

Abstract
Human cytomegalovirus (HCMV) is a widespread pathogen in the human population, affecting many immunologically immature and immunocompromised patients, and can result in severe complications, such as interstitial pneumonia and mental retardation. Current chemotherapies for the treatment of HCMV infections include ganciclovir (GCV), foscarnet, and cidofovir. However, the high incidences of adverse effects (neutropenia and nephrotoxicity) limit the use of these drugs. Cyclopropavir (CPV), a guanosine nucleoside analog, is 10-fold more active against HCMV than GCV (50% effective concentrations [EC50s] = 0.46 and 4.1 μM, respectively). We hypothesize that the mechanism of action of CPV is similar to that of GCV: phosphorylation to a monophosphate by viral pUL97 protein kinase with further phosphorylation to a triphosphate by endogenous kinases, resulting in inhibition of viral DNA synthesis. To test this hypothesis, we isolated a CPV-resistant virus, sequenced its genome, and discovered that bp 498 of UL97 was deleted. This mutation caused a frameshift in UL97 resulting in a truncated protein that lacks a kinase domain. To determine if this base pair deletion was responsible for drug resistance, the mutation was engineered into the wild-type viral genome, which was then exposed to increasing concentrations of CPV. The results demonstrate that the engineered virus was approximately 72-fold more resistant to CPV (EC50 = 25.8 ± 3.1 μM) than the wild-type virus (EC50 = 0.36 ± 0.11 μM). We conclude, therefore, that this mutation is sufficient for drug resistance and that pUL97 is involved in the mechanism of action of CPV.
INTRODUCTION
Human cytomegalovirus (HCMV), a betaherpesvirus, is a widespread pathogen affecting a majority of the world's population (1). Individuals at risk for symptomatic HCMV disease—which includes interstitial pneumonia, encephalitis, and retinitis—are those with deficiencies in T-cell immunity. In addition, with more than 4,000 cases reported each year, HCMV is the most common congenital infection in the United States and can result in severe mental retardation, hearing loss, and/or vision loss (2). The drugs currently approved by the FDA for the prophylaxis or treatment of systemic HCMV infections are ganciclovir (GCV) (Fig. 1) and its oral prodrug valganciclovir, foscarnet, and cidofovir (3–5). GCV is the most common therapy option for the treatment of HCMV infections among patients with impaired immunity, particularly those with advanced HIV/AIDS, those undergoing cancer chemotherapy, and recipients of solid-organ or bone marrow transplants (6–8). GCV, an acyclic analog of the nucleoside guanosine, is converted intracellularly to a monophosphate by a viral protein kinase encoded by the HCMV gene UL97 (9, 10). Upon further phosphorylation by cellular enzymes (11, 12), GCV triphosphate competes with endogenous dGTP for incorporation into progeny viral genomes, resulting in pUL54 (viral DNA polymerase) inhibition (13, 14). Once incorporated, GCV also acts as a chain terminator, further inhibiting viral DNA synthesis (15, 16). However, long-term therapy is typically required due to recurrence of infection and reactivation of latent HCMV upon cessation of therapy. Drug resistance and adverse effects, such as neutropenia (affecting as many as 30% of patients) and nephrotoxicity (affecting as many as 50% of patients), are prevalent and limit the use of GCV, foscarnet, and cidofovir (3, 17–19). With the increased use of immunosuppression for cancer chemotherapy and organ transplantation, there is an increasing need for more-effective and less-toxic drugs to treat HCMV.
Fig 1.
Structures of ganciclovir (GCV) and cyclopropavir (CPV).
Cyclopropavir (CPV) (Fig. 1), a methylenecyclopropane guanosine nucleoside analog, is currently being explored as a viable chemotherapy option for HCMV due to its high potency and low incidence of adverse effects in vivo (20, 21). Previous studies have demonstrated that CPV is approximately 10-fold more active in vitro (50% effective concentration [EC50] = 0.46 μM) than GCV (EC50 = 4.1 μM), with no observed increase in cytotoxicity (22). Furthermore, CPV elicits a 2- to 5-log reduction in murine cytomegalovirus titers in vivo, resulting in reduced mortality in SCID mice (21). Although the mechanism of action of CPV has been hypothesized in previous studies, the exact mechanism by which CPV elicits an antiviral effect has not been determined (23). We have established that both CPV and GCV are phosphorylated to monophosphates by viral pUL97 protein kinase (24). Additional phosphorylation to a triphosphate by endogenous kinases is also similar for CPV and GCV (25). However, viral DNA polymerase inhibition and incorporation into HCMV are known for GCV (13–16) but have not been established for CPV. Furthermore, the necessity of phosphorylation by pUL97 for antiviral activity is unknown. Thus, the goal of this study is to determine whether the viral protein kinase pUL97 is involved in the mechanism of action of CPV.
MATERIALS AND METHODS
Chemicals.
CPV was synthesized in the laboratory of J. Zemlicka as described previously (22). GCV was kindly provided by Hoffmann-La Roche (Palo Alto, CA).
Cell culture procedures.
Human foreskin fibroblasts (HFF) were grown in minimal essential medium with Earle's salts and 10% fetal bovine serum. They were grown at 37°C under a humidified atmosphere of 3 to 5% CO2 and 97 to 95% air and were regularly passaged at 1:2 dilutions using conventional procedures with 0.05% trypsin and 0.02% EDTA in HEPES-buffered saline (26).
Viral strain.
The HCMV strain Towne was kindly provided by M. F. Stinski, University of Iowa. A bacterial artificial chromosome (BAC) clone of strain AD169 (AD169rv) was generously provided by U. H. Koszinowski, Ludwig-Maximilians University (Munich, Germany) (27).
HCMV plaque reduction assay.
HFF were seeded at 85,000 per well in a 24-well cluster dish and were infected 3 days later with HCMV at 100 PFU per well. Two hours postinfection, media containing serial dilutions of drug, fetal bovine serum (final concentration, 3%), and 0.5% methylcellulose were added. After incubation at 37°C for 9 to 11 days, cell monolayers were stained with crystal violet, and plaques were enumerated by light microscopy. The number of plaques observed in the presence of each drug concentration was compared to the number observed in the absence of the drug in order to determine the drug effect. The percentage of inhibition of plaque numbers was plotted against the log10 drug concentration. Fifty percent effective concentrations (EC50s) were interpolated from the linear portions of the regression lines. All drug dilutions were tested at least in duplicate.
Selection of CPV-resistant virus.
HFF were infected with HCMV at a multiplicity of infection (MOI) of 0.01 and were grown in a 25-cm2 tissue culture flask in the presence of 0.5 μM CPV for 2 weeks. Supernatant progeny virus was then passaged in the presence of first 1.0 μM and then 2.0 μM concentrations of CPV; the duration of each passage was 2 weeks. The resulting virus was further passaged in 2.0 μM CPV for 3 months and was then frozen in liquid nitrogen, and the titer was determined. The resulting virus stock was purified using the Klein limiting-dilution method (28) and was termed 2696r.
DNA sequencing.
Primers for PCR amplification and sequencing were determined on the basis of the published sequence of the Towne strain of HCMV. After PCR amplification, products were run on a 0.8% agarose gel, extracted, and purified using a QIAquick gel extraction kit (Qiagen, Valencia, CA). Sequencing PCR was carried out with the BigDye Terminator cycle-sequencing ready-reaction kit (Applied Biosystems, Foster City, CA), and sequencing reaction products were separated and detected with an ABI Prism 310 genetic analyzer. Sequences were aligned and edited using Sequencher software (Gene Codes, Ann Arbor, MI).
Western blot analysis.
HFF were infected with wild-type HCMV or 2696r at an MOI of approximately 1.0 PFU/ml and were grown in a 75-cm2 tissue culture flask. After 4 days, cells were harvested and were lysed using lysis buffer (2% NP-40, 0.1 M Tris [pH 8.8], 0.1% SDS, 0.3 M NaCl), and protein levels were determined using the DC protein assay (Bio-Rad Corp., Hercules, CA). Fifty micrograms of protein per sample was electrophoresed under reducing conditions and was blotted electrophoretically onto nitrocellulose paper (Bio-Rad Corp., Hercules, CA). pUL97 was detected using a previously characterized polyclonal antibody developed against full-length pUL97 (a generous gift of Don Coen, Harvard University, Boston, MA) (29) and the SuperSignal West Pico chemiluminescent substrate imaging system (Thermo Scientific, Rockford, IL). A monoclonal antibody against human actin was used as a loading control.
Marker transfer studies.
“En passant” mutagenesis of BACs was performed as described elsewhere (30–32) to remove bp 498 of the UL97 open reading frame from a BAC clone of HCMV strain AD169, AD169rv (a generous gift of Ulrich Koszinowski) (27). All manipulations were performed in Escherichia coli strain GS1783 (a generous gift of Greg Smith, Northwestern University, Chicago, IL). Briefly, an excisable kanamycin resistance marker (I-SceI-aphAI) was amplified from a plasmid template by using oligonucleotide primers d_bp498_F (5′-AAA CTT CGG CCA TGT GGT CGT TCG AGT ACG ATC GCG ACG GGA CGT GAC CAG CG T ACG CCG TAG GGA TAA CAG GGT AAT CGA TTT-3′) and d_bp498_Rv (5′-TGC CGC CGG TGA AGA GAG CGC GGC GTA CGC TGG TCA CGT CCC GTC GCG ATC GT A CTC GAA GCC AGT GTT ACA ACC AAT TAA CC-3′), which had been custom synthesized, and was purified via polyacrylamide gel electrophoresis (PAGE) by Integrated DNA Technologies, Inc. (Coralville, IA). The resulting PCR product was gel purified using a NucleoSpin gel and PCR cleanup kit (Macherey-Nagel, Inc., Bethlehem, PA) and was then electroporated into E. coli containing the AD169rv BAC. Integrated colonies were resolved to remove the kanamycin resistance marker. The resulting recombinant BAC, AD169rv d498, was confirmed by restriction enzyme digestion analysis and by DNA sequencing of the modified region (Genewiz, Inc., South Plainfield, NJ).
Virus reconstitution.
Infectious virus was reconstituted from BAC DNA as described previously (32), by cotransfection of ∼0.7 × 105 human foreskin fibroblasts in a 24-well cluster plate (Corning, Inc.) with a total of 1 μg DNA containing 750 ng BAC DNA, 200 ng of the pp71 expression plasmid pSG5-pp71 (33) (a gift of Robert Kalejta, University of Wisconsin, Madison, WI), and 50 ng of the Cre recombinase expression plasmid pCAGGS-nlsCre (34) (a gift of Michael I. Kotlikoff, Cornell University, Ithaca, NY), by using 7 μl of Superfect transfection reagent (Qiagen, Inc., Valencia, CA) according to the manufacturer's recommendations.
HCMV growth assay.
HFF were seeded and grown to confluence in a 25-cm2 tissue culture flask at 37°C. Cells were then infected with virus at an MOI of 0.02. Two hours postinfection, medium was added to the flask, and samples were collected every 24 h over the course of 10 days (collected samples were stored at −80°C until titers could be determined). The titer of virus for each sample was determined by first plating a 96-well plate with HFF grown to confluence. Serial dilutions (1:3) of samples (total volume, 200 μl) were then plated, and the virus was allowed to grow for 7 to 9 days at 37°C. Finally, cell monolayers were stained with crystal violet and plaques were enumerated by light microscopy. The virus titer was calculated as 5 × 3n × the number of plaques, where n is the serial dilution in which the plaques were enumerated.
RESULTS
Initial isolation and characterization of 2696r (CPV-resistant HCMV).
We hypothesize that the mechanism of action of CPV is a multiphase process: initial phosphorylation by viral pUL97 to a monophosphate, followed by phosphorylation by endogenous kinases to a triphosphate, resulting in the inhibition of viral DNA synthesis. Although we have demonstrated previously that CPV is phosphorylated by pUL97 to a monophosphate (the initial step of our hypothesis) (24), it has not been determined whether this enzymatic conversion is necessary for CPV to elicit an antiviral effect. Therefore, to test the first step of our hypothesis, HFF infected with a CPV-resistant HCMV strain (2696r) were exposed to increasing concentrations of the drug in order to confirm resistance (Fig. 2; Table 1). As expected, 2696r was approximately 25-fold more resistant to CPV (EC50 = 22.5 ± 3.5 μM) than the wild-type (Towne strain) virus (EC50 = 0.91 ± 0.15 μM). In addition, HFF infected with 2696r were exposed to increasing concentrations of GCV in order to determine if the virus is also resistant to this drug. As shown in Fig. 2, 2696r is approximately 28-fold more resistant to GCV (EC50 = 41.3 ± 2.8 μM) than the wild-type virus (EC50 = 1.5 ± 0.16 μM). These results demonstrate that 2696r, which is resistant to CPV, exhibits cross-resistance with GCV.
Fig 2.
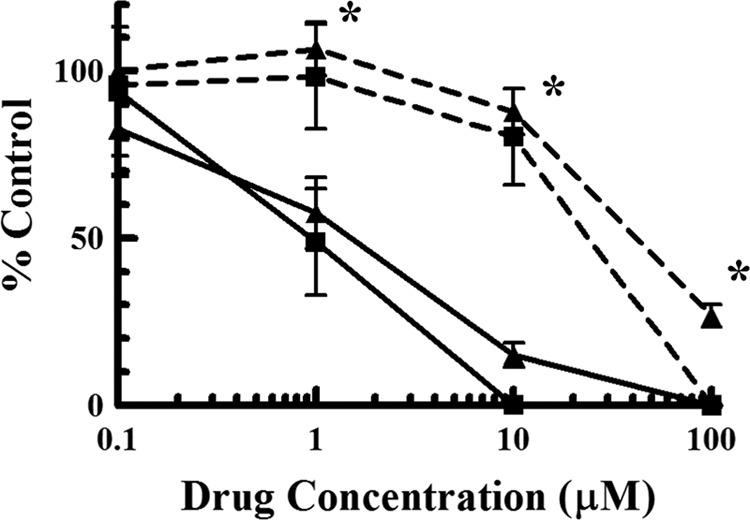
Initial characterization of a CPV-resistant HCMV isolate (2696r). HFF infected with the CPV-resistant HCMV isolate (dashed lines) were exposed to increasing concentrations of either CPV (■) or GCV (▲) and were compared to HFF infected with wild-type virus (solid lines) exposed to the same concentrations of drugs. Values are means ± standard deviations from at least two experiments. Asterisks indicate that the replication of 2696r is significantly different (P < 0.01) from that of the wild-type virus (with both drugs).
Table 1.
Inhibition of 2696r and HCMV UL97Δ498 replication by ganciclovir and cyclopropavir
| Compound | Comparison of Towne and 2696r |
Comparison of AD169rv and HCMV UL97Δ498 |
||||
|---|---|---|---|---|---|---|
| EC50 (μM)a for: |
Fold resistance of 2696r | EC50 (μM)b for: |
Fold resistance of HCMV UL97Δ498 | |||
| Towne | 2696r | AD169rv | HCMV UL97Δ498 | |||
| GCV | 1.5 ± 0.16 | 41.3 ± 2.8 | 28 | 2.0 ± 0.24 | 28.1 ± 6.6 | 14 |
| CPV | 0.91 ± 0.15 | 22.5 ± 3.5 | 25 | 0.36 ± 0.11 | 25.8 ± 3.1 | 72 |
Values are means ± standard deviations from at least two experiments.
Values are means ± standard deviations from at least four experiments.
DNA sequencing of 2696r.
Since 2696r was resistant to GCV, and GCV resistance has been mapped to mutations in UL54 and UL97 (17, 18), the open reading frames of UL54 and UL97 in 2696r were sequenced. The results demonstrated that the open reading frame of UL54 in 2696r contained no mutations, whereas bp 498 of the 2696r gene UL97 was deleted (Fig. 3). This deletion results in a codon frameshift and the introduction of a stop codon at bp 502 to 504 (corresponding to bp 503 to 505 of wild-type HCMV UL97). The protein product of this mutated gene would be considerably truncated (167 amino acids versus 707 amino acids for wild-type [Towne strain] pUL97), lacking both an ATP binding site and a substrate pocket (10, 35, 36). To confirm that pUL97 is truncated in 2696r compared to pUL97 in the wild-type virus, Western blot analysis was performed (Fig. 4). The results depict a significantly lower molecular mass reactive species (molecular mass, 17.8 kDa) in the 2696r lane that is absent from the wild-type lane. In addition, the reactive species corresponding to pUL97 in the wild-type lane (molecular mass, 78.3 kDa) is absent from the 2696r lane. These results demonstrate that pUL97 is indeed truncated in 2696r.
Fig 3.
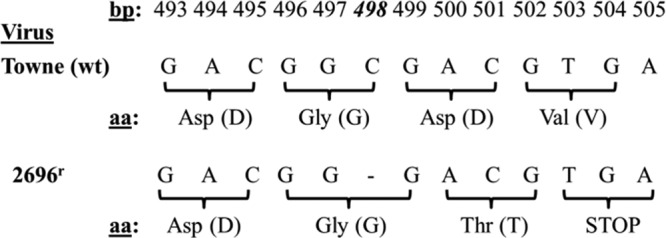
DNA sequencing of a CPV-resistant HCMV isolate (2696r). The genome of 2696r was isolated and sequenced using an ABI Prism 310 genetic analyzer. The results are shown as a comparison of the wild-type and mutant DNA sequences, with corresponding codon translation.
Fig 4.
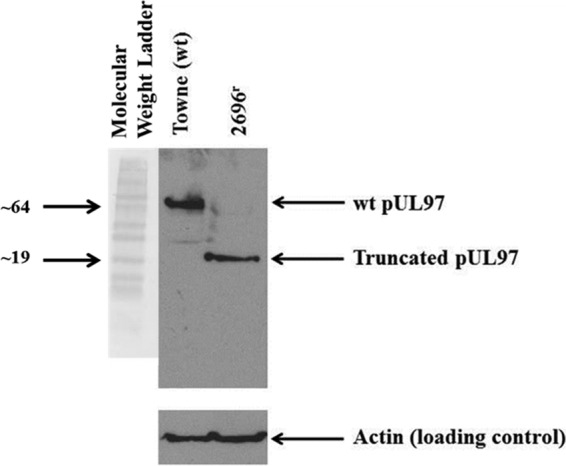
Western blot analysis of 2696r and wild-type (wt) HCMV pUL97. HFF infected with 2696r or the wild type were harvested 4 days postinfection (MOI, ∼1). Proteins from the cells were extracted and were analyzed via Western blotting using an anti-pUL97 antibody. The locations of pUL97 from the wild-type virus and of truncated pUL97 from 2696r are indicated. An anti-actin antibody was used as a loading control. The positions of molecular weight markers (in thousands) are indicated on the left.
Recombinant HCMV UL97Δ498 exhibits resistance to both CPV and GCV.
To confirm that the bp 498 deletion of 2696r is the mechanism by which this virus resists the antiviral effects of CPV, this mutation was engineered into a wild-type virus (producing HCMV UL97Δ498). Since previous studies have demonstrated that UL97-null mutants have a 1- to 2-log growth defect that is attributable to the loss of kinase activity (29, 37), and thus the mutation discovered in 2696r would result in a virus without an active pUL97 kinase, both 2696r and HCMV UL97Δ498 were assayed for replication competency, and their growth was compared to the growth of a wild-type virus. As shown in Fig. 5, in agreement with previous results, 2696r (1.0 × 105 ± 0.17 × 105 PFU/ml) and HCMV UL97Δ498 (1.5 × 105 ± 0.11 × 105 PFU/ml) exhibited growth defects in excess of 1 log unit compared to wild-type (BAC-derived AD169rv) virus (2.8 × 106 ± 0.17 × 106 PFU/ml).
Fig 5.
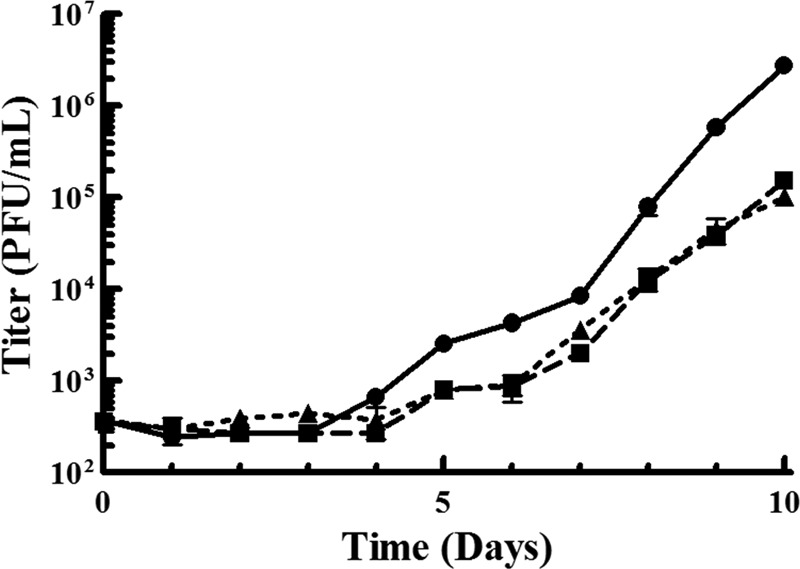
Replication competency of HCMV UL97Δ498. HFF were infected with a virus (wild-type AD169rv [●], HCMV UL97Δ498 [■], or 2696r [▲]) at an MOI of 0.02. Samples were taken every 24 h for 10 days; titers were determined by plaque assays following serial dilution of samples. Values are means ± standard deviations from at least three experiments.
Following confirmation that the engineered virus replicated (albeit at a lower rate than that of the wild-type virus), cells infected with HCMV UL97Δ498 were exposed to increasing concentrations of either CPV or GCV and were compared to wild-type virus under the same conditions. The results, shown in Fig. 6 and Table 1, demonstrate that HCMV UL97Δ498 was approximately 72-fold more resistant to CPV (EC50 = 25.8 ± 3.1 μM) and 14-fold more resistant to GCV (EC50 = 28.1 ± 6.6 μM) than the wild-type virus (BAC-derived AD169rv) (EC50s = 0.36 ± 0.11 and 2.0 ± 0.24 μM, respectively). We conclude, therefore, that the base pair deletion mutation in the open reading frame of the HCMV gene UL97 is sufficient for drug resistance and that the protein product of this gene is involved in the mechanism of action of CPV.
Fig 6.
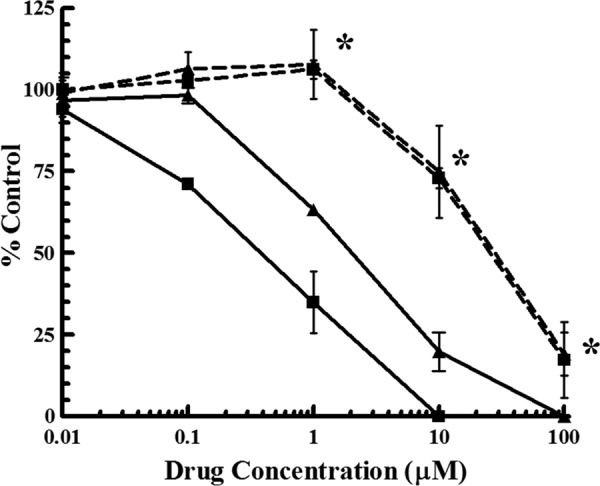
Resistance of HCMV UL97Δ498 to CPV and GCV. HFF infected with a wild-type HCMV engineered with the deletion of UL97 bp 498 (dashed lines) were exposed to increasing concentrations of either CPV (■) or GCV (▲) and were compared to cells infected with a wild-type virus (solid lines) exposed to the same concentrations of drugs. Values are means ± standard deviations from at least four experiments. Asterisks indicate that the replication of HCMV UL97Δ498 is significantly different (P < 0.01) from that of the wild-type virus (with both drugs).
DISCUSSION
CPV is a known inhibitor of HCMV replication (23). The hypothesized mechanism by which CPV elicits this effect involves the phosphorylation of CPV to a monophosphate by the viral enzyme pUL97. Our current results demonstrate that a previously uncharacterized mutation in the open reading frame of UL97, which would result in the expression of a truncated pUL97 lacking an active kinase domain, renders the virus resistant to both CPV and GCV. These results establish that pUL97 is necessary for CPV to elicit an antiviral effect and, taken together with our previous results (24), establish that pUL97 is the enzyme responsible for the initial phosphorylation of CPV to a monophosphate.
Previous studies have identified mutations within the HCMV genome that confer resistance to CPV on the virus. The first example is a frameshift mutation in UL27, developed as a result of exposure to CPV, in a virus with wild-type UL97 (38). This mutation, presumably, compensates for the loss of pUL97 enzymatic activity, since CPV exhibits some pUL97-inhibitory activity, and a mutation responsible for HCMV resistance to maribavir, a potent pUL97 inhibitor, occurs in UL27 (38–40). Next, a recombinant virus lacking a functional pUL97 kinase is 21-fold more resistant to CPV than wild-type virus (23). The previously uncharacterized mutation that we present in this study, which causes a frameshift with a concomitant premature translational termination of the viral kinase pUL97 and, presumably, a complete loss of enzymatic function, behaves similarly to the previously characterized UL97-null mutant with regard to growth characteristics (Fig. 5) and CPV resistance (Fig. 6). Finally, some mutations generated as a result of exposure to CPV confer resistance to the drug without the complete loss of enzymatic function (41–43). These mutations confer resistance not only to CPV but to GCV as well. The mutation described here (UL97Δ498) also confers cross-resistance with GCV, since GCV requires initial phosphorylation by pUL97 as part of its mechanism of action (9).
Although there are many mutations that confer cross-resistance between GCV and CPV, including the mutation we present above, several mutations confer resistance to GCV but not to CPV (41, 44, 45). This suggests that these two drugs either interact differently or occupy different, though close, sites within the substrate binding pocket of the pUL97 kinase. We therefore cannot rule out the possibility that certain mutations in UL97 may confer resistance to CPV without conferring resistance to GCV. In addition, mutations that confer resistance to maribavir (46), a potent pUL97 inhibitor (47), on HCMV demonstrate no cross-resistance with GCV (48). This suggests that these two nucleoside analogs either interact differently or occupy distinct sites within the substrate binding pocket of pUL97. Given that CPV demonstrates characteristics of both GCV (substrate of pUL97 [24]) and maribavir (inhibitor of pUL97 [38]), we hypothesize that CPV binds to a site central to these two nucleoside analogs, occupying space that would otherwise be occupied by either GCV or maribavir. We further speculate that, given the possibility that CPV and maribavir occupy similar sites within the substrate binding pocket of pUL97, there could be mutations that confer resistance to both CPV and maribavir. Given that maribavir and CPV are currently being evaluated as candidates for clinical trials, an examination of mutations that could confer cross-resistance to these nucleoside analogs is warranted.
The use of CPV for the treatment or prophylaxis of HCMV disease is promising. Our current results demonstrate that pUL97 is an integral part of the mechanism of action of CPV. As such, it is possible that cross-resistance between CPV and currently approved chemotherapeutics for the treatment of HCMV may occur. However, this has not been observed in clinic. In fact, clinical strains of HCMV resistant to GCV were sensitive to earlier analogs of CPV (49). Also, the increased antiviral activity with no observed increase in toxicity (22) and the ability to achieve therapeutic concentrations in vivo without prodrug modification (5, 20) are two reasons why CPV should be superior to GCV for the treatment of HCMV disease. Further examination of the mechanism of action and preclinical development of this compound is warranted.
ACKNOWLEDGMENTS
This work was supported by grants from the NIH (CA32779, AI26077, GM103433-10), Microbiotix, Inc., and the American Heart Association (12GRNT11890012), as well as by funds from the University of Michigan and Drake University.
Footnotes
Published ahead of print 1 July 2013
REFERENCES
- 1.Ho M. 2008. The history of cytomegalovirus and its diseases. Med. Microbiol. Immunol. 197:65–73 [DOI] [PubMed] [Google Scholar]
- 2.Manicklal S, Emery VC, Lazzarotto T, Boppana SB, Gupta RK. 2013. The “silent” global burden of congenital cytomegalovirus. Clin. Microbiol. Rev. 26:86–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Andrei G, De Clercq E, Snoeck R. 2009. Drug targets in cytomegalovirus infection. Infect. Disord. Drug Targets 9:201–222 [DOI] [PubMed] [Google Scholar]
- 4.Biron KK. 2006. Antiviral drugs for cytomegalovirus diseases. Antiviral Res. 71:154–163 [DOI] [PubMed] [Google Scholar]
- 5.Cocohoba JM, McNickoll IR. 2002. Valganciclovir: an advance in cytomegalovirus therapeutics. Ann. Pharmacother. 36:1075–1079 [DOI] [PubMed] [Google Scholar]
- 6.Faulds D, Heel RC. 1990. Ganciclovir: a review of its antiviral activity, pharmacokinetic properties, and therapeutic efficacy in cytomegalovirus infections. Drugs 39:597–638 [DOI] [PubMed] [Google Scholar]
- 7.Noble S, Faulds D. 1998. Ganciclovir: an update of its use in the prevention of cytomegalovirus infection and disease in transplant recipients. Drugs 56:115–146 [DOI] [PubMed] [Google Scholar]
- 8.Whitley RJ, Jacobson MA, Friedberg DN, Holland GN, Jabs DA, Dieterich DT, Hardy WD, Polis MA, Deutsch TA, Feinberg J, Spector SA, Walmsley S, Drew WL, Powderly WG, Griffiths PD, Benson CA, Kessler HA. 1998. Guidelines for the treatment of cytomegalovirus diseases in patients with AIDS in the era of potent antiretroviral therapy: recommendations of an international panel. International AIDS Society—USA. Arch. Intern. Med. 158:957–969 [DOI] [PubMed] [Google Scholar]
- 9.Littler E, Stuart AD, Chee MS. 1992. Human cytomegalovirus UL97 open reading frame encodes a protein that phosphorylates the antiviral nucleoside analogue ganciclovir. Nature 358:160–162 [DOI] [PubMed] [Google Scholar]
- 10.He Z, He YS, Kim Y, Chu L, Ohmstede C, Biron KK, Coen DM. 1997. The human cytomegalovirus UL97 protein is a protein kinase that autophosphorylates on serines and threonines. J. Virol. 71:405–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Biron KK, Stanat SC, Sorrell JB, Fyfe JA, Keller PM, Lambe CU, Nelson DJ. 1985. Metabolic activation of the nucleoside analog 9-[(2-hydroxy-1-(hydroxymethyl)ethoxy]methyl)guanine in human diploid fibroblasts infected with human cytomegalovirus. Proc. Natl. Acad. Sci. U. S. A. 82:2473–2477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Boehme RE. 1984. Phosphorylation of the antiviral precursor 9-(1,3-dihydroxy-2-propoxymethyl)guanine monophosphate by guanylate kinase isozymes. J. Biol. Chem. 259:12346–12349 [PubMed] [Google Scholar]
- 13.Freitas VR, Smee DF, Chernow M, Boehme R, Matthews TR. 1985. Activity of 9-(1,3-dihydroxy-2-propoxymethyl)guanine compared with that of acyclovir against human, monkey, and rodent cytomegaloviruses. Antimicrob. Agents Chemother. 28:240–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Matthews T, Boehme R. 1988. Antiviral activity and mechanism of action of ganciclovir. Rev. Infect. Dis. 10:S490–S494 [DOI] [PubMed] [Google Scholar]
- 15.Hamzeh FM, Lietman PS, Gibson W, Hayward GS. 1990. Identification of the lytic origin of DNA replication in human cytomegalovirus by a novel approach utilizing ganciclovir-induced chain termination. J. Virol. 64:6184–6195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hamzeh FM, Lietman PS. 1991. Intranuclear accumulation of subgenomic noninfectious human cytomegalovirus DNA in infected cells in the presence of ganciclovir. Antimicrob. Agents Chemother. 35:1818–1823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Baldanti F, Lurain N, Gerna G. 2004. Clinical and biologic aspects of human cytomegalovirus resistance to antiviral drugs. Hum. Immunol. 65:403–409 [DOI] [PubMed] [Google Scholar]
- 18.Gilbert C, Bestman-Smith J, Boivin G. 2002. Resistance of herpesviruses to antiviral drugs: clinical impacts and molecular mechanisms. Drug Resist. Updat. 5:88–114 [DOI] [PubMed] [Google Scholar]
- 19.Gilbert C, Boivin G. 2005. Human cytomegalovirus resistance to antiviral drugs. Antimicrob. Agents Chemother. 49:873–883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bowlin TL, Brooks JL, Zemlicka J. 2009. Preclinical pharmacokinetic, toxicokinetic and toxicology results for cyclopropavir, a promising new agent for the treatment of beta- and gamma-herpesviruses. Antiviral Res. 82:A46–A47 [Google Scholar]
- 21.Kern ER, Bidanset DJ, Harline CB, Yan Z, Zemlicka J, Quenelle DC. 2004. Oral activity of a methylenecyclopropane analog, cyclopropavir, in animal models for cytomegalovirus infections. Antimicrob. Agents Chemother. 48:4745–4753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhou S, Breitenbach JM, Borysko KZ, Drach JC, Kern ER, Gullen E, Cheng YC, Zemlicka J. 2004. Synthesis and antiviral activity of (Z)- and (E)-2,2-[bis(hydroxymethyl)cyclopropylidene]methylpurines and -pyrimidines: second-generation methylenecyclopropane analogues of nucleosides. J. Med. Chem. 47:566–575 [DOI] [PubMed] [Google Scholar]
- 23.Kern ER, Kushner NL, Hartline CB, Williams-Aziz SL, Harden EA, Zhou S, Zemlicka J, Prichard MN. 2005. In vitro activity and mechanism of action of methylenecyclopropane analogs of nucleosides against herpesvirus replication. Antimicrob. Agents Chemother. 49:1039–1045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gentry BG, Kamil JP, Coen DM, Zemlicka J, Drach JC. 2010. Stereoselective phosphorylation of cyclopropavir by pUL97 and competitive inhibition by maribavir. Antimicrob. Agents Chemother. 54:3093–3098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gentry BG, Gentry SN, Jackson TL, Zemlicka J, Drach JC. 2011. Phosphorylation of antiviral and endogenous nucleotides to di- and triphosphates by guanosine monophosphate kinase. Biochem. Pharmacol. 81:43–49 [DOI] [PubMed] [Google Scholar]
- 26.Shipman C., Jr 1969. Evaluation of 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) as a tissue culture buffer. Proc. Soc. Exp. Biol. Med. 130:305–310 [DOI] [PubMed] [Google Scholar]
- 27.Hobom U, Brune W, Messerle M, Hahn G, Koszinowski UH. 2000. Fast screening procedures for random transposon libraries of cloned herpesvirus genomes: mutational analysis of human cytomegalovirus envelope glycoprotein genes. J. Virol. 74:7720–7729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Klein RJ. 1975. Isolation of herpes simplex virus clones and drug resistant mutants in microcultures. Arch. Virol. 49:73–80 [DOI] [PubMed] [Google Scholar]
- 29.Kamil JP, Coen DM. 2007. Human cytomegalovirus protein kinase UL97 forms a complex with the tegument phosphoprotein pp65. J. Virol. 81:10659–10668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tischer BK, von Einem J, Kaufer B, Osterrieder N. 2006. Two-step red-mediated recombination for versatile high-efficiency markerless DNA manipulation in Escherichia coli. Biotechniques 40:191–197 [DOI] [PubMed] [Google Scholar]
- 31.Tischer BK, Smith GA, Osterrieder N. 2010. En passant mutagenesis: a two step markerless red recombination system. Methods Mol. Biol. 634:421–430 [DOI] [PubMed] [Google Scholar]
- 32.Wang D, Li G, Schauflinger M, Nguyen CC, Hall ED, Yurochko AD, von Einem J, Kamil JP. 2013. The ULb′ region of the human cytomegalovirus genome confers an increased requirement for the viral protein kinase UL97. J. Virol. 87:6359–6376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kalejta RF, Bechtel JT, Shenk T. 2003. Human cytomegalovirus pp71 stimulates cell cycle progression by inducing the proteasome-dependent degradation of the retinoblastoma family of tumor suppressors. Mol. Cell. Biol. 23:1885–1895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jarosinski KW, Hunt HD, Osterrieder N. 2010. Down-regulation of MHC class I by the Marek's disease virus (MDV) UL49.5 gene product mildly affects virulence in a haplotype-specific fashion. Virology. 405:457–463 [DOI] [PubMed] [Google Scholar]
- 35.Michel D, Pavic I, Zimmermann A, Haupt E, Wunderlich K, Heuschmid M, Mertens T. 1996. The UL97 gene product of human cytomegalovirus is an early-late protein with a nuclear localization but is not a nucleoside kinase. J. Virol. 70:6340–6346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sullivan V, Talarico CL, Stanat SC, Davis M, Coen DM, Biron KK. 1992. A protein kinase homologue controls phosphorylation of ganciclovir in human cytomegalovirus-infected cells. Nature 358:162–164 [DOI] [PubMed] [Google Scholar]
- 37.Prichard MN, Gao N, Jairath S, Mulamba G, Krosky P, Coen DM, Parker BO, Pari GS. 1999. A recombinant human cytomegalovirus with a large deletion in UL97 has a severe replication deficiency. J. Virol. 73:5663–5670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.James SH, Hartline CB, Harden EA, Driebe EM, Schupp JM, Engelthaler DM, Keim PS, Bowlin TL, Kern ER, Prichard MN. 2011. Cyclopropavir inhibits the normal function of the human cytomegalovirus UL97 kinase. Antimicrob. Agents Chemother. 55:4682–4691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Komazin G, Ptak RG, Emmer BT, Townsend LB, Drach JC. 2003. Resistance of human cytomegalovirus to the benzimidazole l-ribonucleoside maribavir maps to UL27. J. Virol. 77:11499–11506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Prichard MN. 2009. Function of human cytomegalovirus UL97 kinase in viral infection and its inhibition by maribavir. Rev. Med. Virol. 19:215–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chou S, Bowlin TL. 2011. Cytomegalovirus UL97 mutations affecting cyclopropavir and ganciclovir susceptibility. Antimicrob. Agents Chemother. 55:382–384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Marschall M, Stein-Gerlach M, Freitag M, Kupfer R, van Den Bogaard M, Stamminger T. 2001. Inhibitors of human cytomegalovirus replication drastically reduce the activity of the viral protein kinase pUL97. J. Gen. Virol. 82(Part 6):1439–1450 [DOI] [PubMed] [Google Scholar]
- 43.Michel D, Schaarschmidt P, Wunderlich K, Heuschmid M, Simoncini L, Muhlberger D, Zimmermann A, Pavic I, Mertens T. 1998. Functional regions of the human cytomegalovirus protein pUL97 involved in nuclear localization and phosphorylation of ganciclovir and pUL97 itself. J. Gen. Virol. 79(Part 9):2105–2112 [DOI] [PubMed] [Google Scholar]
- 44.Chou S, Marousek G, Bowlin TL. 2012. Cyclopropavir susceptibility of cytomegalovirus DNA polymerase mutants selected after antiviral drug exposure. Antimicrob. Agents Chemother. 56:197–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Erice A. 1999. Resistance of human cytomegalovirus to antiviral drugs. Clin. Microbiol. Rev. 12:286–297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chou S, Wechel LC, Marousek GI. 2007. Cytomegalovirus UL97 kinase mutations that confer maribavir resistance. J. Infect. Dis. 196:91–94 [DOI] [PubMed] [Google Scholar]
- 47.Evers DL, Komazin G, Ptak RG, Shin D, Emmer BT, Townsend LB, Drach JC. 2004. Inhibition of human cytomegalovirus replication by benzimidazole nucleosides involves three distinct mechanisms. Antimicrob. Agents Chemother. 48:3918–3927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chou S. 2008. Cytomegalovirus UL97 mutations in the era of ganciclovir and maribavir. Rev. Med. Virol. 18:233–246 [DOI] [PubMed] [Google Scholar]
- 49.Baldanti F, Sarasini A, Drach JC, Zemlicka J, Gerna G. 2002. Z-isomers of 2-hydroxymethylcyclopropylidenemethyl adenine (synadenol) and guanine (synguanol) are active against ganciclovir- and foscarnet-resistant human cytomegalovirus UL97 mutants. Antiviral Res. 56:273–278 [DOI] [PubMed] [Google Scholar]