

Abstract
As opposed to Enterococcus faecalis, which is intrinsically resistant to lincosamides, streptogramins A, and pleuromutilins (LSAP phenotype) by production of the ABC protein Lsa(A), Enterococcus faecium is naturally susceptible. Since this phenotype may be selected for in vivo by quinupristin-dalfopristin (Q-D), the aim of this study was to investigate the molecular mechanism of acquired LSAP resistance in E. faecium. Six LSAP-resistant in vitro mutants of E. faecium HM1070 as well as three different pairs of clinical isolates (pre- and postexposure to Q-D) were studied. The full genome sequence of an in vitro mutant (E. faecium UCN90B) was determined by using 454 sequencing technology and was compared with that of the parental strain. Single-nucleotide replacement was carried out to confirm the role of this mutation. By comparative genomic analysis, a point mutation was found within a 1,503-bp gene coding for an ABC homologue showing 66% amino acid identity with Lsa(A). This mutation (C1349T) led to an amino acid substitution (Thr450Ile). An identical mutation was identified in all in vitro and in vivo resistant strains but was not present in susceptible strains. The wild-type allele was named eat(A) (for Enterococcus ABC transporter), and its mutated allelic variant was named eat(A)v. The introduction of eat(A)v from UCN90B into HM1070 conferred the LSAP phenotype, whereas that of eat(A) from HM1070 into UCN90B restored susceptibility entirely. This is the first description of the molecular mechanism of acquired LSAP resistance in E. faecium. Characterization of the biochemical mechanism of resistance and the physiological role of this ABC protein need further investigations.
INTRODUCTION
Enterococci, particularly Enterococcus faecalis and Enterococcus faecium, are responsible for numerous infections worldwide, being the second or third most common pathogens in hospitals (1). For several years, these opportunistic pathogens have become more and more resistant to antibiotics with the emergence of vancomycin-resistant E. faecium (VREF) isolates belonging mostly to clonal complex 17 (CC17) (2). Since VREF isolates are resistant to β-lactam and glycopeptide antibiotics, only a few molecules are still active against these isolates, such as quinupristin-dalfopristin (Q-D), linezolid, and daptomycin (2, 3).
Q-D is an injectable streptogramin approved by the U.S. FDA for the treatment of severe VREF infections associated with bacteremia (3). Streptogramins form with macrolides (e.g., erythromycin) and lincosamides (e.g., lincomycin and clindamycin), a group of structurally distinct antibiotics (referred to as MLS) that present similar mechanism of action and cross-resistance patterns (4). Actually, streptogramins correspond to a mixture of two compounds that act synergically: streptogramins A (e.g., dalfopristin) and streptogramins B (e.g., quinupristin). In addition, pleuromutilins (e.g., tiamulin) are also a class of protein synthesis inhibitors that share ribosomal binding sites with lincosamide and streptogramin A antibiotics (5).
In enterococci, MLS resistance is due mainly to a ribosomal alteration mediated by a ribosomal methylase encoded by the erm(B) or erm(A) [formerly designated erm(TR)] gene, which is responsible for cross-resistance to all macrolides, lincosamides, and streptogramins B (MLSB phenotype) that can be constitutive or inducible (4). Besides this common mechanism of resistance, there is a peculiar phenotype exhibiting cross-resistance to lincosamides, streptogramins A, and pleuromutilins, called the LSAP phenotype (formerly the LSA phenotype) due to ABC proteins. For instance, intrinsic LSAP resistance in E. faecalis is due to the production of the ABC (ATP-binding cassette) homologue Lsa(A) (6–8). Other Lsa-like proteins have been involved in LSAP resistance: Lsa(B) and Lsa(E), identified in Staphylococcus spp. (9–12), and Lsa(C), described in Streptococcus agalactiae (13, 14). As opposed to E. faecalis, E. faecium is intrinsically susceptible to all macrolides and related compounds, but this LSAP phenotype may be selected in vivo after Q-D therapy exposure (15). Even if the support of resistance is very likely mediated through a chromosomal mutation(s), the biochemical and genetic basis of this resistance was not elucidated.
The aim of the study was then to (i) identify the molecular mechanism of LSAP resistance in E. faecium mutants selected both in vitro and in vivo, (ii) confirm the role of a single point mutation in LSAP resistance by allelic replacement, and (iii) evaluate the prevalence of this resistance mechanism among a collection of well-characterized VREF clinical isolates.
(A preliminary report of this work was presented at the 22nd European Congress of Clinical Microbiology and Infectious Diseases, London, United Kingdom, 31 March to 3 April 2012 [16].)
MATERIALS AND METHODS
Bacterial strains and antimicrobial susceptibility testing.
Strains selected both in vitro and in vivo were studied (Table 1). Six LSAP-resistant mutants of E. faecium HM1070 were selected in vitro with a frequency of ca. 10−9 by using lincomycin, dalfopristin, or tiamulin (at 1 μg/ml, 16 μg/ml, and 2 μg/ml, respectively). Approximately 109 CFU of exponentially growing bacteria was plated onto brain heart infusion (BHI) agar containing antibiotic concentrations (i.e., 4× MIC), and mutants were recovered after 48 h of incubation in ambient air at 37°C. Also, three pairs of clinical isolates collected from three different patients who were treated with Q-D were included (15). For each patient, one LSAP-susceptible isolate (preexposure to Q-D) and one LSAP-resistant isolate (postexposure to Q-D) were obtained. Noteworthy, strains from each pair were indistinguishable by pulsed-field gel electrophoresis (PFGE) (15).
Table 1.
Bacterial strains and plasmids used in this study
| Strain or plasmid | Relevant characteristic(s) and antibiotic resistance(s)a | Reference or source |
|---|---|---|
| Strains | ||
| E. faecium HM1070 | Wild-type parental strain; Fusr Rifr | 31 |
| E. faecium UCN90A | LSAPr mutant derived from HM1070 (selected in vitro with Lin) | This study |
| E. faecium UCN90B | LSAPr mutant derived from HM1070 (selected in vitro with Lin) | This study |
| E. faecium UCN91A | LSAPr mutant derived from HM1070 (selected in vitro with Da) | This study |
| E. faecium UCN91B | LSAPr mutant derived from HM1070 (selected in vitro with Da) | This study |
| E. faecium UCN92A | LSAPr mutant derived from HM1070 (selected in vitro with Tia) | This study |
| E. faecium UCN92B | LSAPr mutant derived from HM1070 (selected in vitro with Tia) | This study |
| E. faecium UCN94 | HM1070 derivative with allelic replacement of eat(A) by eat(A)v from UCN90B (LSAP phenotype) | This study |
| E. faecium UCN95 | UCN90B derivative with allelic replacement of eat(A)v by eat(A) from HM1070 (wild-type phenotype) | This study |
| E. faecium UCN80 | Susceptible strain (isolated from patient B) | 15 |
| E. faecium UCN80-1 | LSAPr mutant derived from UCN80 (selected in vivo with Q-D) | 15 |
| E. faecium UCN81 | Susceptible strain (isolated from patient C) | 15 |
| E. faecium UCN81-1 | LSAPr mutant derived from UCN81 (selected in vivo with Q-D) | 15 |
| E. faecium UCN82 | Susceptible strain (isolated from patient F) | 15 |
| E. faecium UCN82-1 | LSAPr mutant derived from UCN82 (selected in vivo with Q-D) | 15 |
| E. coli TOP10 | Cloning strain | Invitrogen |
| Plasmids | ||
| pCR2.1-TOPO | General AT cloning vector; Kanr Ampr | Invitrogen |
| pG1KT | Derivative of the thermosensitive shuttle plasmid pG(+)host5 (containing promoterless and terminatorless Kanr cassette AphA-3); used for mutagenesis; Eryr | 19 |
| pG1KTΩeat(A) | pG1KT derivative carrying the 3′ end of eat(A); used for allelic replacement in UCN90B | This study |
| pG1KTΩeat(A)v | pG1KT derivative carrying the 3′ end of eat(A)v; used for allelic replacement in HM1070 | This study |
Ampr, ampicillin resistance; Da, dalfopristin; Eryr, erythromycin resistance; Fusr, fusidic acid resistance; Kanr, kanamycin resistance; Lin, lincomycin; LSAP, lincosamides-streptogramins A-pleuromutilins (LSAP phenotype); Q-D, quinupristin-dalfopristin; Rifr, rifampin resistance; Tia, tiamulin; eat(A), wild-type allele; eat(A)v, allele of eat(A) with a single mutation (C1349T) leading to an amino acid substitution (Thr450Ile).
A collection of 60 clonally unrelated (different PFGE profiles) VREF clinical isolates were also screened for the presence of the putative point mutation responsible for LSAP resistance. These strains were received at the French Reference Centre for Enterococci between 2006 and 2008 and were extensively characterized by phenotypic and genotypic methods (17).
E. faecium HM1070 and E. faecium UCN90B were used for allelic replacement experiments. Staphylococcus aureus ATCC 29213 and Escherichia coli TOP10 were used as a control for antimicrobial susceptibility testing and as a cloning strain, respectively.
MICs of erythromycin, lincomycin, clindamycin, dalfopristin, quinupristin, Q-D, and tiamulin were determined by the broth microdilution method (tested range, 0.06 to 64 μg/ml) according to CLSI guidelines (18).
Whole-genome sequencing.
Genomic DNA was extracted from mid-log-phase cultures of E. faecium HM1070 (LSAP-susceptible strain) and E. faecium UCN90B (LSAP-resistant mutant derived from E. faecium HM1070) by using NucleoBond buffer set III and the NucleoBond AX-G 100 system (Macherey-Nagel, Hoerdt, France) according to the manufacturer's instructions. High-throughput sequencing was performed by using a 454 Life Sciences (Roche) GS-FLX system (DNAVision, Charleroi, Belgium). Shotgun sequencing for E. faecium HM1070 led to an assembly of 138 contigs with sizes ranging from 619 to 83,932 bp, with an aggregate genome size of 2,591,399 bp and an 11.3× average coverage of the genome, while data for E. faecium UCN90B were as follows: 268 contigs (sizes from 557 to 91,723 bp), an aggregate genome of 2,562,062 bp, and an 8.5× average genomic coverage. Comparative genomic analysis of the two strains was performed by using the Mosaik (http://code.google.com/p/mosaik-aligner/) and Samtools (http://samtools.sourceforge.net/) bioinformatics software tools (DNAVision, Charleroi, Belgium). The nucleotide and deduced protein sequences for each contig were analyzed with the BLASTN and BLASTX programs available on the Internet at the National Center for Biotechnology Information website (http://blast.ncbi.nlm.nih.gov/Blast.cgi).
PCR amplification and sequencing.
Bacterial genomic DNA was extracted by using the QIAamp DNA minikit (Qiagen, Courtaboeuf, France). PCR experiments for the detection of the point mutation putatively involved in LSAP resistance were carried out under standard conditions by using primers synthesized by Sigma-Aldrich France (Table 2). Briefly, 5 μl of total DNA was subjected to PCR in a 50-μl reaction mixture containing 1× PCR buffer (10 mM Tris-HCl [pH 8.3], 50 mM KCl), 1.5 mM MgCl2, 200 μM each deoxynucleotide triphosphate, 0.4 μM each primer, and 1 U of GoTaq Flexi DNA polymerase (Promega, Charbonnières-les-Bains, France). PCR amplifications were performed by using a Mastercycler gradient thermal cycler (Eppendorf, Le Pecq, France) as follows: (i) an initial denaturation step for 5 min at 95°C; (ii) 30 cycles of PCR, with 1 cycle consisting of 30 s at 95°C, 30 s at 55°C, and 1 min at 72°C; and (iii) a final extension step for 5 min at 72°C. Purified PCR products were then directly sequenced with the same sets of primers in both directions (GATC Biotech, Konstanz, Germany).
Table 2.
Deoxynucleotide primers used in this study
| Primera | Nucleotide sequence (5′–3′) | Positionsb | Purpose |
|---|---|---|---|
| lsaF-Efm-F | TTTGAACAACCTCCGAAAGC | 660490–660509 | Detection of the point mutation in eat(A) |
| lsaF-Efm-R | TTTCTGTGCCTGCATCTGTC | 660927–660946 | |
| lsaF-Efm-mut-F | GCAATCGTGAATCGGATGG | 660098–660116 | Allelic replacement of eat(A) and checking of pG1KT derivatives |
| lsaF-Efm-mut-R | GGAATCGTACAGCGAACGC | 661148–661166 | |
| M13-Fm | GTTGTAAAACGACGGCCAG | NA | |
| M13-Rm | GGATAACAATTTCACACAGG | NA | |
| kana-R | GCTTATATACCTTAGCAGGAG | NA | |
| lsaF-Efm-3′-R | TCCCCTTAACCATACCTTGTTG | 660791–660812 | |
| lsaF-Efm-cont-F | TCGAGCAGATGAGTATGGG | 660543–660561 | |
| lsaF-Efm-cont-R | TTCTAATTGTTCTTGATTGAAG | 660649–660670 | |
| lsaF-Efm-GSP-R1 | TTGCATCAATTGGAGCT | 659565–659581 | Determination of start transcription site of eat(A) by 5′ RACE |
| lsaF-Efm-GSP-R2 | AGGCAAATTCTTGCTGATGG | 659455–659474 | |
| lsaF-Efm-GSP-R3 | TTGTCTTTCCTCGACCGTTT | 659386–659405 |
cont, control; Efm, Enterococcus faecium; F, forward primer; GSP, gene-specific primer; mut, mutagenesis; R, reverse primer.
Primer positions were determined according to the genome sequence of E. faecium Aus0004 (GenBank accession no. NC_017022). NA, not applicable.
Allelic replacement by site-directed mutagenesis.
Fragments containing the 3′ ends of the eat(A) gene and a variant of the eat(A) gene [eat(A)v] were amplified by using primers lsa-Efm-mut-F and lsa-Efm-mut-R (Table 2) from E. faecium HM1070 and E. faecium UCN90B, respectively. PCR products were cloned into the pCR2.1-TOPO plasmid (Invitrogen) and transformed into E. coli TOP10 cells. Recombinant plasmids were extracted by using a QIAprep Spin Miniprep kit (Qiagen, Courtaboeuf, France), according to the manufacturer's instructions, and then digested with the EcoRI enzyme (New England BioLabs, Evry, France). Restriction products were then cloned into the thermosensitive plasmid pG1KT, which is a derivative of the shuttle plasmid pG(+)host5 containing the promoterless and terminatorless kanamycin resistance cassette AphA-3 (19), as previously described (20). Briefly, E. coli transformants were selected on medium containing erythromycin (150 μg/ml), and construction was checked by specific PCR amplifications (Table 2). Recombinant plasmids pG1KTΩeat(A) and pG1KTΩeat(A)v were then introduced by electrotransformation into E. faecium UCN90B and E. faecium HM1070, respectively (Table 1). E. faecium transformants were selected on medium containing kanamycin (500 μg/ml) and erythromycin (15 μg/ml) after an incubation step at 42°C, allowing plasmid integration into the chromosome. The spontaneous loss of pG1KT was obtained by daily subculture in tryptone-soy broth (AES Laboratories, Combourg, France) at 30°C with no antibiotic. Candidate colonies were tested for their susceptibility to MLS and checked by PCR sequencing.
5′ RACE.
Total RNAs were extracted from cultures of E. faecium HM1070 by using the ZR Fungal/Bacterial RNA Miniprep kit (Zymo Research, Irvine, CA). The transcription start site (TSS) and promoter sequences were then determined by using the 5′ rapid amplification of cDNA ends (RACE) system kit (Invitrogen), according to the manufacturer's instructions, with different specific primers (Table 2).
Multiple alignment and phylogenetic analysis.
Sequence comparison and phylogenetic analysis were performed by using the neighbor-joining algorithm with ClustalX software (version 1.83).
Nucleotide sequence accession numbers.
The nucleotide sequences of eat(A) and eat(A)v were deposited in the GenBank database under accession no. KF010778 and KF010779, respectively.
RESULTS
Identification of a single mutation in an lsa-like gene.
By comparing the entire genome of E. faecium HM1070 (LSAP-susceptible strain) and that of E. faecium UCN90B (LSAP-resistant mutant derived from E. faecium HM1070), we found 50 different mutations, including one within a 1,503-bp gene coding for a 500-amino-acid (ca. 58-kDa) ABC protein homologue of Lsa-like proteins. This mutation was responsible for a transition (C1349T) leading to the amino acid substitution Thr450Ile (Fig. 1). By using specific PCR primers, a strictly identical mutation (C1349T) was identified among all in vitro and in vivo resistant strains but was not present in susceptible strains. According to the recommendations for tetracycline and MLS nomenclature (http://faculty.washington.edu/marilynr/), the wild-type gene was named eat(A) (for E nterococcus ABC transporter), whereas the allele with the single point mutation C1349T was designated eat(A)v. A BLAST analysis showed that eat(A) was present in all sequenced E. faecium genomes but not in other enterococcal species (data not shown), suggesting that eat(A) is species specific for E. faecium and is an intrinsic gene of this species. The Eat(A) protein displayed 66%, 44%, 43%, and 42% amino acid identities with other proteins conferring LSAP-type resistance in various Gram-positive organisms, Lsa(A), Lsa(E), Lsa(B), and Lsa(C), respectively. Like other Lsa-like proteins, the structure of Eat(A) showed duplications of Walker A and B motifs, the ABC signature, and the H-loop switch (Fig. 1).
Fig 1.

Amino acid sequence comparison of the Eat(A) and Eat(A)v ABC proteins with Lsa-like proteins involved in MLS resistance, Lsa(A), Lsa(B), Lsa(C), and Lsa(E). The two copies of Walker A and B motifs and ABC signatures are boxed. Similarities in amino acid sequences are marked by asterisks (same amino acid), colons (strong similarity), and dots (family similarity). Multiple-sequence alignment was done with ClustalX 1.83 software. The location of the Thr450Ile substitution is indicated by an arrow.
Drug susceptibility patterns conferred by the single mutation C1349T.
By single-nucleotide allelic replacement, we demonstrated that the unique substitution Thr450Ile was responsible for the LSAP phenotype (Table 3). Indeed, the introduction of the mutated allele eat(A)v from E. faecium UCN90B into susceptible E. faecium strain HM1070 (also known as strain UCN94) conferred the LSAP phenotype with an increase of MICs of lincomycin (from 0.25 to 8 μg/ml), clindamycin (from 0.12 to 2 μg/ml), dalfopristin (from 4 to >64 μg/ml), Q-D (from 0.5 to 2 μg/ml), and tiamulin (from 0.5 to 64 μg/ml), whereas MICs of erythromycin and quinupristin did not change (Table 3). Conversely, the introduction of the wild-type allele eat(A) from E. faecium HM1070 into LSAP-resistant E. faecium strain UCN90B (also known as strain UCN95) restored entire susceptibility to lincomycin (from 8 to 0.25 μg/ml), clindamycin (from 2 to 0.06 μg/ml), dalfopristin (from >64 to 4 μg/ml), Q-D (from 2 to 0.5 μg/ml), and tiamulin (from 32 to 0.5 μg/ml), with no alteration of MICs of erythromycin and quinupristin (Table 3).
Table 3.
MICs of macrolides and related compounds for E. faecium strains
| Strain | MIC (μg/ml)a |
||||||
|---|---|---|---|---|---|---|---|
| ERY | LIN | CLI | DAL | QUI | Q-D | TIA | |
| HM1070 | 0.12 | 0.25 | 0.12 | 4 | 2 | 0.5 | 0.5 |
| In vitro mutants | |||||||
| UCN90A | 0.12 | 8 | 2 | >64 | 2 | 2 | 32 |
| UCN90B | 0.12 | 8 | 2 | >64 | 2 | 2 | 32 |
| UCN91A | 0.12 | 8 | 2 | >64 | 2 | 2 | 32 |
| UCN91B | 0.12 | 8 | 2 | >64 | 2 | 2 | 32 |
| UCN92A | 0.12 | 8 | 2 | >64 | 2 | 2 | 32 |
| UCN92B | 0.12 | 8 | 2 | >64 | 2 | 2 | 32 |
| UCN94 | 0.12 | 8 | 2 | >64 | 2 | 2 | 64 |
| UCN95 | 0.12 | 0.25 | 0.06 | 4 | 2 | 0.5 | 0.5 |
| Clinical isolates (including in vivo mutants) | |||||||
| UCN80 | >64 | >64 | >64 | 2 | >64 | 1 | 0.5 |
| UCN80-1 | >64 | >64 | >64 | 16 | >64 | 4 | 32 |
| UCN81 | >64 | >64 | >64 | 2 | >64 | 1 | 0.5 |
| UCN81-1 | >64 | >64 | >64 | 16 | >64 | 4 | 32 |
| UCN82 | >64 | >64 | >64 | 2 | >64 | 0.5 | 1 |
| UCN82-1 | >64 | >64 | >64 | 32 | >64 | 4 | 64 |
CLI, clindamycin; DAL, dalfopristin; ERY, erythromycin; LIN, lincomycin; Q-D, quinupristin-dalfopristin; QUI, quinupristin; TIA, tiamulin.
In addition, all in vitro mutants harboring the C1349T mutation presented the same LSAP phenotype as strain UCN90B, with MICs of lincomycin, clindamycin, dalfopristin, Q-D, and tiamulin at 8, 2, >64, 2, and 32 μg/ml, respectively (Table 3). Concerning clinical isolates, all were highly resistant to erythromycin, lincomycin, clindamycin, and quinupristin due to a MLSB phenotype mediated by the erm(B) gene (data not shown), but only in vivo mutants with the C1349T mutation presented the LSAP phenotype (Table 3).
Genetic environment of eat(A) in E. faecium Aus0004.
In the genome of E. faecium Aus0004, the eat(A) gene corresponded to the gene EFAU004_00630 (positions 659270 to 660772). This gene was surrounded by the two genes EFAU004_00629 (positions 657891 to 659012) and EFAU004_00631 (positions 659270 to 660772), coding for a glycerate kinase (373 amino acids) and a phosphoglycerate mutase (175 amino acids), respectively (Fig. 2). Bioinformatically, no operon structure was predicted, and we unambiguously determined the TSS to be 221 bp upstream of the start codon of eat(A), revealing a long 5′ untranslated region (UTR) (Fig. 2).
Fig 2.
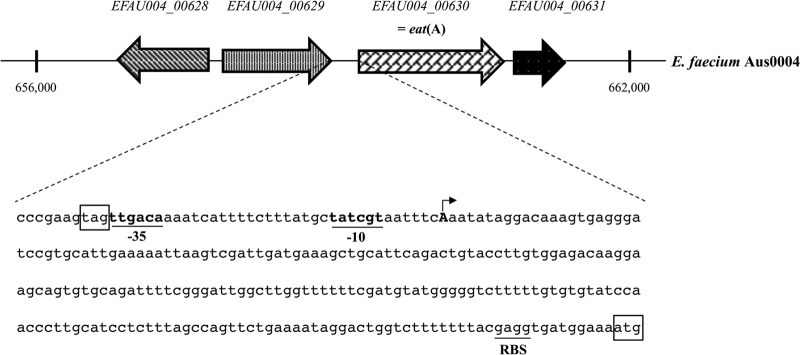
Schematic map of genetic environment of eat(A) (i.e., EFAU004_00630) in the genome of E. faecium Aus0004 (GenBank accession no. NC_017022). Open reading frames are shown as arrows indicating the orientation of their coding sequence. The genes EFAU004_00628, EFAU004_00629, and EFAU004_00631 code for a 2-dehydropentoate 2-reductase, a glycerate kinase, and a phosphoglycerate mutase, respectively. The nucleotide sequence corresponding to the upstream region of the eat(A) gene is represented in detail. The −35 and −10 promoter boxes are underlined, and the transcription start site is represented by an arrow. The start codon of eat(A) and its putative ribosome-binding site (RBS) as well as the stop codon of EFAU004_00629 are also indicated.
Prevalence of the mutated allele eat(A)v among VREF clinical isolates.
Out of the 60 VREF isolates, the point mutation C1349T was detected in 14 (23.3%) strains by PCR sequencing. All the VREF isolates were highly resistant to macrolides, lincosamides, and quinupristin (MLSB phenotype). On the other hand, all eat(A)v-positive strains were also resistant to quinupristin-dalfopristin and tiamulin, whereas all susceptible strains (except two) remained entirely susceptible to these antibiotics (data not shown). This suggests that these molecules may be used as phenotypic markers of resistance in the case of combined resistance mechanisms.
DISCUSSION
ABC systems constitute one of the largest families of proteins, with most of them being involved in import and export, often called ABC transporters (21). These transporters share an organization with two hydrophobic transmembrane domains (TMDs) and two intracytoplasmic nucleotide-binding domains (NBDs) implicated in ATP hydrolysis and comprising specific motifs (Walker A and B motifs and the ABC signature) that are conserved in all ABC proteins (22). Besides importers and exporters, there is a third group of ABC proteins (named class 2) that lack TMDs, consisting of two NBDs fused into a single protein (21–23). Several of these class 2 ABC systems have been involved in MLS resistance, such as Msr-, Vga-, or Lsa-like proteins (21).
The observed profile of cross-resistance to lincosamides, streptogramins A, and pleuromutilins conferred by Eat(A)v was similar to those conferred by other Lsa-like proteins. In Enterococcus faecalis, Lsa(A) is responsible for intrinsic LSAP resistance (6, 14). Lsa(B), encoded by a plasmid-borne gene from Staphylococcus sciuri, confers an increase in MICs of lincosamides, whereas streptogramins A and pleuromutilins had not been tested; however, a LSAP phenotype is very likely (9). A chromosomal gene, lsa(C), was demonstrated to be responsible for acquired LSAP resistance in Streptococcus agalactiae clinical isolates (14). Very recently, a novel gene, named lsa(E), which likely originated from E. faecalis, was found in both methicillin-resistant and -susceptible Staphylococcus aureus isolates of animal and human origins (11, 12). Interestingly, all Lsa-like proteins conferring a LSAP phenotype [i.e., Lsa(A), Lsa(B), Lsa(C), Lsa(E), and Eat(A)v] possess an isoleucine (a hydrophobic amino acid) instead of a threonine (a polar neutral amino acid) (Fig. 1). This suggests an important role of position 450 in LSAP resistance, since it is located within the Walker B motif of the second NBD, a domain known to be involved in ATP binding (24).
Another important question that remains poorly elucidated is the biochemical mechanism of resistance. Indeed, even if class 2 ABC proteins are presumed to act as efflux pump systems, only one study of Msr(A) suggests that these proteins might be able to hijack the TMDs of ABC transporters to mediate efflux (25). However, no membrane partners for Msr(A) have clearly been identified so far (26). A ribosomal protection mechanism of resistance might also be hypothesized. Since eat(A) is an innate gene in E. faecium, it is obvious that it has a physiological role in the bacterial cell, and Eat(A) might be involved in protein translation since Lsa-like proteins are homologous to the eukaryotic elongation factor eEF-3 from the fungus Saccharomyces cerevisiae (27).
Concerning the expression of the eat(A) gene, a long 5′ UTR has been identified, suggesting either a transcriptional mechanism or a posttranscriptional (translational) regulation mechanism. Transcriptional attenuation may occur since a Rho-independent transcription terminator has been bioinformatically predicted (data not shown). This regulatory strategy is largely used by bacteria for sensing intracellular signals (28) and has already been described for the regulation of erm(K), another MLS resistance determinant (29). Also, a 44-amino-acid putative peptide preceding the eat(A) start codon was identified, which may be part of a mechanism of translational attenuation; however, no obvious inverted repeat sequences have been found (data not shown). The presence of a leader peptide in the 5′ UTR has been reported or postulated to be involved in the posttranscriptional regulation of several MLS resistance genes, such as erm(A), erm(B), erm(C), msr(A), lsa(A), and lsa(B) (6, 9, 30). Further investigations are currently in progress to determine the exact mechanism of eat(A) expression regulation.
In conclusion, this is the first characterization of the molecular mechanism of the acquired LSAP resistance phenotype in E. faecium. Even though the LSAP phenotype could be phenotypically detected, PCR detection using specific primers may be needed, especially for E. faecium strains with combined resistance mechanisms (e.g., the MLSB phenotype). As for other ABC proteins of this class, the biochemical mechanism of resistance and the physiological role will need to be further investigated.
ACKNOWLEDGMENTS
This work was funded by a grant from the Ministère de l'Enseignement Supérieur et de la Recherche (EA4655), Université Caen Basse-Normandie, France.
We gratefully acknowledge the technical assistance of Michel Auzou. We kindly thank Marilyn C. Roberts for providing us the gene name according to the nomenclature center for MLS genes (http://faculty.washington.edu/marilynr/).
Footnotes
Published ahead of print 8 July 2013
This paper is dedicated to the memory of Brigitte Malbruny.
REFERENCES
- 1.Arias CA, Murray BE. 2012. The rise of the Enterococcus: beyond vancomycin resistance. Nat. Rev. Microbiol. 10:266–278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cattoir V, Leclercq R. 2013. Twenty-five years of shared life with vancomycin-resistant enterococci: is it time to divorce? J. Antimicrob. Chemother. 68:731–742 [DOI] [PubMed] [Google Scholar]
- 3.Eliopoulos GM. 2003. Quinupristin-dalfopristin and linezolid: evidence and opinion. Clin. Infect. Dis. 36:473–481 [DOI] [PubMed] [Google Scholar]
- 4.Leclercq R. 2002. Mechanisms of resistance to macrolides and lincosamides: nature of the resistance elements and their clinical implications. Clin. Infect. Dis. 34:482–492 [DOI] [PubMed] [Google Scholar]
- 5.Novak R, Shlaes DM. 2010. The pleuromutilin antibiotics: a new class for human use. Curr. Opin. Investig. Drugs 11:182–191 [PubMed] [Google Scholar]
- 6.Singh KV, Weinstock GM, Murray BE. 2002. An Enterococcus faecalis ABC homologue (Lsa) is required for the resistance of this species to clindamycin and quinupristin-dalfopristin. Antimicrob. Agents Chemother. 46:1845–1850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dina J, Malbruny B, Leclercq R. 2003. Nonsense mutations in the lsa-like gene in Enterococcus faecalis isolates susceptible to lincosamides and streptogramins A. Antimicrob. Agents Chemother. 47:2307–2309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Singh KV, Murray BE. 2005. Differences in the Enterococcus faecalis lsa locus that influence susceptibility to quinupristin-dalfopristin and clindamycin. Antimicrob. Agents Chemother. 49:32–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kehrenberg C, Ojo KK, Schwarz S. 2004. Nucleotide sequence and organization of the multiresistance plasmid pSCFS1 from Staphylococcus sciuri. J. Antimicrob. Chemother. 54:936–939 [DOI] [PubMed] [Google Scholar]
- 10.Kehrenberg C, Aarestrup FM, Schwarz S. 2007. IS21-558 insertion sequences are involved in the mobility of the multiresistance gene cfr. Antimicrob. Agents Chemother. 51:483–487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wendlandt S, Lozano C, Kadlec K, Gómez-Sanz E, Zarazaga M, Torres C, Schwarz S. 2013. The enterococcal ABC transporter gene lsa(E) confers combined resistance to lincosamides, pleuromutilins and streptogramin A antibiotics in methicillin-susceptible and methicillin-resistant Staphylococcus aureus. J. Antimicrob. Chemother. 68:473–475 [DOI] [PubMed] [Google Scholar]
- 12.Li B, Wendlandt S, Yao J, Liu Y, Zhang Q, Shi Z, Wei J, Shao D, Schwarz S, Wang S, Ma Z. 2013. Detection and new genetic environment of the pleuromutilin-lincosamide-streptogramin A resistance gene lsa(E) in methicillin-resistant Staphylococcus aureus of swine origin. J. Antimicrob. Chemother. 68:1251–1255 [DOI] [PubMed] [Google Scholar]
- 13.Malbruny B, Werno AM, Anderson TP, Murdoch DR, Leclercq R. 2004. A new phenotype of resistance to lincosamide and streptogramin A-type antibiotics in Streptococcus agalactiae in New Zealand. J. Antimicrob. Chemother. 54:1040–1044 [DOI] [PubMed] [Google Scholar]
- 14.Malbruny B, Werno AM, Murdoch DR, Leclercq R, Cattoir V. 2011. Cross-resistance to lincosamides, streptogramins A, and pleuromutilins due to the lsa(C) gene in Streptococcus agalactiae UCN70. Antimicrob. Agents Chemother. 55:1470–1474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dowzicky M, Talbot GH, Feger C, Prokocimer P, Etienne J, Leclercq R. 2000. Characterization of isolates associated with emerging resistance to quinupristin/dalfopristin (Synercid) during a worldwide clinical program. Diagn. Microbiol. Infect. Dis. 37:57–62 [DOI] [PubMed] [Google Scholar]
- 16.Malbruny B, Leclercq R, Cattoir V. 2012. Abstr 22nd Eur. Congr. Clin. Microbiol. Infect. Dis., London, United Kingdom, 31 March to 3 April 2012, abstr P1284 [Google Scholar]
- 17.Bérenger R, Bourdon N, Auzou M, Leclercq R, Cattoir V. 2011. In vitro activity of new antimicrobial agents against glycopeptide-resistant Enterococcus faecium clinical isolates from France between 2006 and 2008. Med. Mal. Infect. 41:405–409 [DOI] [PubMed] [Google Scholar]
- 18.Clinical and Laboratory Standards Institute 2013. Performance standards for antimicrobial susceptibility testing; approved standard, 23rd ed., M100-S23 Clinical and Laboratory Standards Institute, Wayne, PA [Google Scholar]
- 19.Lalioui L, Pellegrini E, Dramsi S, Baptista M, Bourgeois N, Doucet-Populaire F, Rusniok C, Zouine M, Glaser P, Kunst F, Poyart C, Trieu-Cuot P. 2005. The SrtA sortase of Streptococcus agalactiae is required for cell wall anchoring of proteins containing the LPXTG motif, for adhesion to epithelial cells, and for colonization of the mouse intestine. Infect. Immun. 73:3342–3350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Arsène S, Leclercq R. 2007. Role of a qnr-like gene in the intrinsic resistance of Enterococcus faecalis to fluoroquinolones. Antimicrob. Agents Chemother. 51:3254–3258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Davidson AL, Dassa E, Orelle C, Chen J. 2008. Structure, function, and evolution of bacterial ATP-binding cassette systems. Microbiol. Mol. Biol. Rev. 72:317–364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Biemans-Oldehinkel E, Doeven MK, Poolman B. 2006. ABC transporter architecture and regulatory roles of accessory domains. FEBS Lett. 580:1023–1035 [DOI] [PubMed] [Google Scholar]
- 23.Kos V, Ford RC. 2009. The ATP-binding cassette family: a structural perspective. Cell. Mol. Life Sci. 66:3111–3126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Falcón-Pérez JM, Martínez-Burgos M, Molano J, Mazón MJ, Eraso P. 2001. Domain interactions in the yeast ATP binding cassette transporter Ycf1p: intragenic suppressor analysis of mutations in the nucleotide binding domains. J. Bacteriol. 183:4761–4770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ross JI, Eady EA, Cove JH, Cunliffe WJ, Baumberg S, Wootton JC. 1990. Inducible erythromycin resistance in staphylococci is encoded by a member of the ATP-binding transport super-gene family. Mol. Microbiol. 4:1207–1214 [DOI] [PubMed] [Google Scholar]
- 26.Reynolds E, Ross JI, Cove JH. 2003. Msr(A) and related macrolide/streptogramin resistance determinants: incomplete transporters? Int. J. Antimicrob. Agents 22:228–236 [DOI] [PubMed] [Google Scholar]
- 27.Andersen CB, Becker T, Blau M, Anand M, Halic M, Balar B, Mielke T, Boesen T, Pedersen JS, Spahn CM, Kinzy TG, Andersen GR, Beckmann R. 2006. Structure of eEF3 and the mechanism of transfer RNA release from the E-site. Nature 443:663–668 [DOI] [PubMed] [Google Scholar]
- 28.Merino E, Yanofsky C. 2005. Transcription attenuation: a highly conserved regulatory strategy used by bacteria. Trends Genet. 21:260–265 [DOI] [PubMed] [Google Scholar]
- 29.Kwak JH, Choi EC, Weisblum B. 1991. Transcriptional attenuation control of ermK, a macrolide-lincosamide-streptogramin B resistance determinant from Bacillus licheniformis. J. Bacteriol. 173:4725–4735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Depardieu F, Podglajen I, Leclercq R, Collatz E, Courvalin P. 2007. Modes and modulations of antibiotic resistance gene expression. Clin. Microbiol. Rev. 20:79–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bozdogan B, Leclercq R. 1999. Effects of genes encoding resistance to streptogramins A and B on the activity of quinupristin-dalfopristin against Enterococcus faecium. Antimicrob. Agents Chemother. 43:2720–2725 [DOI] [PMC free article] [PubMed] [Google Scholar]