

Abstract
Undecaprenyl pyrophosphate synthase (UppS) catalyzes the formation of the C55 lipid carrier (UPP) that is essential for bacterial peptidoglycan biosynthesis. We selected here a vancomycin (VAN)-resistant derivative of Bacillus subtilis W168 that contains a single-point mutation in the ribosome-binding site of the uppS gene designated uppS1. Genetic reconstruction experiments demonstrate that the uppS1 allele is sufficient to confer low-level VAN resistance and causes reduced UppS translation. The decreased level of UppS renders B. subtilis slightly more susceptible to many late-acting cell wall antibiotics, including β-lactams, but significantly more resistant to fosfomycin and d-cycloserine, antibiotics that interfere with the very early steps of cell wall synthesis. We further show that the uppS1 allele leads to slightly elevated expression of the σM regulon, possibly helping to compensate for the stress caused by a decrease in UPP levels. Notably, the uppS1 mutation increases resistance to VAN, fosfomycin, and d-cycloserine in wild-type cells, but this effect is greatly reduced or eliminated in a sigM mutant background. Our findings suggest that, although UppS is an attractive antibacterial target, incomplete inhibition of UppS function may lead to increased resistance to some cell wall-active antibiotics.
INTRODUCTION
The selective pressure of current antibiotics, including vancomycin (VAN), has led to the emergence of drug-resistant bacteria, calling for an urgent need for novel antibacterial drugs (1, 2). Undecaprenyl pyrophosphate synthase (UppS) is a potential target for antibiotics due to its essential role in peptidoglycan synthesis (3, 4). UppS is a cis-prenyltransferase that catalyzes the condensation of farnesyl diphosphate with eight molecules of isopentenyl diphosphate to generate undecaprenyl pyrophosphate (UPP, C55-PP). C55-PP is first dephosphorylated to undecaprenyl phosphate (C55-P) and then converted to the lipid I and lipid II species needed for bacterial cell wall biosynthesis. C55-P also serves as a carrier in the synthesis of peptidoglycan as well as various other cell wall components such as wall teichoic acids, lipopolysaccharide, and O-antigens (5–7). Considerable effort has been devoted to the identification of selective UppS inhibitors as lead compounds for antibiotic development (8–12).
VAN is a member of the glycopeptide antibiotic family and interacts with the d-alanyl-d-alanine terminus of the pentapeptide of lipid II, thereby inhibiting the transglycosylation and transpeptidation reactions in peptidoglycan assembly (13, 14). Although VAN is not effective against most Gram-negative bacteria because of an inability to penetrate their outer membrane (15), it has been widely used as a last-line antibiotic to combat methicillin-resistant Staphylococcus aureus (16). However, resistance to VAN emerged and was first reported in 1988 for an isolate of Enterococcus faecium (17), and resistance has also emerged in other bacteria, including S. aureus (18–21).
The most common mechanism of resistance to VAN in enterococci is the conversion of the terminal d-Ala-d-Ala in the peptidoglycan pentapeptide to d-alanyl-d-lactate (VanA, VanB, and VanD type) or to d-alanyl-d-serine (VanC, VanE, and VanG type), to which VAN exhibits low binding affinities (22, 23). Expression of all six types of resistance is regulated by the similar two-component signal transduction system (TCS) composed of a membrane-bound VanS histidine kinase and a cytoplasmic VanR response regulator (19, 24). Recently, three novel gene clusters conferring VAN resistance have also been discovered in Enterococcus species (25–27). In S. aureus, another type of resistance mechanism has been identified through point mutations of endogenous TCSs that control normal cell wall metabolism (WalRK) (28, 29) or the cellular response to cell wall damage (VraRS and GraRS) (30, 31). Members of the genus Bacillus are generally sensitive to VAN (32), and the mutant strain of the model bacterium B. subtilis lacking the extracytoplasmic function sigma factor σM is slightly more sensitive than the wild type to VAN (33). However, naturally occurring VAN-resistant strains of B. subtilis have not been reported.
The aim of the present study was to identify genes that might be involved in the emergence of VAN resistance in B. subtilis, a strain lacking any known VAN resistance mechanisms. We used whole-genome resequencing and identified a single-point mutation (A to C) in the ribosome-binding site (RBS) of B. subtilis uppS. This uppS1 allele confers low-level VAN resistance (an ∼2-fold-higher MIC than the susceptible parental strain). We further demonstrate that reduced UppS expression has unexpected and differential effects on the susceptibility of B. subtilis to cell wall antibiotics.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
The bacterial strains, plasmids, and primers used in the present study are listed in Table 1. Double mutants were generated through chromosomal transformation (34). Cells were routinely cultured in Luria-Bertani (LB) medium at 37°C. The following antibiotics were used when appropriate: spectinomycin (Spec; 100 μg/ml), tetracycline (5 μg/ml), kanamycin (15 μg/ml), chloramphenicol (10 μg/ml), or macrolide-lincosamide-streptogramin B (MLS) (contains 1 μg of erythromycin/ml and 25 μg of lincomycin/ml).
Table 1.
Bacterial strains, plasmids, and primers used in this study
| Strain, plasmid, or primer | Genotype, description, or sequence (5′–3′)a | Source or referenceb |
|---|---|---|
| Strains | ||
| W168 | trpC2 | BGSC no. 1A1 |
| HB13564 | W168 uppS1 yqbO::Tn7 (VAN-resistant isolate) | Tn7 library with W168 |
| HB13647 | W168 kan-uppS+ | LFH PCR with W168 |
| HB13648 | W168 kan-uppS1 | LFH PCR with W168 |
| HB13649 | W168 kan::uppS1 amyE::Pspac(hy)-uppS (cat) | pYH003 with HB13648 |
| HB13650 | W168 amyE::Pspac(hy)-uppS+-FLAG (cat) | pYH004 with W168 |
| HB13651 | W168 amyE::Pspac(hy)-uppS1-FLAG (cat) | pYH005 with W168 |
| HB13591 | W168 sigM::tet | 43 |
| HB13592 | W168 sigX::spec | 43 |
| HB13549 | W168 sigW::mls | 60 |
| HB13654 | W168 sigM::tet kan-uppS+ | HB13591 chr DNA with HB13647 |
| HB13655 | W168 sigM::tet kan-uppS1 | HB13591 chr DNA with HB13648 |
| HB13656 | W168 sigX::spec kan-uppS+ | HB13592 chr DNA with HB13647 |
| HB13657 | W168 sigX::spec kan-uppS1 | HB13592 chr DNA with HB13648 |
| HB13658 | W168 sigW::mls kan-uppS+ | HB13549 chr DNA with HB13647 |
| HB13659 | W168 sigW::mls kan-uppS1 | HB13549 chr DNA with HB13648 |
| Plasmids | ||
| pPL82 | IPTG-inducible expression vector (amyE integration) | 42 |
| pYH003 | Pspac(hy)-uppS in pPL82 | This study |
| PYH004 | Pspac(hy)-uppS+-FLAG in pPL82 | This study |
| PYH005 | Pspac(hy)-uppS1-FLAG in pPL82 | This study |
| Primers (no. [name]) | ||
| 6011 (uppS up-F) | TGAATCAGCTGTTAAGTATGAATCAC | |
| 6012 [uppS up-R(kan)] | CCTATCACCTCAAATGGTTCGCTGTGTACATAGTTTTTCATTAAACTTCCA | |
| 6014 [uppS do-F(kan)] | CGAGCGCCTACGAGGAATTTGTATCGACTGTTGATTACATTGATTATCAGCA | |
| 6015 (uppS do-R) | TACTCAGCCTTATCTCGCCG | |
| 6019 (uppS Pspac-F) | GCGCCCCGGGAACCTTTTTGGGTGACGGAG | |
| 6020 (uppS Pspac-R) | GCGCTCTAGATTTGTTTCATGTCCACCATCC | |
| 6021 (uppS-FLAG Pspac-R) | GCGCTCTAGATTACTTATCGTCGTCATCCTTGTAATCAATTCCGCCAAACCTCC |
Restriction sites are underlined, and the sequences complementary to antibiotic resistance cassettes for LFH PCR are in boldface.
chr, chromosomal.
Transposon mutagenesis and screening.
The modified Tn7 transposon mutagenesis system mTn7SX was used to isolate mutants with increased resistance to VAN (35, 36). The amplified Tn7 library DNA was transformed into the wild-type B. subtilis W168 (Bacillus Genetic Stock Center [BGSC] accession number 1A1), and the resulting transposants were selected on LB agar plates containing Spec (100 μg/ml) and VAN (0.3 or 0.5 μg/ml). To determine whether the VAN resistance is linked to a transposon insertion, the chromosomal DNA of the Tn7 mutant was transformed into the wild-type strain and selected with 100 μg of Spec/ml. The 10 transformants were tested for susceptibility to VAN using a Bioscreen C microbial growth analyzer (Growth Curves USA, Piscataway, NJ) at 37°C with vigorous shaking. The Tn7 insertion site in the chromosome was determined by arbitrary PCR and subsequent DNA sequencing (37).
Whole-genome resequencing.
Genomic DNA was extracted from B. subtilis wild-type and HB13564 mutant strains grown in LB medium to an optical density at 600 nm (OD600) of 0.4 using the Qiagen DNeasy blood and tissue kit. The quantity and purity of DNA was determined using a NanoDrop spectrophotometer (NanoDrop Technologies, Inc., Wilmington, DE) and sequenced using an Illumina HiSeq 2000 at the Cornell University Life Sciences Core Laboratories Center. The resulting genomic sequence data were assembled with MOSAIC, using the reference sequence (38), under GenBank accession number ABQK00000000.
Genetic reconstruction.
The single-point mutation in the RBS of the uppS gene was moved into the wild-type chromosome using long-flanking homology (LFH) PCR as described previously (39, 40). The upstream fragment, ending after the stop codon of the frr gene, was amplified from the wild-type B. subtilis strain using the primers 6011 (uppS up-F) and 6012 [uppS up-R(kan)]. The downstream fragment containing the promoter and RBS of uppS1 was amplified from HB13564 using the primers 6014 [uppS do-F(kan)] and 6015 (uppS do-R). The upstream and downstream fragments were joined to a kanamycin resistance cassette using primers 6011 and 6015. The final PCR products were transformed into the wild-type B. subtilis strain, and the correct introduction of the point mutation was confirmed by DNA sequencing.
Plasmid construction.
Gene cloning and molecular biology procedures were performed as described by Sambrook and Russell (41). Plasmid pPL82 was used for the expression of UppS and UppS-FLAG under the control of isopropyl-β-d-thiogalactopyranoside (IPTG)-inducible promoter Pspac(hy) (42). For the construction of pYH003 (pPL82-uppS+), the uppS coding region and the native RBS was amplified from B. subtilis wild-type chromosomal DNA by PCR using the primers 6019 (uppS Pspac-F) and 6020 (uppS Pspac-R). pYH004 (pPL82-uppS+-FLAG) and pYH005 (pPL82-uppS1-FLAG) were constructed in a similar manner using a pair of primers, 6019 (uppS Pspac-F) and 6021 (uppS-FLAG Pspac-R). Construct integrity was verified by DNA sequencing. The resulting constructs were then integrated by a double-crossover event into the amyE locus.
Antibiotic susceptibility tests.
The Etest method and disk diffusion assay were performed on Mueller-Hinton (MH) agar plates as described previously (43). Etest strips (bioMérieux, Durham, NC) impregnated with VAN (concentrations ranging from 0.016 to 256 μg/ml) were used to determine the MIC. The following antibiotics and quantities were used in the disk diffusion assays: ramoplanin, 300 μg; moenomycin, 500 μg; bacitracin, 2.5 mg; cefuroxime, 50 μg; ampicillin, 50 μg; penicillin G, 50 μg; fosfomycin, 500 μg; and d-cycloserine, 500 μg. For the MIC test, strains were grown to an OD600 of 0.4 and then diluted 1:400 in MH broth. Aliquots (200 μl) of the diluted culture were dispensed in 100-well Bioscreen C microplates, and a range of at least nine antibiotic concentrations close to the MIC were added to each well. Growth was measured spectrophotometrically (OD600) every 30 min for 24 h using a Bioscreen C microbial growth analyzer at 37°C with continuous shaking. MIC was defined as the lowest concentration of antibiotic that completely inhibited growth at the 16-h time point. Representative antibiotic concentrations tested included the following: ramoplanin (0, 0.25, 0.3, 0.35, 0.4, 0.45, 0.5, 0.55, 0.6, and 0.7 μg/ml), moenomycin (0, 1, 2, 3, 4, 5, 6, 8, 10, and 12 μg/ml), bacitracin (0, 500, 600, 700, 800, 900, 1,000, 1,100, 1,200, and 1,300 μg/ml), cefuroxime (0, 1.5, 2, 2.5, 3, 3.5, 4, 4.5, 5, and 6 μg/ml), ampicillin (0, 0.5, 0.55, 0.6, 0.65, 0.7, 0.75, 0.8, 0.9, and 1 μg/ml), penicillin G (0, 0.1, 0.2, 0.3, 0.4, 0.5, 0.6, 0.7, 0.8, and 1 μg/ml), fosfomycin (0, 25, 50, 75, 100, 125, 150, 175, 200, and 250 μg/ml), and d-cycloserine (0, 50, 75, 100, 125, 150, 175, 200, 225, and 250 μg/ml).
Immunoblot analysis.
Strains expressing FLAG-tagged UppS were grown in LB medium to an OD600 of 0.4 in the presence or absence of 0.1 mM IPTG. Whole-cell lysates were prepared as described by Smaldone et al. (44). Bradford assays were used for protein quantification, and then 20 μg of total protein was resolved by electrophoresis in a 12% sodium dodecyl sulfate-polyacrylamide gel and transferred onto an immunoblot polyvinylidene difluoride membrane (Bio-Rad). Membranes were blocked with blocking solution (TBS-T containing 5% skim milk) and immunoblotted with anti-FLAG rabbit antibody (Sigma-Aldrich). Alkaline phosphatase-linked anti-rabbit IgG was used as a secondary antibody. Immunoreactive bands were visualized by adding the BCIP (5-bromo-4-chloro-3-indolylphosphate)/nitroblue tetrazolium substrate (Bio-Rad). The relative levels of UppS-FLAG were compared by using ImageJ software.
Microarray analyses.
Two microarrays were performed in biological triplicates with a dye swap. Total RNA was isolated from HB13647 (kan-uppS+) and HB13648 (kan-uppS1) grown in LB medium to an OD600 of 0.4, using the RNeasy minikit (Qiagen), followed by DNase treatment with Turbo DNA free (Ambion). The quantity and purity of RNA was determined using a NanoDrop spectrophotometer. cDNA labeling and microarray analysis were performed as described previously (45). The GenePix Pro software package (version 6.0) was used for image processing and analysis. The normalized microarray data sets were filtered to remove genes that were not expressed at levels significantly above background under either condition (sum of mean fluorescence intensities, <20). In addition, the means and standard deviations of the fluorescence intensities were computed for each gene, and those for which the standard deviation was greater than the mean value were ignored. The fold change was calculated by using the average signal intensities for HB13648 divided by those for HB13647.
Microarray data accession number.
The microarray data set is available in the NCBI GEO database under accession number GSE45305.
RESULTS AND DISCUSSION
Identification of a single point mutation in a B. subtilis mutant with low-level resistance to VAN.
To identify genes whose inactivation or modification leads to the development of VAN resistance in B. subtilis, we selected for VAN-resistant mutants. We introduced an amplified mTn7SX library into the wild-type B. subtilis strain by transformation (35), and isolated 19 resistant strains that were able to grow on LB agar supplemented with VAN (0.3 or 0.5 μg/ml). To determine the relative resistance levels of isolated strains, we monitored their growth rates in the presence of VAN using a Bioscreen C growth analyzer. Upon rescreening, 18 of 19 isolates were only slightly more resistant to VAN than the wild type. The single remaining isolate (HB13564) showed an ∼2-fold increase in the MIC of VAN in both liquid growth assays (Fig. 1A) and using the Etest assay (Fig. 1B). The mTn7SX insertion was found to be located in yqbO, encoding a putative lytic transglycosylase. However, linkage tests demonstrated that the VAN resistance phenotype was not linked to the transposon insertion, suggesting that other spontaneous mutations were responsible for the VAN resistance of HB13564.
Fig 1.

Whole-genome resequencing of a spontaneous mutant (HB13564) of B. subtilis with low-level resistance to VAN. (A) Liquid growth assays, in the absence or presence of VAN, were performed in MH medium using a Bioscreen C growth analyzer. The data are representative of at least three independent experiments. (B) Determination of MICs of VAN using an Etest assay. The Etest strips (bioMérieux) with VAN concentration of 0.016 to 256 μg/ml were applied onto MH agar plates, and then the plates were incubated at 37°C for 18 h. The MIC (in μg/ml) was determined by identifying where bacterial growth intersects the Etest strip. (C) Schematic representation of the location of the single nucleotide polymorphism (SNP) identified by Illumina whole-genome resequencing. The cluster of uppS-cdsA-dxr-rseP-proS genes comprises a single transcription unit. (D) Sanger sequencing of genomic DNA confirms the A-to-C substitution in the RBS of the uppS gene in the HB13564 strain. The RBS sequence and start codon are underlined, and the mutated residue is indicated with an arrow.
To identify genomic changes responsible for the increased VAN resistance, we performed whole-genome resequencing of the HB13564 strain and its vancomycin-susceptible parent. Comparison of the whole-genome sequences revealed a single-base substitution (A to C) in the RBS of the uppS gene that encodes an undecaprenyl pyrophosphate synthase, a key enzyme in cell wall peptidoglycan biosynthesis. The uppS gene is essential for the survival of B. subtilis (46), and the five-gene cluster, including uppS comprises a single operon (Fig. 1C) (47). This point mutation (designated uppS1) was further confirmed by PCR and DNA sequencing (Fig. 1D).
The uppS1 allele confers resistance to VAN and reduces UppS translation.
To determine whether the uppS1 allele is necessary and sufficient for VAN resistance, we moved the point mutation into the parental wild-type strain as outlined in Fig. 2A. A kanamycin antibiotic resistance cassette was introduced as a selectable marker after the stop codon of the frr gene, located directly upstream of the uppS gene. The correct introduction of the point mutation in strain HB13648 (kan-uppS1) was confirmed by PCR and DNA sequencing. As a control, we also constructed strain HB13647 (kan-uppS+) carrying a wild-type RBS sequence. We next compared the VAN susceptibility of the reconstructed HB13647 (kan-uppS+) and HB13648 (kan-uppS1) strains using a Bioscreen C growth analyzer. Indeed, the reconstructed strain carrying the uppS1 allele exhibited a reduced susceptibility to VAN compared to the wild type (Fig. 2B), indicating that the VAN resistance of the HB13564 mutant is due to the uppS1 allele.
Fig 2.

Evaluation of the effect of the uppS1 allele on low-level VAN resistance. (A) Genetic reconstruction of the uppS1 allele in the wild-type background. The uppS1 allele, indicated by a thick gray vertical line, was amplified from the HB13564 mutant. A kanamycin antibiotic resistance cassette was introduced after the stop codon of the frr gene, followed by a 715-bp downstream fragment containing the promoter and RBS of the uppS gene. The uppS1 allele was introduced into the parental wild-type strain by transformation and was confirmed by DNA sequencing. (B) IPTG induction of uppS restores VAN sensitivity to wild-type levels. Complementation of uppS1 by Pspac(hy)-uppS was observed with addition of 0.1 mM IPTG.
Since a point mutation in the RBS can cause a weak interaction between the RBS sequence and the 3′ end of 16S rRNA, we hypothesized that this mutation might reduce UppS translation. To test whether VAN resistance is associated with decreased UppS synthesis, the uppS gene was expressed under the control of the IPTG-inducible Pspac(hy) promoter at the amyE locus of the reconstructed mutant. As expected, IPTG induction of uppS restored VAN sensitivity to wild-type levels (Fig. 2B). Control experiments showed that IPTG itself does not increase VAN sensitivity (data not shown).
To determine whether the RBS substitution decreased UppS translation, we performed an immunoblot analysis of total protein extracts from the B. subtilis wild-type strains harboring Pspac(hy)-uppS+-FLAG and Pspac(hy)-uppS1-FLAG at the amyE locus. C-terminally FLAG-tagged UppS (UppS-FLAG) was expressed under the control of the IPTG-inducible promoter. As shown in Fig. 3, UppS-FLAG levels were ∼2-fold decreased by the RBS mutation, possibly indicating an impaired mRNA-ribosome pairing efficiency. These results suggest that the uppS1 mutation reduces translation, which we suggest confers low-level resistance to VAN in B. subtilis.
Fig 3.
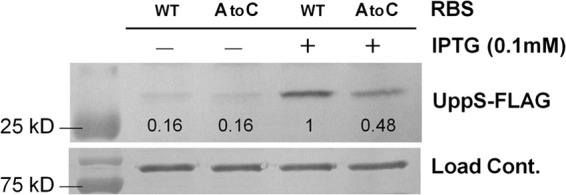
Effect of the uppS1 allele on the UppS protein level. Strains containing Pspac(hy)-uppS+-FLAG and Pspac(hy)-uppS1-FLAG at the neutral amyE chromosomal locus were grown in LB medium at 37°C in the absence or presence of 0.1 mM IPTG. At an OD600 of 0.4, cells were harvested for protein extraction. A total of 20 μg of protein from each strain was electrophoresed on a 12% acrylamide gel and transferred to PVDF membrane. The signal intensities were quantified using ImageJ software. A representative data set is shown.
Reduced UppS levels affect susceptibility of B. subtilis to cell wall antibiotics.
Newly synthesized UPP (C55-PP) is dephosphorylated to C55-P and then converted to lipid I and lipid II, intermediates for peptidoglycan biosynthesis (Fig. 4A). Thus, lowering UppS protein levels is predicted to lead to less UPP and thereby results in the reduced synthesis of lipid II. To examine how the uppS1 allele affects susceptibility of B. subtilis to cell wall antibiotics that inhibit other steps in peptidoglycan biosynthesis, we performed disk diffusion assays on MH agar plates. The uppS1 mutation made B. subtilis slightly more susceptible to most of the antibiotics tested (Fig. 4B). In contrast, the uppS1 mutant exhibited significantly increased resistance to fosfomycin and d-cycloserine, which interfere with very early steps of cell wall synthesis before the utilization of UPP (Fig. 5). Moreover, IPTG induction of uppS restored sensitivity to fosfomycin and d-cycloserine to wild-type levels. We also defined the MICs of these cell wall antibiotics for the uppS1 mutant (HB13648) and wild-type (HB13647) strains using a liquid medium growth inhibition assay (Table 2). These results suggest that decreased UppS synthesis is associated with altered susceptibility of B. subtilis to several cell wall antibiotics.
Fig 4.

Decreased level of UppS increases susceptibility to late-acting cell wall antibiotics. (A) Schematic representation of the bacterial peptidoglycan synthesis pathway, related genes, and cell wall antibiotics that interfere with specific steps. (B) Effect of the uppS1 allele on susceptibility to late-acting cell wall antibiotics. Disk diffusion assays were performed on MH agar plates with filter paper disks containing different types of antibiotics. Each bar represents the average zone of inhibition, expressed as total diameter minus diameter of the filter paper disk (6.5 mm). Three independent experiments were performed for each strain. Error bars indicate standard deviations.
Fig 5.
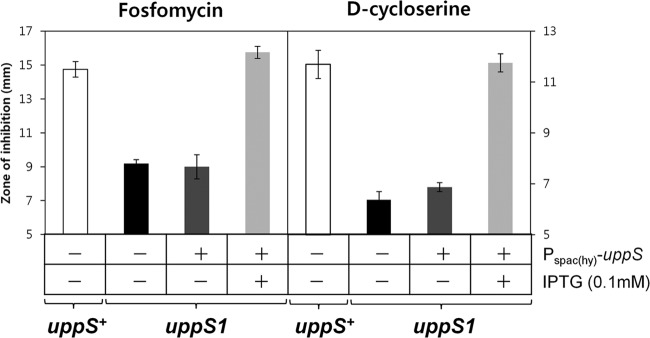
Decreased level of UppS reduces susceptibility to early-acting cell wall antibiotics. Disk diffusion assays were performed on MH agar plates with fosfomycin and d-cycloserine. Overexpression of uppS was induced by the addition of 0.1 mM IPTG. Three independent experiments were performed for each strain. Error bars indicate standard deviations.
Table 2.
MICs for cell wall-active antibiotics
| Strain or fold change | MICa (μg/ml) |
|||||||
|---|---|---|---|---|---|---|---|---|
| Ramoplanin | Moenomycin | Bacitracin | Cefuroxime | Ampicillin | Penicillin G | Fosfomycin | d-Cycloserineb | |
| uppS+ strain | 0.5 | 5.0 | 1,000 | 4.0 | 1.0 | 0.6 | 100 | 125 |
| uppS1 mutant | 0.3 | 2.0 | 700 | 3.5 | 0.8 | 0.4 | 200 | 225 |
| Fold changec | 0.60 | 0.40 | 0.70 | 0.87 | 0.80 | 0.66 | 2.00 | 1.80 |
MICs were determined by the growth inhibition assay in MH broth. Data are reported as median values from at least three independent experiments.
The MIC endpoint was established after 24 h of incubation. All others were determined after 16 h.
The fold change was calculated by dividing the MIC for the uppS1 mutant (HB13648) by the MIC for the wild type (HB13647).
uppS1 leads to elevated expression of the σM regulon and thereby affects antibiotic susceptibility.
Chemical or genetic inhibition of cell wall synthesis often leads to the activation of adaptive responses that can confer antibiotic resistance. We used microarray-based gene expression profiling to identify differences in gene expression patterns between the uppS1 mutant (HB13648) and wild-type (HB13647) strains in the absence of antibiotic exposure. A regulon-based expression analysis revealed that genes in the σM regulon were upregulated between 1.5- and ∼3-fold in the uppS1 mutant. The B. subtilis σM factor is known to be activated in response to the inhibition of cell wall synthesis (33, 48–50), and previous studies have shown that σM-regulated genes are often upregulated between 2- and 4-fold by VAN treatment.
The σM regulon includes several cell wall biosynthesis enzymes (49), and contributes to the maintenance of cell wall integrity in response to cell envelope stress (51). In the uppS1 mutant, the (p)ppGpp synthetase gene ywaC was induced ∼3-fold. This is consistent with a previous report showing that the ywaC promoter is activated by lesions in teichoic acid biosynthesis (postulated to sequester lipid II), by depletion of an enzyme required for polyisoprenoid (undecaprenol) synthesis, and by vancomycin, ramoplanin, fosfomycin, and bacitracin (52). In addition to its direct targets, activation of σM indirectly affects the expression of numerous genes through the action of the Spx transcription factor (49) and likely due to effects of YwaC on (p)ppGpp, which is a global regulator of gene expression during stringent response (53, 54).
To determine whether σM is involved in the enhanced resistance to VAN, fosfomycin, and d-cycloserine, we introduced a null allele of sigM into the uppS1 strain and compared the VAN susceptibility of the strains using a Bioscreen C growth analyzer. Consistent with the known role of σM in VAN resistance (33), the disruption of sigM led to VAN sensitivity, but no effects were attributed to disruption of two other cell wall-related σ factors, sigX and sigW (Fig. 6B). Significantly, in the sigM uppS1 strain, there was no increase in VAN resistance relative to the sigM single mutant. Thus, the uppS1 mutation not only leads to a modest upregulation of σM activity (Fig. 6A), but the increased resistance to VAN depends on σM (Fig. 6B). Similarly, the increased resistance to fosfomycin due to the uppS1 mutation was dependent on σM (Fig. 6C). Indeed, the sigM uppS1 strain was more sensitive to fosfomycin than the sigM single mutant. The uppS1 mutation does slightly increase d-cycloserine resistance even in the sigM background (Fig. 6C), but most of the observed resistance to d-cycloserine appears to require σM. These results support a model in which uppS1 leads to an increased expression of the σM regulon (Fig. 6A), and this contributes to the observed resistance to VAN, fosfomycin, and d-cycloserine. It is not yet known which gene(s) in the σM regulon are important for this effect. Since these same antibiotics can also induce the σM regulon in wild-type cells, the effects here may reflect the advantage of having one or more genes already upregulated prior to antibiotic exposure. The σW-regulated fosB gene is critical for fosfomycin resistance (55). However, it is not significantly upregulated in the uppS1 strain, and thus other genes likely contribute to the observed resistance.
Fig 6.

The compensatory-adaptive response of B. subtilis through σM to a decrease in UppS levels. (A) Microarray transcriptional analysis of strain HB13648 (kan-uppS1) and parental strain HB13647 (kan-uppS+). Black triangles indicate the σM regulon genes whose expression changed at least 1.5-fold. The data shown are mean fluorescence intensities from two independent experiments. Each comparison was performed with dye swaps on each of three biological replicates. (B) Effect of inactivation of sigM, sigX, and sigW on the susceptibility of the HB13648 (kan-uppS1) strain to VAN. The MIC (in μg/ml) for VAN was determined in MH broth by using a Bioscreen C growth analyzer. (C) Effect of sigM inactivation on the susceptibility of the HB13648 (kan-uppS1) strain to fosfomycin and d-cycloserine. Disk diffusion assays were performed on MH agar plates with fosfomycin and d-cycloserine. Three independent experiments were performed for each strain. Error bars indicate standard deviations.
Flux-based model for effects of uppS1 on antibiotic susceptibility.
Altered expression of the σM regulon likely accounts for the increased resistance to some antibiotics noted in the uppS1 mutant. However, increased expression of the σM regulon does not explain the increased susceptibility to late-acting cell wall synthesis inhibitors (Fig. 4B). We hypothesize that these effects may result from an altered flux of intermediates in the peptidoglycan biosynthesis pathway. Specifically, we hypothesize that reduced availability of C55-P (due to uppS1) may decrease the cellular demand for UDP-MurNAc-pentapeptide (Fig. 4A). As a result, the cell may tolerate higher levels of inhibition by fosfomycin (a suicide substrate for MurA) and d-cycloserine (a competitive inhibitor of d-Ala d-Ala ligase). We further propose that the uppS1 allele may lead to a reduction in lipid II levels, and this might account for increase in susceptibility to ramoplanin, moenomycin, bacitracin, and β-lactams (Fig. 4B). Since binding of moenomycin (to the transglycosylase active site) and β-lactams (to the transpeptidase site) is competitive with binding of lipid II, we propose that reduced lipid II levels leave these enzymes more susceptible to antibiotic binding and inactivation. Similarly, since bacitracin inhibits the regeneration of C55-P from UPP, uppS1 cells (impaired in de novo synthesis of C-55-P) are predicted to have increased bacitracin sensitivity, as observed (Fig. 4B).
Conclusions.
Our data demonstrate that reduced synthesis of UppS resulting from the uppS1 ribosome-binding site mutation alters susceptibility to several mechanistically distinct inhibitors of peptidoglycan synthesis. The uppS1 allele was selected for its ability to confer resistance to VAN. Our results indicate that uppS1 leads to an increased activity of σM, a known VAN-activated σ factor. Further, σM is required for increased resistance to VAN and fosfomycin, and for most of the uppS1-dependent d-cycloserine resistance. This suggests that higher-level expression of the σM regulon in the uppS1 mutant activates an adaptive response that provides protection against some cell wall antibiotics. σM is also known to be a major determinant for resistance to moenomycin (50), bacitracin (50, 56), and cefuroxime (48). However, despite increased expression of the σM regulon (Fig. 6A), the uppS1 strain has an increased sensitivity to these antibiotics (Fig. 4B). We suggest that, for moenomycin and these β-lactams, the major effects of the uppS1 allele are due to reduced lipid II concentrations, and the consequent lack of substrate protection of the penicillin-binding protein active sites targeted by these inhibitors. In the case of bacitracin, we suggest that the reduced levels of UPP synthesis in the uppS1 strain compounds the effects of reduced UPP recycling in the presence of bacitracin.
UppS is of considerable interest as a new antibacterial target due to its essential role in synthesizing the C55 lipid carrier for peptidoglycan biosynthesis. Our results suggest that incomplete inhibition of UppS synthesis may cause an unexpected decrease in susceptibility to some cell wall antibiotics. It should also be noted that the human UppS homolog, dehydrodolichyl diphosphate synthase, is essential for the biosynthesis of dolichols (57), and defects are responsible for certain variants of retinitis pigmentosa (58, 59). These factors should be considered during efforts to develop new antibacterial drugs targeting the undecaprenyl pyrophosphate synthase.
ACKNOWLEDGMENT
This study was supported by a grant from the National Institutes of Health (GM047446).
Footnotes
Published ahead of print 24 June 2013
REFERENCES
- 1.Fischbach MA, Walsh CT. 2009. Antibiotics for emerging pathogens. Science 325:1089–1093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chambers HF, Deleo FR. 2009. Waves of resistance: Staphylococcus aureus in the antibiotic era. Nat. Rev. Microbiol. 7:629–641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Payne DJ, Gwynn MN, Holmes DJ, Pompliano DL. 2007. Drugs for bad bugs: confronting the challenges of antibacterial discovery. Nat. Rev. Drug Discov. 6:29–40 [DOI] [PubMed] [Google Scholar]
- 4.Sinko W, de Oliveira C, Williams S, Van Wynsberghe A, Durrant JD, Cao R, Oldfield E, McCammon JA. 2011. Applying molecular dynamics simulations to identify rarely sampled ligand-bound conformational states of undecaprenyl pyrophosphate synthase, an antibacterial target. Chem. Biol. Drug Design 77:412–420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ogura K, Koyama T. 1998. Enzymatic aspects of isoprenoid chain elongation. Chem. Rev. 98:1263–1276 [DOI] [PubMed] [Google Scholar]
- 6.Takahashi I, Ogura K. 1982. Prenyltransferases of Bacillus subtilis: undecaprenyl pyrophosphate synthetase and geranylgeranyl pyrophosphate synthetase. J. Biochem. 92:1527–1537 [DOI] [PubMed] [Google Scholar]
- 7.Teng KH, Liang PH. 2012. Structures, mechanisms and inhibitors of undecaprenyl diphosphate synthase: a cis-prenyltransferase for bacterial peptidoglycan biosynthesis. Bioorg. Chem. 43:51–57 [DOI] [PubMed] [Google Scholar]
- 8.Li H, Huang J, Jiang X, Seefeld M, McQueney M, Macarron R. 2003. The effect of triton concentration on the activity of undecaprenyl pyrophosphate synthase inhibitors. J. Biomol. Screening 8:712–715 [DOI] [PubMed] [Google Scholar]
- 9.Guo RT, Cao R, Liang PH, Ko TP, Chang TH, Hudock MP, Jeng WY, Chen CK, Zhang Y, Song Y, Kuo CJ, Yin F, Oldfield E, Wang AH. 2007. Bisphosphonates target multiple sites in both cis- and trans-prenyltransferases. Proc. Natl. Acad. Sci. U. S. A. 104:10022–10027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Peukert S, Sun Y, Zhang R, Hurley B, Sabio M, Shen X, Gray C, Dzink-Fox J, Tao J, Cebula R, Wattanasin S. 2008. Design and structure-activity relationships of potent and selective inhibitors of undecaprenyl pyrophosphate synthase (UPPS): tetramic, tetronic acids, and dihydropyridin-2-ones. Bioorg. Med. Chem. Lett. 18:1840–1844 [DOI] [PubMed] [Google Scholar]
- 11.Kuo CJ, Guo RT, Lu IL, Liu HG, Wu SY, Ko TP, Wang AH, Liang PH. 2008. Structure-based inhibitors exhibit differential activities against Helicobacter pylori and Escherichia coli undecaprenyl pyrophosphate synthases. J. Biomed. Biotechnol. 2008:841312. 10.1155/2008/841312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Inokoshi J, Nakamura Y, Hongbin Z, Uchida R, Nonaka K, Masuma R, Tomoda H. 2013. Spirohexalines, new inhibitors of bacterial undecaprenyl pyrophosphate synthase, produced by Penicillium brasilianum FKI-3368. J. Antibiot. 66:37–41 [DOI] [PubMed] [Google Scholar]
- 13.Nieto M, Perkins HR. 1971. Modifications of the acyl-d-alanyl-d-alanine terminus affecting complex formation with vancomycin. Biochem. J. 123:789–803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reynolds PE. 1989. Structure, biochemistry, and mechanism of action of glycopeptide antibiotics. Eur. J. Clin. Microbiol. Infect. Dis. 8:943–950 [DOI] [PubMed] [Google Scholar]
- 15.Murray PR, Baron EJ, Pfaller MA, Tenover FC, Yolken RH. (ed). 1995. Manual of clinical microbiology, 6th ed. ASM Press, Washington, DC [Google Scholar]
- 16.Walsh C. 1999. Deconstructing vancomycin. Science 284:442–443 [DOI] [PubMed] [Google Scholar]
- 17.Leclercq R, Derlot E, Duval J, Courvalin P. 1988. Plasmid-mediated resistance to vancomycin and teicoplanin in Enterococcus faecium. N. Engl. J. Med. 319:157–161 [DOI] [PubMed] [Google Scholar]
- 18.Gould IM. 2010. VRSA-doomsday superbug or damp squib? Lancet Infect. Dis. 10:816–818 [DOI] [PubMed] [Google Scholar]
- 19.Courvalin P. 2006. Vancomycin resistance in gram-positive cocci. Clin. Infect. Dis. 42(Suppl 1):S25–S34 [DOI] [PubMed] [Google Scholar]
- 20.Weigel LM, Clewell DB, Gill SR, Clark NC, McDougal LK, Flannagan SE, Kolonay JF, Shetty J, Killgore GE, Tenover FC. 2003. Genetic analysis of a high-level vancomycin-resistant isolate of Staphylococcus aureus. Science 302:1569–1571 [DOI] [PubMed] [Google Scholar]
- 21.Noble WC, Virani Z, Cree RG. 1992. Cotransfer of vancomycin and other resistance genes from Enterococcus faecalis NCTC 12201 to Staphylococcus aureus. FEMS Microbiol. Lett. 72:195–198 [DOI] [PubMed] [Google Scholar]
- 22.Depardieu F, Podglajen I, Leclercq R, Collatz E, Courvalin P. 2007. Modes and modulations of antibiotic resistance gene expression. Clin. Microbiol. Rev. 20:79–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pootoolal J, Neu J, Wright GD. 2002. Glycopeptide antibiotic resistance. Annu. Rev. Pharmacol. Toxicol. 42:381–408 [DOI] [PubMed] [Google Scholar]
- 24.Arthur M, Molinas C, Courvalin P. 1992. The VanS-VanR two-component regulatory system controls synthesis of depsipeptide peptidoglycan precursors in Enterococcus faecium BM4147. J. Bacteriol. 174:2582–2591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lebreton F, Depardieu F, Bourdon N, Fines-Guyon M, Berger P, Camiade S, Leclercq R, Courvalin P, Cattoir V. 2011. d-Ala-d-Ser VanN-type transferable vancomycin resistance in Enterococcus faecium. Antimicrob. Agents Chemother. 55:4606–4612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Boyd DA, Willey BM, Fawcett D, Gillani N, Mulvey MR. 2008. Molecular characterization of Enterococcus faecalis N06-0364 with low-level vancomycin resistance harboring a novel d-Ala-d-Ser gene cluster, vanL. Antimicrob. Agents Chemother. 52:2667–2672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xu X, Lin D, Yan G, Ye X, Wu S, Guo Y, Zhu D, Hu F, Zhang Y, Wang F, Jacoby GA, Wang M. 2010. vanM, a new glycopeptide resistance gene cluster found in Enterococcus faecium. Antimicrob. Agents Chemother. 54:4643–4647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jansen A, Turck M, Szekat C, Nagel M, Clever I, Bierbaum G. 2007. Role of insertion elements and yycFG in the development of decreased susceptibility to vancomycin in Staphylococcus aureus. Int. J. Med. Microbiol. 297:205–215 [DOI] [PubMed] [Google Scholar]
- 29.Mwangi MM, Wu SW, Zhou Y, Sieradzki K, de Lencastre H, Richardson P, Bruce D, Rubin E, Myers E, Siggia ED, Tomasz A. 2007. Tracking the in vivo evolution of multidrug resistance in Staphylococcus aureus by whole-genome sequencing. Proc. Natl. Acad. Sci. U. S. A. 104:9451–9456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Howden BP, Stinear TP, Allen DL, Johnson PD, Ward PB, Davies JK. 2008. Genomic analysis reveals a point mutation in the two-component sensor gene graS that leads to intermediate vancomycin resistance in clinical Staphylococcus aureus. Antimicrob. Agents Chemother. 52:3755–3762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kato Y, Suzuki T, Ida T, Maebashi K. 2010. Genetic changes associated with glycopeptide resistance in Staphylococcus aureus: predominance of amino acid substitutions in YvqF/VraSR. J. Antimicrob. Chemother. 65:37–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kervick GN, Flynn HW, Jr, Alfonso E, Miller D. 1990. Antibiotic therapy for Bacillus species infections. Am. J. Ophthalmol. 110:683–687 [DOI] [PubMed] [Google Scholar]
- 33.Cao M, Wang T, Ye R, Helmann JD. 2002. Antibiotics that inhibit cell wall biosynthesis induce expression of the Bacillus subtilis σW and σM regulons. Mol. Microbiol. 45:1267–1276 [DOI] [PubMed] [Google Scholar]
- 34.Harwood CR, Cutting SM. 1990. Molecular biological methods for Bacillus. Wiley, New York, NY [Google Scholar]
- 35.Bordi C, Butcher BG, Shi Q, Hachmann AB, Peters JE, Helmann JD. 2008. In vitro mutagenesis of Bacillus subtilis by using a modified Tn7 transposon with an outward-facing inducible promoter. Appl. Environ. Microbiol. 74:3419–3425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Salipante SJ, Barlow M, Hall BG. 2003. GeneHunter, a transposon tool for identification and isolation of cryptic antibiotic resistance genes. Antimicrob. Agents Chemother. 47:3840–3845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Peters JE, Craig NL. 2000. Tn7 transposes proximal to DNA double-strand breaks and into regions where chromosomal DNA replication terminates. Mol. Cell 6:573–582 [DOI] [PubMed] [Google Scholar]
- 38.Srivatsan A, Han Y, Peng J, Tehranchi AK, Gibbs R, Wang JD, Chen R. 2008. High-precision, whole-genome sequencing of laboratory strains facilitates genetic studies. PLoS Genet. 4:e1000139. 10.1371/journal.pgen.1000139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mascher T, Margulis NG, Wang T, Ye RW, Helmann JD. 2003. Cell wall stress responses in Bacillus subtilis: the regulatory network of the bacitracin stimulon. Mol. Microbiol. 50:1591–1604 [DOI] [PubMed] [Google Scholar]
- 40.Wach A. 1996. PCR-synthesis of marker cassettes with long flanking homology regions for gene disruptions in Saccharomyces cerevisiae. Yeast 12:259–265 [DOI] [PubMed] [Google Scholar]
- 41.Sambrook J, Russell DW. 2001. Molecular cloning: a laboratory manual, 3rd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 42.Quisel JD, Burkholder WF, Grossman AD. 2001. In vivo effects of sporulation kinases on mutant Spo0A proteins in Bacillus subtilis. J. Bacteriol. 183:6573–6578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lee YH, Nam KH, Helmann JD. 2013. A mutation of the RNA polymerase beta' subunit (rpoC) confers cephalosporin resistance in Bacillus subtilis. Antimicrob. Agents Chemother. 57:56–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Smaldone GT, Antelmann H, Gaballa A, Helmann JD. 2012. The FsrA sRNA and FbpB protein mediate the iron-dependent induction of the Bacillus subtilis lutABC iron-sulfur-containing oxidases. J. Bacteriol. 194:2586–2593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hachmann AB, Angert ER, Helmann JD. 2009. Genetic analysis of factors affecting susceptibility of Bacillus subtilis to daptomycin. Antimicrob. Agents Chemother. 53:1598–1609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Inaoka T, Ochi K. 2012. Undecaprenyl pyrophosphate involvement in susceptibility of Bacillus subtilis to rare earth elements. J. Bacteriol. 194:5632–5637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Price MN, Huang KH, Alm EJ, Arkin AP. 2005. A novel method for accurate operon predictions in all sequenced prokaryotes. Nucleic Acids Res. 33:880–892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Luo Y, Helmann JD. 2012. Analysis of the role of Bacillus subtilis sigma(M) in beta-lactam resistance reveals an essential role for c-di-AMP in peptidoglycan homeostasis. Mol. Microbiol. 83:623–639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Eiamphungporn W, Helmann JD. 2008. The Bacillus subtilis σM regulon and its contribution to cell envelope stress responses. Mol. Microbiol. 67:830–848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mascher T, Hachmann AB, Helmann JD. 2007. Regulatory overlap and functional redundancy among Bacillus subtilis extracytoplasmic function sigma factors. J. Bacteriol. 189:6919–6927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Thackray PD, Moir A. 2003. SigM, an extracytoplasmic function sigma factor of Bacillus subtilis, is activated in response to cell wall antibiotics, ethanol, heat, acid, and superoxide stress. J. Bacteriol. 185:3491–3498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.D'Elia MA, Millar KE, Bhavsar AP, Tomljenovic AM, Hutter B, Schaab C, Moreno-Hagelsieb G, Brown ED. 2009. Probing teichoic acid genetics with bioactive molecules reveals new interactions among diverse processes in bacterial cell wall biogenesis. Chem. Biol. 16:548–556 [DOI] [PubMed] [Google Scholar]
- 53.Magnusson LU, Farewell A, Nystrom T. 2005. ppGpp: a global regulator in Escherichia coli. Trends Microbiol. 13:236–242 [DOI] [PubMed] [Google Scholar]
- 54.Artsimovitch I, Patlan V, Sekine S, Vassylyeva MN, Hosaka T, Ochi K, Yokoyama S, Vassylyev DG. 2004. Structural basis for transcription regulation by alarmone ppGpp. Cell 117:299–310 [DOI] [PubMed] [Google Scholar]
- 55.Cao M, Bernat BA, Wang Z, Armstrong RN, Helmann JD. 2001. FosB, a cysteine-dependent fosfomycin resistance protein under the control of σW, an extracytoplasmic-function sigma factor in Bacillus subtilis. J. Bacteriol. 183:2380–2383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cao M, Helmann JD. 2002. Regulation of the Bacillus subtilis bcrC bacitracin resistance gene by two extracytoplasmic function sigma factors. J. Bacteriol. 184:6123–6129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Endo S, Zhang YW, Takahashi S, Koyama T. 2003. Identification of human dehydrodolichyl diphosphate synthase gene. Biochim. Biophys. Acta 1625:291–295 [DOI] [PubMed] [Google Scholar]
- 58.Zuchner S, Dallman J, Wen R, Beecham G, Naj A, Farooq A, Kohli MA, Whitehead PL, Hulme W, Konidari I, Edwards YJ, Cai G, Peter I, Seo D, Buxbaum JD, Haines JL, Blanton S, Young J, Alfonso E, Vance JM, Lam BL, Pericak-Vance MA. 2011. Whole-exome sequencing links a variant in DHDDS to retinitis pigmentosa. Am. J. Hum. Genet. 88:201–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zelinger L, Banin E, Obolensky A, Mizrahi-Meissonnier L, Beryozkin A, Bandah-Rozenfeld D, Frenkel S, Ben-Yosef T, Merin S, Schwartz SB, Cideciyan AV, Jacobson SG, Sharon D. 2011. A missense mutation in DHDDS, encoding dehydrodolichyl diphosphate synthase, is associated with autosomal-recessive retinitis pigmentosa in Ashkenazi Jews. Am. J. Hum. Genet. 88:207–215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lee YH, Kingston AW, Helmann JD. 2012. Glutamate dehydrogenase affects resistance to cell wall antibiotics in Bacillus subtilis. J. Bacteriol. 194:993–1001 [DOI] [PMC free article] [PubMed] [Google Scholar]