

Abstract
Development of persistent hepatitis C virus (HCV) infection may be mediated by HCV NS3 · 4A protease-dependent inhibition of host innate immunity. When double-stranded RNA (dsRNA) is detected in virus-infected cells, host innate immunity mounts an antiviral response by upregulating production of type I interferons (α/β interferon [IFN-α/β]); HCV counters by cleaving the IFN-β stimulator 1 (IPS-1) adaptor protein, decreasing synthesis of IFN-α/β. We evaluated HCV protease (telaprevir, boceprevir, and TMC435350), polymerase (HCV-796 and VX-222), and NS5A (BMS-790052) inhibitors for the ability to restore IPS-1-mediated Rig-I signaling by measuring Sendai virus-induced IFN-β promoter activation in HCV replicon cells after various exposure durations. All direct-acting HCV antivirals tested restored mitochondrial localization of IPS-1 and rescued Sendai virus-induced IRF3 signaling after 7 days by inhibiting HCV replication, thereby reducing the abundance of HCV NS3 · 4A protease. With 4-day treatment, HCV protease inhibitors, but not polymerase inhibitors, restored mitochondrial localization of IPS-1 and rescued IFN-β promoter activation in the presence of equivalent levels of NS3 protein in protease or polymerase inhibitor-treated cells. The concentrations of HCV protease and polymerase inhibitors needed to rescue IRF3-mediated signaling in vitro were in the range of those observed in vivo in the plasma of treated HCV patients. These findings suggest that (i) HCV protease, polymerase, and NS5A inhibitors can restore virus-induced IRF3 signaling by inhibiting viral replication, thereby reducing NS3 protease levels, and (ii) HCV protease inhibitors can restore innate immunity by directly inhibiting NS3 protease-mediated cleavage of IPS-1 at clinically achievable concentrations.
INTRODUCTION
Hepatitis C virus (HCV) is a hepatotropic virus that belongs to the family Flaviviridae. Chronic HCV infection can result in fibrosis, cirrhosis, and hepatocellular carcinoma (1). It is estimated that approximately 2% of the world population (∼170 million people) is infected with HCV, making it a major world health concern (2–6). The HCV genome consists of a positive-strand RNA molecule of approximately 9,600 nucleotides encoding both structural and nonstructural proteins. Nonstructural proteins 3 (NS3) and 5B (NS5B) have serine protease and RNA-dependent RNA polymerase functions, respectively, which are essential for proteolytic processing of the nonstructural-protein region of the HCV polyprotein and for viral RNA replication.
Because the HCV polymerase and protease are necessary for the viral replication cycle, both have been attractive targets for anti-HCV drug development (7). Several inhibitors of the NS3 · 4A protease (e.g., Incivek [8] and Victrelis [9]), in combination with pegylated alpha interferon (PEG-IFN) and ribavirin, are approved for treatment of genotype 1 chronic HCV infection in adult patients. Additional direct-acting antivirals (DAAs) targeting HCV polymerase (PSI-7977 [K. V. Kowdley, E. Lawitz, I. Crespo, T. Hassanein, M. Davis, M. DeMicco, D. R. Nelson, D. Bernstein, N. H. Afdhal, I. Jacobson, J. Vierling, S. Gordon, J. K. Anderson, R. H. Hyland, R. G. Hindes, C. Baker, R. Sorensen, E. Albanis, W. T. Symonds, and M. M. Berrey, presented at the 47th Annual Meeting of the European Association for the Study of the Liver {EASL}, Barcelona, Spain, 18 to 22 April 2012], ABT-333 [F. Poordad, E. Lawitz, E. DeJesus, K. Kowdley, I. Gaultier, D. Cohen, W. Xie, L. Larsen, T. Pilot-Matias, G. Koev, E. Dumas, T. Podsadecki, and B. Bernstein, presented at the 47th Annual Meeting of the European Association for the Study of Liver Diseases {EASL}, Barcelona, Spain, 2012], and NS5A protein [10, 11]) or a combination of multiple DAAs (anti-NS5A and anti-NS3) (12; M. Sulkowski, D. Gardiner, E. Lawitz, F. Hinestrosa, D. Nelson, P. Thuluvath, M. Rodriguez-Torres, A. Lok, H. Schwartz, K. R. Reddy, T. Eley, M. Wind-Rotolo, S.-P. Huang, M. Gao, F. McPhee, R. Hindes, W. Symonds, C. Pasquinelli, and D. Grasela, presented at the 47th Annual Meeting of the European Association for the Study of Liver Diseases [EASL], Barcelona, Spain, 2012) with promising efficacy have been described recently.
In addition to being essential for proteolytic cleavage of the viral polyprotein, the HCV NS3 · 4A protease has also been shown to suppress the innate immune response by cleaving beta interferon (IFN-β) stimulator 1 (IPS-1) (13) and Toll/interleukin 1 (IL-1) receptor domain-containing adapter inducing IFN-β (TRIF) (14) in cultured cells. In response to viral replication intermediates, such as viral RNA and proteins, IPS-1 and TRIF adaptor protein act as signaling intermediates in retinoic acid-inducible gene (Rig-I) and/or Toll-like receptor 3 (TLR3) pathways (15–17). Activation of these pathways induces IRF3 activation and subsequent transcriptional activation of type I interferons (IFN-α/β) and interferon-sensitive genes (ISGs) (18). Type I interferons and ISGs play a major role in combating viral infections (19). Interferon production and ISG expression through Rig-I-mediated recognition of HCV RNA has been demonstrated in vitro in Huh7 cells and in vivo in mice (20–22). The role of HCV RNA in IFN pathway stimulation was further demonstrated by Rig-I stimulation in HEK293 cells expressing functional (competent for RNA synthesis) HCV NS5B protein and its blockage by HCV polymerase inhibitor (23). In this system, expression of NS5A inhibited NS5B-mediated RIG-I-dependent luciferase production from the IFN-β promoter. However, these studies were conducted in the absence of other HCV proteins (such as NS3 · 4A protease, NS4B, and NS5A and -B) that have been shown to modulate the host innate immune system (13, 24).
Cleavage of IPS-1 and TRIF by HCV NS3 · 4A blocks the downstream signaling pathway, resulting in inefficient activation of IRF3 and severely reducing the host innate immune response against the viral infection (13, 14). It is possible that HCV protease inhibitors (PI) can play a dual role, referred to as a “double-whammy” effect (25), in countering viral infection through a direct antiviral mechanism, as well as by abrogating the HCV protease-mediated downregulation of innate immunity pathways, such as the Rig-I and TLR3 pathways (26).
HCV patients can be treated with telaprevir or boceprevir HCV PI for 12 to 44 weeks (27, 28). Due to the inhibition of HCV replication, the levels of NS3 · 4A protein will ultimately be insufficient to cleave newly synthesized IPS-1 and TRIF, restoring the IFN signaling pathway. However, a PI can directly limit the efficiency with which IPS-1 and TRIF are cleaved by NS3 · 4A and could restore the IFN signaling pathway. It has been reported that high concentrations (>100-fold over the antiviral 50% effective concentrations [EC50]) of the HCV PI TMC435350 and its analog, TMC380765, are necessary to restore the Rig-I pathway (29) in HCV replicon cells. Because it was unknown whether these high concentrations of HCV PI could be achieved in patients, the clinical relevance of restoration of innate immunity has been a subject of debate in the field (29). Both TMC435350 and TMC380765 were shown to be capable of rescuing IFN-β levels at much higher concentrations (>100-fold over the antiviral EC50 for genotype 1 HCV) (29). However, as recent clinical data suggest (30), the amount of TMC435350 needed for restoration of innate immunity (IFN-α/β) and ISGs in vitro was in the range required for clinical efficacy.
In this study, we evaluated the direct and indirect effects of HCV protease, polymerase, and NS5A inhibitors on innate immunity (IRF3 signaling) in HCV replicon cells. Sendai virus induction of IFN-β promoter transcription and immunofluorescence were used to explore the effects of the dose and duration of treatment on restoration of IPS-1 mitochondrial localization and signaling in HCV replicon cells. We show that short-term exposure to HCV PI, but not HCV polymerase inhibitors, could restore IRF3 signaling, most likely through direct inhibition of the HCV protease. In contrast, prolonged exposure to either HCV protease, polymerase, or NS5A inhibitors could rescue IRF3 signaling at concentrations that can be observed in the plasma of treated patients (clinically achievable concentrations), most likely through an indirect reduction of HCV protease levels resulting from viral-replication inhibition. As the cleavage of IPS-1 is associated with reduced activation of the endogenous IFN system (31), rescue of the IRF3 signaling pathway in vivo might contribute to the significant efficacy observed in HCV clinical trials of DAAs.
MATERIALS AND METHODS
Cell lines.
HCV genotype 1b (HCV-1b) mADE replicon cells (Con1 cells with adaptive mutations) were used in the current study. These cells were generated by transfection of Huh7 cells with RNA from the pBR322-HCV-neo-mADE plasmid with adaptive mutations in HCV nonstructural proteins (32). IFN-α-cured HCV replicon cells were generated by continuous treatment of HCV-1b replicon cells with 10 IU/ml of IFN-α-2b (Calbiochem, San Diego, CA). After 8 weeks, HCV RNA levels were determined by branched-DNA (bDNA) assay and by the absence of G418-selectable colonies. Both HCV replicon cells and IFN-cured replicon cells were propagated in Dulbecco's modified Eagle's medium (DMEM), obtained from JRH Biosciences (Lenexa, KS) and supplemented with 10% heat-inactivated fetal bovine serum (FBS), 2 mM l-glutamine, and non-essential amino acids (JRH).
Antiviral compounds.
All test compounds were synthesized at Vertex Pharmaceuticals Inc. (Cambridge, MA). Telaprevir (8), boceprevir (9), and TMC435350 (33) are peptidomimetic NS3 · 4A PI developed by Vertex, Merck (Whitehouse Station, NJ), and Janssen (Titusville, NJ), respectively. HCV-796 (P. Chandra, D. Raible, D. Harper, J. Speth, S. Villano, and G. Bichier, presented at Digestive Disease Week [DDW], Los Angeles, CA, 2006) and VX-222 (34) were developed by Wyeth and Vertex, respectively, as nonnucleoside inhibitors of HCV NS5B polymerase. BMS-790052 (11) is an inhibitor of HCV NS5A developed by Bristol-Myers Squibb (New York, NY).
HCV RNA analysis.
HCV replicon RNA from cells was measured using a bDNA assay (Quantigene Discover XL kit; Affymetrix Inc., Santa Clara, CA) according to the manufacturer's instructions. Briefly, compound-treated replicon cells were lysed, and HCV RNA was immobilized on capture plates using HCV-specific oligonucleotides (designed based on the 5′ untranslated region [UTR] of the HCV-1b genome sequence [GenBank accession no. AJ238799]) overnight at 53°C, and the relative amounts of captured RNA were measured using oligonucleotide probe sets. The EC50 and EC90 are the concentrations of the compound at which the HCV replicon RNA level in cells is reduced by 50% or 90%, respectively, compared to the untreated replicon cell controls. These values were generated from four-parameter curve-fitting analysis, using Softmax (Molecular Devices, Inc., Sunnyvale, CA). At the end of treatment, with effective inhibition of viral replication by HCV inhibitors, the level of HCV protein was too low to be accurately quantitated. Therefore, the HCV RNA level was quantified and used as a surrogate measure.
IFN-β promoter luciferase reporter assay.
Huh7 replicon cells and the interferon-cured replicon cells were seeded at a density of 2 × 104 cells/well in 48-well plates. The cells were treated with various concentrations of HCV inhibitor, as indicated. At the indicated time points, cells were transfected using Lipofectamine 2000 (Invitrogen, Carlsbad, CA) in triplicate with 200 ng of plasmid (pLUC-IFN-β; a kind gift from Tom Maniatis, Harvard University, Cambridge, MA) encoding an IFN-β promoter fused to the firefly luciferase (FLuc) coding sequence. Ten nanograms of a plasmid carrying the Renilla luciferase (RLuc) coding sequence (pRL-SV40 vector; Promega, Madison, WI) were included as a transfection control. After 24 h, the cells were infected with Sendai virus (20 hemagglutination [HA] units/well) for 18 h. FLuc and RLuc activities were measured using the Dual-Glo luciferase assay system (Promega). To compensate for transfection efficiency, FLuc activity was normalized to RLuc activity for each well. IFN-β promoter activity from Sendai virus-stimulated wells was expressed as the fold increase over unstimulated cells. IFN-β promoter activity in compound-treated HCV replicon cells was expressed as the fold increase over untreated HCV replicon cells (background). The cells were maintained in the presence of the compounds by refreshing the medium containing compounds during transfection and throughout the experiment.
Immunofluorescence.
HCV replicon cells and IFN-cured replicon cells were seeded at a density of 2 × 104 cells/well in 4-well BD Falcon culture slides (Fisher Scientific, Pittsburgh, PA) and were treated with various concentrations of HCV inhibitors as indicated. At the indicated time points posttreatment, the cells were gently fixed with methanol at room temperature, blocked with 10% normal goat serum for 30 min, and stained with NS3 (Axxora LLC, San Diego, CA) and IPS-1 specific antibodies (Abcam Inc., Cambridge, MA) for 1 h. After washing three times with PBS containing 0.1% Triton X-100, the cells were stained with goat anti-rabbit and goat anti-mouse antibodies labeled with fluorescein isothiocyanate (FITC) (Life Technologies, Grand Island, NY) and Alexa 594 (Life Technologies, Grand Island, NY) fluorophores, respectively, for 1 h. The cells were washed three times with PBS containing 0.1% Triton X100, and the nuclei were stained with 2.5 μg/ml of Hoechst 332258. The cells were embedded with Prolong Gold Anti-Fade reagent (Invitrogen, Carlsbad, CA), and staining was detected by fluorescence microscopy.
High-content automated image acquisition and analysis for quantitation of HCV NS3 and IPS-1.
Images were acquired on a Cellomics ArrayScan VTI instrument (ThermoScientific, Pittsburgh, PA). Briefly, 25 fields were captured from each well of a 24-well plate using a 10× objective across three fluorescent channels. For image analysis, field and cell intensities were quantified using the Spot Detector BioApplication, with at least 10,000 events measured per well.
RESULTS
Similar inhibition of HCV replication by HCV protease and polymerase inhibitors at early time points.
The effect of telaprevir, an HCV PI (35), or HCV-796, a polymerase inhibitor (36), on HCV replication was evaluated in HCV replicon cells treated with different concentrations of the inhibitors for 1, 2, or 3 days (Fig. 1A). Both dose- and time-dependent reductions in HCV replicon RNA levels were observed with telaprevir (Fig. 1B) and HCV-796 (Fig. 1D). Consistent with previous reports (35, 36), 3 days of treatment with both telaprevir and HCV-796 decreased HCV replicon RNA levels, with EC50s of 0.9 μM and 0.034 μM, respectively. Furthermore, 3 days of treatment with the highest tested concentration of either inhibitor resulted in a greater than 95% reduction in HCV RNA levels and analogous decreases in NS3 protein levels (Fig. 1B and D and data not shown).
Fig 1.

IFN-β promoter activation after 4-day treatment. (A) Experimental design. (B to E) Time- and dose-dependent inhibition of HCV replication and recovery of IFN-β activity were evaluated by analyzing HCV RNA levels and IFN-β promoter-driven luciferase expression (means ± standard deviations [SD] from three replicates) in the presence of the HCV protease inhibitor telaprevir (B and C) or a polymerase inhibitor (HCV-796) (D and E) in HCV replicon cells.
An HCV PI, but not a polymerase inhibitor, can rescue IFN-β promoter activity with a shorter incubation time.
The effects of HCV inhibitors on HCV RNA and protein levels and on the restoration of HCV IRF3-dependent IFN-β promoter activity were examined using Sendai virus infection of HCV replicon cells. In cells that have been previously cured of HCV replicon using IFN, IFN-β promoter activity can be stimulated 4.7- ± 1.5-fold above background levels compared to only a 1.3- ± 0.04-fold activation of IFN-β promoter in HCV replicon cells (background levels) (data not shown). As shown in Fig. 1C, there was a time- and dose-dependent increase in the ability of telaprevir to restore IRF3 signaling. At day 4, robust (5.5- ± 0.3-fold over background) and moderate (3.6- ± 0.3-fold over background) activation of IFN-β promoter activity and 98% and 90% decreases in HCV RNA replication were observed with 10 μM and 3 μM telaprevir, respectively. In contrast the HCV polymerase inhibitor HCV-796 only partially restored IFN-β promoter activity to 1.3- ± 0.2-fold at day 4 (Fig. 1E), despite a 99% reduction in HCV RNA at 0.5 and 1.5 μM. Thus, although 4 days of treatment of HCV replicon cells with either telaprevir or HCV-796 resulted in similar inhibition of HCV RNA replication (Fig. 1B and D) and a concordant decrease in NS3 protein levels (data not shown), only telaprevir was able to significantly restore IFN-β promoter activity.
Restoration of the IRF3 signaling pathway at early time points may be a result of inhibition of IPS-1 cleavage by an HCV PI.
To further explore whether the difference in IFN-β promoter restoration observed with a protease versus a PI was due to a difference in the amount of IPS-1 cleavage, we evaluated NS3 and IPS-1 expression and intracellular distribution in HCV replicon cells treated with telaprevir and HCV-796 for 3 days. As expected, in the absence of treatment, we observed punctate staining of HCV NS3 protein in HCV replicon cells (Fig. 2B) accompanied by diffuse IPS-1 staining (Fig. 2C) (37), suggesting the existence of cleaved IPS-1 in the cytoplasm. HCV RNA and NS3 protein levels in the cells treated with both inhibitors were affected in parallel. Although HCV replicon RNA levels were reduced more than 95% in cells treated with an HCV PI (telaprevir) or an HCV polymerase inhibitor (HCV-796) (Fig. 1B and 1D), HCV NS3 proteins were still detected (Fig. 2J and N), albeit at a lower level than in untreated HCV replicon cells (Fig. 2B). The sustained presence of HCV NS3 is consistent with a reported 26-hour half-life (38). IPS-1 is a mitochondrial protein and typically appears punctate by immunohistochemistry (Fig. 2G). In HCV replicon cells treated with telaprevir for 3 days, a shift in the localization of cytoplasmic cleaved IPS-1 to mitochondrion-associated full-length IPS-1 was detected as a change from a diffuse (Fig. 2C) to a punctate (Fig. 2K) staining pattern. Quantitative analyses showed that telaprevir decreased the number of HCV protease (NS3)-positive cells but did not eliminate them, and it also increased the amount of full-length IPS-1 punctate staining (Fig. 3A). These data suggest that at early times after the initiation of treatment, telaprevir was able to rescue IPS-1 from cleavage by the HCV protease remaining in HCV replicon cells.
Fig 2.

Restoration of mitochondrial association of IPS-1 following 3-day treatment. Shown is immunofluorescence visualization of NS3 and IPS-1 expression in HCV replicon cells following 3-day incubation with the HCV protease inhibitor telaprevir or the HCV polymerase inhibitor HCV-796. Nuclei are stained blue with Hoechst 332258.
Fig 3.
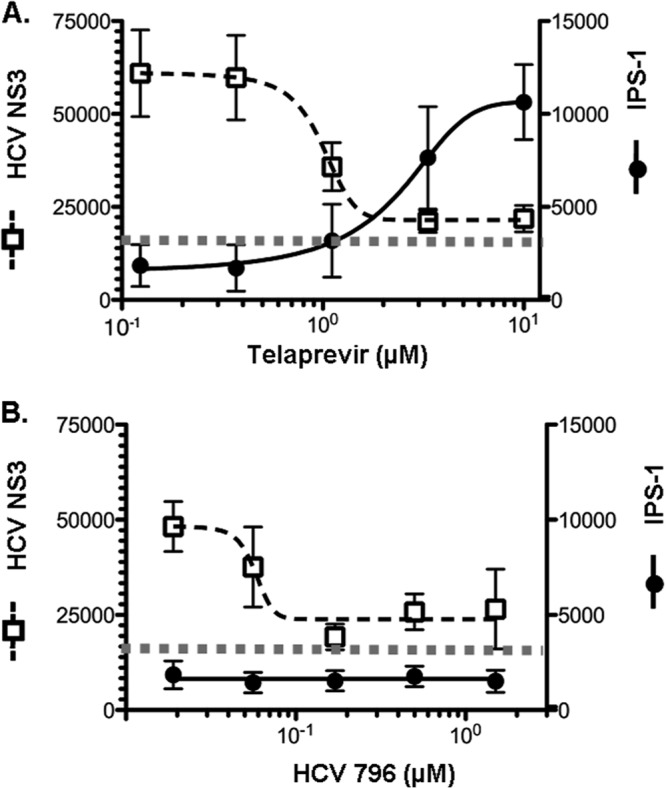
Recovery of mitochondrial IPS-1 association in HCV replicon cells after 3-day treatment with an HCV protease inhibitor, but not an HCV polymerase inhibitor. Field intensity levels of NS3 and IPS-1 immunofluorescence (means ± SD from 25 fields) were quantitated from each image using high-content image analysis (as described in Materials and Methods) after treatment with the HCV protease inhibitor telaprevir (A) or the HCV polymerase inhibitor HCV-796 (B). The horizontal dotted lines represent NS3 background staining.
In HCV replicon cells treated with HCV-796, diffuse IPS-1 staining was observed, consistent with cleaved IPS-1 (Fig. 2O). While the HCV-796-treated cells showed a concomitant reduction in levels of HCV NS3 protease, it was not accompanied by an increase in full-length IPS-1 staining (Fig. 3B). These observations, together with the results shown in Fig. 1, suggest that telaprevir was able to restore the Rig-I pathway and IFN-β promoter activity by a combined effect of inhibiting HCV replication (thereby reducing the total amount of NS3 indirectly) and direct inhibition of HCV protease cleavage of IPS-1.
PI-mediated rescue of IFN-β promoter activation.
To determine whether other PI could rescue IRF3-mediated transcription, boceprevir and TMC435350 were evaluated for the ability to restore the Rig-I pathway and IFN-β promoter activity. Both inhibitors showed a greater than 3 log10 decrease in HCV RNA levels at day 14 in patients when dosed in combination with IFN and ribavirin (39, 40).
In cells treated with telaprevir for 4 days, 50% rescue of IFN-β promoter activity was observed at 3.1 ± 1.1 μM, whereas antiviral activity was observed with an EC50/EC90 of 0.8 ± 0.07 and 1.8 ± 0.07 μM, respectively. HCV replicon cells treated with boceprevir demonstrated suppression of HCV RNA replication with an EC50/EC90 of 1.1 ± 0.07 μM and 2.3 ± 0.07 μM, respectively (Table 1), and a 50% recovery of IFN-β promoter activity at 3.3 ± 0.1 μM. Similarly, TMC435350 showed an EC50/EC90 of 0.003 ± 0.001 μM and 0.01, respectively (Table 1), and a 50% rescue of IFN-β promoter activity was observed at 0.1 ± 0.01 μM. These studies indicate that the rescue of IFN-β promoter activity is not restricted to telaprevir but is also observed with other HCV NS3 inhibitors, suggesting there is a common mechanism for HCV PI that allows the restoration of the Rig-I pathway. In contrast, neither HCV polymerase nor NS5A inhibitors showed rescue of IFN-β promoter activity at the highest concentration tested at day 4 (Table 1).
Table 1.
Inhibition of HCV replication and recovery of IFN-β promoter activation after incubation with HCV inhibitors
| Class | Inhibitor | Anti-HCV activity (HCV-1b replicon)a,b |
IFN-β promoter activation EC50 (μM)b |
Pharmacokinetics data in plasma of treated patientsc |
|||||
|---|---|---|---|---|---|---|---|---|---|
| EC50 (μM) | EC90 (μM) | 4 days | 7 days | Dose (mg) (reference) | Cavg (μM) | Cmax (μM) | Cmin (μM) | ||
| Protease inhibitors | Telaprevir | 0.8 ± 0.07 | 1.8 ± 0.07 | 3.1 ± 1.1 | 1.6 ± 0.7 | 750 q8h (8, 50) | 4.1–4.9 | 5.2 | 3.0–3.8 |
| Boceprevir | 1.1 ± 0.07 | 2.3 ± 0.07 | 3.3 ± 0.1 | NTd | 800 q8h (9, 52) | 1.3–1.9 | 3.3–4.1 | 0.2 | |
| TMC 435350 | 0.003 ± 0.001 | 0.01 ± 0 | 0.1 ± 0.01 | NT | 100 QD (30) | 0.4 | 1.0 | 0.1 | |
| 200 QD (30) | 3.9–11 | 8.2–15.3 | 2.2 | ||||||
| Polymerase inhibitors | HCV-796 | 0.009 ± 0.001 | 0.03 ± 0.01 | >1.5 | 0.3 ± 0.1 | 1,000 BID (P. Chandra, D. Raible, D. Harper, J. Speth, S. Villano, and G. Bichier, presented at Digestive Disease Week [DDW], Los Angeles, CA, 2006) | 3.7 | 4.9 | NA |
| VX-222 | 0.0003 ± 0.0002 | 0.012 ± 0.006 | >10 | 0.02 ± 0.01 | 750 BID (E. L. Rodriguez-Torres, M. B. Conway, K. Kaita, A. M. Sheikh, R. Ghalib, R. Adrover, C. Cooper, M. Silva, M. Rosario, B. Bourgault, L. Proulx, and J. G. McHutchison, presented at the 45th Annual Meeting of the European Association for the Study of Liver disease [EASL], Geneva, Switzerland, 2010) | 11.8 | 23.1 | NA | |
| NS5A inhibitor | BMS-790052 | 5.9 × 10−6 ± 4.7 × 10−6 | 50 ×10−6 ± 2.6 ×10−6 | >0.001 | 108 × 10−6 ± 11.3 × 10−6 | 30 BID (10) | 0.6 | 1.1 | 0.3 |
EC50 and EC90 values for RNA levels were determined in the replicon assay. The EC50 value for IFN-β promoter activation was evaluated in parallel.
Mean ± SD from two independent experiments.
Cmax, maximum concentration in plasma; Cmin, minimum concentration in plasma; Cavg, average concentration in plasma in treated patients.
NT, not tested.
Prolonged inhibition of HCV replication in vitro by HCV inhibitors with different mechanisms of action restores the IFN-α/β signaling pathway.
Previous studies by other investigators using the Sendai virus activation system have shown that treatment of HCV replicon cells for 48 hours with HCV PI, but not HCV polymerase inhibitors, can restore the Rig-I pathway and IFN-β promoter activity (29). However, it can be reasoned that if NS3 levels drop low enough there will not be sufficient NS3 to cleave newly synthesized IPS-1, leading to restoration of the signaling pathway. Therefore, the combination of suppressing new viral replication and increasing the length of treatment to allow NS3 protein to turn over and be eliminated should allow any potent HCV inhibitor to restore IPS-1-mediated signaling. To test this hypothesis, HCV replicon cells were incubated with telaprevir, the HCV NS5B polymerase inhibitors HCV-796 and VX-222, or the NS5A inhibitor BMS-790052 for 6 days. The treated cells were transfected with the IFN-β promoter reporter construct for an additional day, and IFN-β activation was measured 18 h post-Sendai virus infection (Fig. 4A). Sendai virus infection did not lead to a significant induction of IFN-β promoter activity in untreated HCV replicon cells, and in the absence of any inhibitor, HCV replicon cells showed a 3.0- ± 0.6-fold activation of IFN-β promoter activity (considered background). In contrast, a 7.0- ± 2.7-fold activation above background (untreated HCV replicon cells) was observed in IFN-cured cells, where HCV RNA was cleared by IFN treatment.
Fig 4.

Recovery of IFN-β promoter activation following inhibition of HCV replication after 7-day incubation with DAA HCV inhibitors. (A) Experimental design. (B to E) Reduction in RNA levels and restoration of IFN-β activity (means ± SD from three replicates) following treatment with the HCV protease inhibitor telaprevir (B), polymerase inhibitor HCV-796 (C) or VX-222 (D), or NS5A inhibitor BMS-790052 (E).
Consistent with the results shown in Fig. 3, incubation of HCV replicon cells with 10 μM telaprevir for 7 days resulted in significant activation of the IFN-β promoter by 7.1- ± 1.8-fold above background (Fig. 4B). Similarly, cells treated with 1 μM HCV polymerase inhibitor HCV-796 or VX-222 showed a dose-dependent increase in IFN-β promoter activity of 6.8- ± 1.2-fold or 5.0- ± 1.8-fold above background, respectively (Fig. 4C and D). To further extend the analyses of the abilities of different classes of HCV inhibitors to rescue IFN-β promoter activity, HCV replicon cells were treated with BMS-790052, an HCV NS5A inhibitor. In cells treated with 100 and 1,000 pM BMS-790052, there was greater than 95% suppression of HCV RNA, and IFN-β promoter activation was induced to 3.3- ± 0.7-fold and 4.5- ± 1.0-fold above background, respectively, indicating rescue of IFN-β promoter activation.
Extended treatment with an HCV PI restores mitochondrial association of IPS-1.
To investigate whether restoration of IFN-β activity in cells treated for 6 days with HCV-796 coincided with mitochondrial association of IPS-1, changes in the amount and intracellular localization of IPS-1 and NS3 were determined. In the absence of drug, a majority of HCV replicon cells were positive for HCV NS3 staining (Fig. 5B), and IPS-1 appeared as diffuse intracellular staining (Fig. 5C), consistent with the release of IPS-1 from mitochondria after cleavage by HCV protease (41). In contrast, IFN-cured replicon cells do not show cytoplasmic staining for HCV NS3 protein, due to the absence of HCV replicon (Fig. 5F), and IPS-1 demonstrated a punctate cytoplasmic expression pattern (Fig. 5G), which is in agreement with a mitochondrial localization pattern. HCV NS3 protein was not observed in HCV replicon cells incubated for 6 days with 10 μM telaprevir (Fig. 5J) or 0.3 μM HCV-796 (Fig. 5N), and a punctate pattern of IPS-1 staining in the cytoplasm (Fig. 5K and O) was observed, which is consistent with the rescue of IFN-β promoter trans-activation (Fig. 4). IPS-1 displayed a punctate staining pattern matching its mitochondrial localization (Fig. 5L and P), consistent with restoration of the IFN-β signal pathway. A similar staining pattern was observed with 3 μM telaprevir (data not shown). In cells treated with 0.03 μM HCV-796, the HCV NS3 protein was present in some cells (Fig. 6A); however, a mixed pattern of diffuse and punctate IPS-1 staining was seen (Fig. 6B), indicating that the uncleaved form of IPS-1 protein was beginning to appear, which is consistent with a low level of IFN-β promoter activation.
Fig 5.

Restoration of mitochondrial IPS-1 localization after 6-day treatment with HCV protease or polymerase inhibitors. Shown is immunofluorescence visualization of NS3 and IPS-1 expression in HCV replicon cells following 6-day incubation with the HCV protease inhibitor telaprevir or the HCV polymerase inhibitor HCV-796. Nuclei are stained blue with Hoechst 332258.
Fig 6.
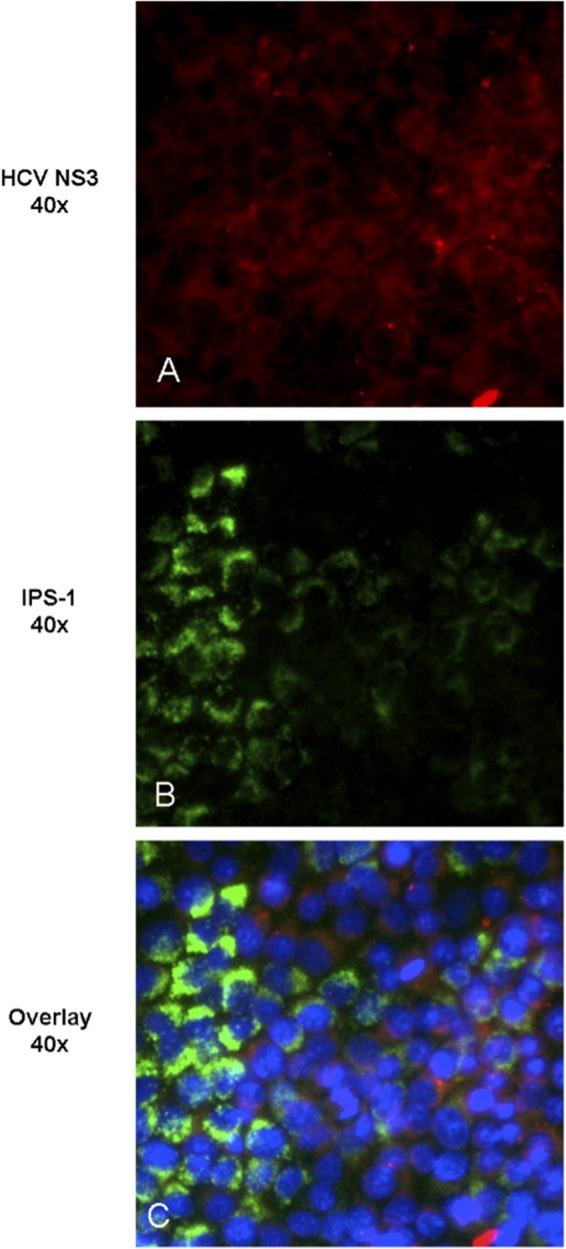
Recovery of IPS-1 mitochondrial association occurs only in replicon cells lacking NS3 after treatment with the HCV polymerase inhibitor HCV-796. Shown is protein localization following long-term (6-day) incubation with 0.03 μM HCV polymerase inhibitor HCV-796. Nuclei are stained blue with Hoechst 332258.
DISCUSSION
Approximately 60 to 80% of individuals exposed to HCV develop a chronic infection (42), indicating that HCV is often successful in subverting the innate and adaptive immune responses. The exact mechanisms underlying the immune evasion, persistence, and nonresponsiveness of chronically infected patients to interferon and ribavirin combination therapy, even in the presence of hepatic IFN-stimulated genes (43), are not well understood, but studies suggest that one mechanism by which HCV may subvert innate immunity is through cleavage of IPS-1.
Recently, type III IFNs (λ), but not type I IFNs (α/β), have been reported to be induced in response to HCV infection of primary human hepatocytes (44), human fetal hepatocytes (45), HCV-infected chimpanzee livers (44), and hepatocytes from liver biopsies of chronically HCV-infected patients (46). Type I IFNs demonstrated more potent antiviral activity than type III IFNs (44) in the in vitro experiments. Surprisingly, neutralization of endogenously produced type III IFNs could further increase the antiviral activity of type I IFN in the primary hepatocyte cultures (44). These results are suggestive of the limited antiviral activity of endogenously produced type III IFNs, as well as the importance of restoration of type I IFN production in response to HCV infection.
Regulation of type I IFN production occurs through coordinated activation of NF-κB and IRF3 (37, 41, 47) via IPS-1, which is a mitochondrial outer membrane protein. However, cleavage of IPS-1 by HCV NS3 · 4A protease at Cys508 dissociates it from its C-terminal membrane anchor in mitochondria and results in redistribution to the cytoplasm, where it is unable to function in the Rig-I signaling pathway (13). Interference in the induction of interferon-stimulated gene synthesis via the Rig-I-mediated pathway may play a significant role in the subversion of the host innate immune response targeted against HCV. Inhibition of HCV NS3 · 4A-catalyzed cleavage of IPS-1 and restoration of the mitochondrial form can be achieved either by inhibiting HCV NS3 · 4A-mediated cleavage of IPS-1 with HCV PI (37) or by lowering the abundance of HCV protease via inhibition of HCV replication by any HCV inhibitor.
Previous studies have largely focused on restoration of innate immune responses in the context of HCV PI (i.e., ITMN-C [37], SCH6 [26], TMC435350 [29], and BILN2061 [48, 49]). Other studies had previously shown that high levels (>100 times the EC50) of TMC435350 were needed to restore the Rig-I pathway in HCV replicon cells in vitro; due to the lack of clinical data at the time of the in vitro studies, it was not known whether such high levels could be achieved in vivo in patients (29). In this report, we show that telaprevir and TMC435350 can restore the Rig-I signaling pathway in HCV replicon cells with drug concentrations in the range of what has been reported in the plasma of treated HCV patients (8, 30, 50).
Consistent with previous studies (29), treatment with an HCV PI such as telaprevir, but not with an HCV polymerase inhibitor, could inhibit the HCV NS3 · 4A protease-mediated cleavage of IPS-1 during a 4-day incubation, despite the presence of equal levels of viral RNA and NS3 protein. It is likely that the rapid restoration of IPS-1 activity in the presence of PI at earlier time points (4 days) is due to the direct inhibition of IPS-1 cleavage by HCV protease. Our observation of HCV PI-mediated mitochondrial reassociation of IPS-1 and rescue of the Rig-I pathway is consistent with earlier reports of restoration of innate immunity by diverse classes of PI, such as ITMN-C (37), SCH6 (26), TMC435350 (29), and BILN2061 (48, 49), excluding a compound-specific effect.
We hypothesized that it may be possible to restore innate immunity by lowering the abundance of HCV protease as a consequence of blocking viral replication. Therefore, we evaluated the abilities of different classes of HCV replication inhibitors to restore the Sendai virus-activated Rig-I pathway and IRF3-dependent IFN-β promoter transcription in HCV replicon cells. These studies revealed that the HCV polymerase inhibitors HCV-796 and VX-222, as well as the NS5A inhibitor BMS-790052, could rescue the Rig-I pathway and that this may likely be the result of the reduction in NS3 protein levels as a consequence of inhibition of viral replication. Furthermore, the concentration of HCV-796 needed to restore 50% IFN-β promoter activity after a 7-day incubation is within the average concentration in plasma (Cavg) of HCV-796 observed in patients (P. Chandra, D. Raible, D. Harper, J. Speth, S. Villano, and G. Bichier, presented at Digestive Disease Week [DDW], Los Angeles, CA, 2006). To our knowledge, this is the first report demonstrating the restoration of the virus-activated Rig-I pathway in vitro in HCV replicon cells (in the presence of all the HCV proteins) by an HCV polymerase inhibitor and an NS5A inhibitor. Our observation of IFN-β pathway restoration by HCV inhibitors could potentially be extended to other classes of HCV inhibitors.
In contrast to the rescue of IPS-1 signaling with HCV PI at 4 days, HCV polymerase or NS5A inhibitor treatment required 7 days of treatment for restoration of the IFN-β pathway. These observed differences could be explained by the contributions of two factors to IPS-1 cleavage: HCV NS3 protease levels and protease activity. A small amount of NS3 · 4A may be sufficient to cleave de novo-synthesized IPS-1. HCV protease inhibitors rescued IPS-1 signaling after 4 days of treatment, whereas HCV polymerase or NS5A inhibitors required 7 days to restore the IFN pathway. The difference may be explained by the level and activity of the HCV NS3 protease, both of which contribute to IPS-1 cleavage. Nonprotease inhibitors indirectly decrease HCV NS3 protease levels by inhibiting viral replication and preventing the synthesis of new HCV protease but do not affect NS3 · 4A existing in cells before treatment. At the start of treatment, it is likely that preexisting NS3 · 4A, which has a half-life of ∼26 h (38), is able to cleave IPS-1, even after polymerase or NS5A inhibitors suppress replication. At later time points, when HCV replication is extensively inhibited by any replication inhibitor, insufficient amounts of NS3 · 4A are synthesized to cleave the de novo-synthesized IPS-1. The formation of native IPS-1 could then mediate signaling of the Rig-I pathway. However, the ability of NS3 PI to inhibit NS3 · 4A protease activity might allow IPS-1 restoration at NS3 levels that normally would be sufficient to cleave all newly synthesized IPS-1. This means that HCV protease inhibitors may have the previously described “double-whammy” effect (25) on IPS-1 recovery: (i) a primary effect on reduction of NS3 protein levels and (ii) a secondary effect on reduction of the cleavage rate of IPS-1, consistent with previous reports (26).
The observed IFN-β promoter activation and staining pattern for IPS-I may indicate that IRF3-dependent transcription can be rescued by extended treatment with HCV protease, polymerase, or NS5A inhibitors. For the polymerase and NS5A inhibitors, this may be due to a reduction of NS3 · 4A protease levels as a consequence of extended inhibition of HCV RNA replication and an incubation time of several times the half-life of the NS3 protease.
In clinical studies, the Cavg of telaprevir was 4.1 to 4.9 μM in patients (8, 50). Taking into consideration the 2-fold serum shift observed for telaprevir antiviral activity in in vitro cell culture studies (unpublished data), the concentrations required to rescue IFN-β promoter activation in HCV replicon cells (Table 1) are similar to the Cavg observed with telaprevir in human plasma. Similarly, the EC50 for stimulation of the IFN-β promoter in Sendai virus-infected replicon cells for TMC435350 is 0.1 μM, which is well within the Cavg observed in the clinic (30) (Table 1).
HCV-infected patients previously treated with PEG-IFN and ribavirin had sustained viral response (SVR) rates of 29% to 83%, depending upon the patient's prior response, upon retreatment with telaprevir in combination with PEG-IFN and ribavirin (51). Additionally, the combination of HCV polymerase inhibitors and ribavirin has shown 47% SVR12 in prior nonresponders (F. Poordad, E. Lawitz, K. V. Kowdley, G. T. Everson, B. Freilich, D. Cohen, S. Siggelkow, M. Heckaman, R. Menon, T. Pilot-Matias, T. Podsadecki, and B. Bernstein, presented at the 47th Annual Meeting of the European Association for the Study of the Liver [EASL], Barcelona, Spain, 18 to 22 April 2012, LB abstr. 1399). Taken together with our in vitro observations, it is possible that DAA-mediated restoration of the IFN pathway might contribute to the improved SVR rates in this patient population with poor IFN responsiveness. However, this remains to be proven in the clinic.
ACKNOWLEDGMENTS
We appreciate the help of Nagraj Mani, Tara Keiffer, and Joshua Leeman (Infectious Diseases, Vertex Pharmaceuticals Inc.) for helpful comments and suggestions on the manuscript.
G.K., C.L., and R.R. are employees of Vertex Pharmaceuticals Inc. and may own stock or options in the company. J.G., K.S., and A.D.K. are former employees of Vertex Pharmaceuticals Inc. and may own or may have owned stock or options in the company at the time this research was performed. Medical writing, editorial, and coordination support was provided by Mrudula Donepudi, Elizabeth Dorn, and Erika D. Reynoso, employees of Vertex Pharmaceuticals Inc. who may own stock or options in the company.
The lead author, Gururaj Kalkeri, wrote the first draft of the manuscript based on an outline of content that was reviewed and agreed upon by all of the authors. All of the authors made substantial contributions to study design, interpretation of data, and critical revision of the manuscript for important intellectual content.
Footnotes
Published ahead of print 8 July 2013
REFERENCES
- 1.Hoofnagle JH. 2002. Course and outcome of hepatitis C. Hepatology 36:S21–S29 [DOI] [PubMed] [Google Scholar]
- 2.Curry MP. 2004. Hepatitis B and hepatitis C viruses in liver transplantation. Transplantation 78:955–963 [DOI] [PubMed] [Google Scholar]
- 3.Purcell RH. 1994. Hepatitis C virus: historical perspective and current concepts. FEMS Microbiol. Rev. 14:181–192 [DOI] [PubMed] [Google Scholar]
- 4.Seeff LB. 2002. Natural history of chronic hepatitis C. Hepatology 36:S35–S46 [DOI] [PubMed] [Google Scholar]
- 5.Strader DB, Wright T, Thomas DL, Seeff LB. 2004. Diagnosis, management, and treatment of hepatitis C. Hepatology 39:1147–1171 [DOI] [PubMed] [Google Scholar]
- 6.Chak E, Talal AH, Sherman KE, Schiff ER, Saab S. 2011. Hepatitis C virus infection in USA: an estimate of true prevalence. Liver Int. 31:1090–1101 [DOI] [PubMed] [Google Scholar]
- 7.Kwong AD, McNair L, Jacobson I, George S. 2008. Recent progress in the development of selected hepatitis C virus NS3.4A protease and NS5B polymerase inhibitors. Curr. Opin. Pharmacol. 8:522–531 [DOI] [PubMed] [Google Scholar]
- 8.Vertex Pharmaceuticals Incorporated 2012. INCIVEK UPI. Vertex Pharmaceuticals Incorporated, Cambridge, MA [Google Scholar]
- 9.Merck and Co., Inc 2011. VICTRELIS UPI. Merck and Co., Inc., Whitehouse Station, NJ [Google Scholar]
- 10.Nettles RE, Gao M, Bifano M, Chung E, Persson A, Marbury TC, Goldwater R, DeMicco MP, Rodriguez-Torres M, Vutikullird A, Fuentes E, Lawitz E, Lopez-Talavera JC, Grasela DM. 2011. Multiple ascending dose study of BMS-790052, a nonstructural protein 5A replication complex inhibitor, in patients infected with hepatitis C virus genotype 1. Hepatology 54:1956–1965 [DOI] [PubMed] [Google Scholar]
- 11.Gao M, Nettles RE, Belema M, Snyder LB, Nguyen VN, Fridell RA, Serrano-Wu MH, Langley DR, Sun JH, O'Boyle DR, II, Lemm JA, Wang C, Knipe JO, Chien C, Colonno RJ, Grasela DM, Meanwell NA, Hamann LG. 2010. Chemical genetics strategy identifies an HCV NS5A inhibitor with a potent clinical effect. Nature 465:96–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lok AS, Gardiner DF, Lawitz E, Martorell C, Everson GT, Ghalib R, Reindollar R, Rustgi V, McPhee F, Wind-Rotolo M, Persson A, Zhu K, Dimitrova DI, Eley T, Guo T, Grasela DM, Pasquinelli C. 2012. Preliminary study of two antiviral agents for hepatitis C genotype 1. N. Engl. J. Med. 366:216–224 [DOI] [PubMed] [Google Scholar]
- 13.Meylan E, Curran J, Hofmann K, Moradpour D, Binder M, Bartenschlager R, Tschopp J. 2005. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature 437:1167–1172 [DOI] [PubMed] [Google Scholar]
- 14.Li K, Foy E, Ferreon JC, Nakamura M, Ferreon AC, Ikeda M, Ray SC, Gale M, Jr, Lemon SM. 2005. Immune evasion by hepatitis C virus NS3/4A protease-mediated cleavage of the Toll-like receptor 3 adaptor protein TRIF. Proc. Natl. Acad. Sci. U. S. A. 102:2992–2997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yoneyama M, Kikuchi M, Natsukawa T, Shinobu N, Imaizumi T, Miyagishi M, Taira K, Akira S, Fujita T. 2004. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat. Immunol. 5:730–737 [DOI] [PubMed] [Google Scholar]
- 16.Bode JG, Brenndorfer ED, Haussinger D. 2007. Subversion of innate host antiviral strategies by the hepatitis C virus. Arch. Biochem. Biophys. 462:254–265 [DOI] [PubMed] [Google Scholar]
- 17.Sumpter R, Jr, Loo YM, Foy E, Li K, Yoneyama M, Fujita T, Lemon SM, Gale M., Jr 2005. Regulating intracellular antiviral defense and permissiveness to hepatitis C virus RNA replication through a cellular RNA helicase, RIG-I. J. Virol. 79:2689–2699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grandvaux N, ten Oever BR, Servant MJ, Hiscott J. 2002. The interferon antiviral response: from viral invasion to evasion. Curr. Opin. Infect. Dis. 15:259–267 [DOI] [PubMed] [Google Scholar]
- 19.Gale M, Jr, Foy EM. 2005. Evasion of intracellular host defence by hepatitis C virus. Nature 436:939–945 [DOI] [PubMed] [Google Scholar]
- 20.Saito T, Owen DM, Jiang F, Marcotrigiano J, Gale M., Jr 2008. Innate immunity induced by composition-dependent RIG-I recognition of hepatitis C virus RNA. Nature 454:523–527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schnell G, Loo YM, Marcotrigiano J, Gale M., Jr 2012. Uridine composition of the poly-U/UC tract of HCV RNA defines non-self recognition by RIG-I. PLoS Pathog. 8:e1002839. 10.1371/journal.ppat.1002839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang YL, Guo YJ, Bin L, Sun SH. 2009. Hepatitis C virus single-stranded RNA induces innate immunity via Toll-like receptor 7. J. Hepatol. 51:29–38 [DOI] [PubMed] [Google Scholar]
- 23.Ranjith-Kumar CT, Wen Y, Baxter N, Bhardwaj K, Cheng Kao C. 2011. A cell-based assay for RNA synthesis by the HCV polymerase reveals new insights on mechanism of polymerase inhibitors and modulation by NS5A. PLoS One 6:e22575. 10.1371/journal.pone.0022575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Moriyama M, Kato N, Otsuka M, Shao RX, Taniguchi H, Kawabe T, Omata M. 2007. Interferon-beta is activated by hepatitis C virus NS5B and inhibited by NS4A, NS4B, and NS5A. Hepatol. Int. 1:302–310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Farley S. 2003. A double whammy for hep C. Nat. Rev. Drug Discov. 2:419 [Google Scholar]
- 26.Foy E, Li K, Wang C, Sumpter R, Jr, Ikeda M, Lemon SM, Gale M., Jr 2003. Regulation of interferon regulatory factor-3 by the hepatitis C virus serine protease. Science 300:1145–1148 [DOI] [PubMed] [Google Scholar]
- 27.Forestier N, Zeuzem S. 2012. Telaprevir for the treatment of hepatitis C. Expert Opin. Pharmacother. 13:593–606 [DOI] [PubMed] [Google Scholar]
- 28.Chang MH, Gordon LA, Fung HB. 2012. Boceprevir: a protease inhibitor for the treatment of hepatitis C. Clin. Therapeut. 34:2021–2038 [DOI] [PubMed] [Google Scholar]
- 29.Liang Y, Ishida H, Lenz O, Lin TI, Nyanguile O, Simmen K, Pyles RB, Bourne N, Yi M, Li K, Lemon SM. 2008. Antiviral suppression vs restoration of RIG-I signaling by hepatitis C protease and polymerase inhibitors. Gastroenterology 135:1710–1718 [DOI] [PubMed] [Google Scholar]
- 30.Tsantrizos YS. 2009. TMC-435, an NS3/4A protease inhibitor for the treatment of HCV infection. Curr. Opin. Investig. Drugs 10:871–881 [PubMed] [Google Scholar]
- 31.Bellecave P, Sarasin-Filipowicz M, Donze O, Kennel A, Gouttenoire J, Meylan E, Terracciano L, Tschopp J, Sarrazin C, Berg T, Moradpour D, Heim MH. 2010. Cleavage of mitochondrial antiviral signaling protein in the liver of patients with chronic hepatitis C correlates with a reduced activation of the endogenous interferon system. Hepatology 51:1127–1136 [DOI] [PubMed] [Google Scholar]
- 32.Lin C, Lin K, Luong YP, Rao BG, Wei YY, Brennan DL, Fulghum JR, Hsiao HM, Ma S, Maxwell JP, Cottrell KM, Perni RB, Gates CA, Kwong AD. 2004. In vitro resistance studies of hepatitis C virus serine protease inhibitors, VX-950 and BILN 2061: structural analysis indicates different resistance mechanisms. J. Biol. Chem. 279:17508–17514 [DOI] [PubMed] [Google Scholar]
- 33.Lin TI, Lenz O, Fanning G, Verbinnen T, Delouvroy F, Scholliers A, Vermeiren K, Rosenquist A, Edlund M, Samuelsson B, Vrang L, de Kock H, Wigerinck P, Raboisson P, Simmen K. 2009. In vitro activity and preclinical profile of TMC435350, a potent hepatitis C virus protease inhibitor. Antimicrob. Agents Chemother. 53:1377–1385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yi G, Deval J, Fan B, Cai H, Soulard C, Ranjith-Kumar CT, Smith DB, Blatt L, Beigelman L, Kao CC. 2012. Biochemical study of the comparative inhibition of hepatitis C virus RNA polymerase by VX-222 and filibuvir. Antimicrob. Agents Chemother. 56:830–837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Perni RB, Almquist SJ, Byrn RA, Chandorkar G, Chaturvedi PR, Courtney LF, Decker CJ, Dinehart K, Gates CA, Harbeson SL, Heiser A, Kalkeri G, Kolaczkowski E, Lin K, Luong YP, Rao BG, Taylor WP, Thomson JA, Tung RD, Wei Y, Kwong AD, Lin C. 2006. Preclinical profile of VX-950, a potent, selective, and orally bioavailable inhibitor of hepatitis C virus NS3-4A serine protease. Antimicrob. Agents Chemother. 50:899–909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kneteman NM, Howe AY, Gao T, Lewis J, Pevear D, Lund G, Douglas D, Mercer DF, Tyrrell DL, Immermann F, Chaudhary I, Speth J, Villano SA, O'Connell J, Collett M. 2009. HCV796: a selective nonstructural protein 5B polymerase inhibitor with potent anti-hepatitis C virus activity in vitro, in mice with chimeric human livers, and in humans infected with hepatitis C virus. Hepatology 49:745–752 [DOI] [PubMed] [Google Scholar]
- 37.Loo YM, Owen DM, Li K, Erickson AK, Johnson CL, Fish PM, Carney DS, Wang T, Ishida H, Yoneyama M, Fujita T, Saito T, Lee WM, Hagedorn CH, Lau DT, Weinman SA, Lemon SM, Gale M., Jr 2006. Viral and therapeutic control of IFN-beta promoter stimulator 1 during hepatitis C virus infection. Proc. Natl. Acad. Sci. U. S. A. 103:6001–6006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wolk B, Sansonno D, Krausslich HG, Dammacco F, Rice CM, Blum HE, Moradpour D. 2000. Subcellular localization, stability, and trans-cleavage competence of the hepatitis C virus NS3-NS4A complex expressed in tetracycline-regulated cell lines. J. Virol. 74:2293–2304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mederacke I, Wedemeyer H, Manns MP. 2009. Boceprevir, an NS3 serine protease inhibitor of hepatitis C virus, for the treatment of HCV infection. Curr. Opin. Investig. Drugs 10:181–189 [PubMed] [Google Scholar]
- 40.Reesink HW, Fanning GC, Abou Farha K, Weegink C, Van Vliet A, van 't Klooster G, Lenz O, Aharchi F, Marien K, Van Remoortere P, de Kock H, Broeckaert F, Meyvisch P, Van Beirendonck E, Simmen K, Verloes R. 2010. Rapid HCV-RNA decline with once-daily TMC435: a phase I study in healthy volunteers and hepatitis C patients. Gastroenterology 138:913–921 [DOI] [PubMed] [Google Scholar]
- 41.Seth RB, Sun L, Ea CK, Chen ZJ. 2005. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell 122:669–682 [DOI] [PubMed] [Google Scholar]
- 42.Nelson PK, Mathers BM, Cowie B, Hagan H, Des Jarlais D, Horyniak D, Degenhardt L. 2011. Global epidemiology of hepatitis B and hepatitis C in people who inject drugs: results of systematic reviews. Lancet 378:571–583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Su AI, Pezacki JP, Wodicka L, Brideau AD, Supekova L, Thimme R, Wieland S, Bukh J, Purcell RH, Schultz PG, Chisari FV. 2002. Genomic analysis of the host response to hepatitis C virus infection. Proc. Natl. Acad. Sci. U. S. A. 99:15669–15674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Thomas E, Gonzalez VD, Li Q, Modi AA, Chen W, Noureddin M, Rotman Y, Liang TJ. 2012. HCV infection induces a unique hepatic innate immune response associated with robust production of type III interferons. Gastroenterology 142:978–988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Marukian S, Andrus L, Sheahan TP, Jones CT, Charles ED, Ploss A, Rice CM, Dustin LB. 2011. Hepatitis C virus induces interferon-lambda and interferon-stimulated genes in primary liver cultures. Hepatology 54:1913–1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jouan L, Chatel-Chaix L, Melancon P, Rodrigue-Gervais IG, Raymond VA, Selliah S, Bilodeau M, Grandvaux N, Lamarre D. 2012. Targeted impairment of innate antiviral responses in the liver of chronic hepatitis C patients. J. Hepatol. 56:70–77 [DOI] [PubMed] [Google Scholar]
- 47.Kato H, Sato S, Yoneyama M, Yamamoto M, Uematsu S, Matsui K, Tsujimura T, Takeda K, Fujita T, Takeuchi O, Akira S. 2005. Cell type-specific involvement of RIG-I in antiviral response. Immunity 23:19–28 [DOI] [PubMed] [Google Scholar]
- 48.Cheng G, Zhong J, Chisari FV. 2006. Inhibition of dsRNA-induced signaling in hepatitis C virus-infected cells by NS3 protease-dependent and -independent mechanisms. Proc. Natl. Acad. Sci. U. S. A. 103:8499–8504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jouan L, Melancon P, Rodrigue-Gervais IG, Raymond VA, Selliah S, Boucher G, Bilodeau M, Grandvaux N, Lamarre D. 2010. Distinct antiviral signaling pathways in primary human hepatocytes and their differential disruption by HCV NS3 protease. J. Hepatol. 52:167–175 [DOI] [PubMed] [Google Scholar]
- 50.Lawitz E, Rodriguez-Torres M, Muir AJ, Kieffer TL, McNair L, Khunvichai A, McHutchison JG. 2008. Antiviral effects and safety of telaprevir, peginterferon alfa-2a, and ribavirin for 28 days in hepatitis C patients. J. Hepatol. 49:163–169 [DOI] [PubMed] [Google Scholar]
- 51.Zeuzem S, Andreone P, Pol S, Lawitz E, Diago M, Roberts S, Focaccia R, Younossi Z, Foster GR, Horban A, Ferenci P, Nevens F, Mullhaupt B, Pockros P, Terg R, Shouval D, van Hoek B, Weiland O, Van Heeswijk R, De Meyer S, Luo D, Boogaerts G, Polo R, Picchio G, Beumont M. 2011. Telaprevir for retreatment of HCV infection. N. Engl. J. Med. 364:2417–2428 [DOI] [PubMed] [Google Scholar]
- 52.Hulskotte E, Gupta S, Xuan F, van Zutven M, O'Mara E, Feng HP, Wagner J, Butterton J. 2012. Pharmacokinetic interaction between the HCV protease inhibitor boceprevir and cyclosporine and tacrolimus in healthy volunteers. Hepatology 56:1622–1630 [DOI] [PubMed] [Google Scholar]