

Abstract
Chagas disease affects more than 10 million people worldwide, and yet, as it has historically been known as a disease of the poor, it remains highly neglected. Two currently available drugs exhibit severe toxicity and low effectiveness, especially in the chronic phase, while new drug discovery has been halted for years as a result of a lack of interest from pharmaceutical companies. Although attempts to repurpose the antifungal drugs posaconazole and ravuconazole (inhibitors of fungal sterol 14α-demethylase [CYP51]) are finally in progress, development of cheaper and more efficient, preferably Trypanosoma cruzi-specific, chemotherapies would be highly advantageous. We have recently reported that the experimental T. cruzi CYP51 inhibitor VNI cures with 100% survival and 100% parasitological clearance both acute and chronic murine infections with the Tulahuen strain of T. cruzi. In this work, we further explored the potential of VNI by assaying nitro-derivative-resistant T. cruzi strains, Y and Colombiana, in highly stringent protocols of acute infection. The data show high antiparasitic efficacy of VNI and its derivative (VNI/VNF) against both forms of T. cruzi that are relevant for mammalian host infection (bloodstream and amastigotes), with the in vivo potency, at 25 mg/kg twice a day (b.i.d.), similar to that of benznidazole (100 mg/kg/day). Transmission electron microscopy and reverse mutation tests were performed to explore cellular ultrastructural and mutagenic aspects of VNI, respectively. No mutagenic potential could be seen by the Ames test at up to 3.5 μM, and the main ultrastructural damage induced by VNI in T. cruzi was related to Golgi apparatus and endoplasmic reticulum organization, with membrane blebs presenting an autophagic phenotype. Thus, these preliminary studies confirm VNI as a very promising trypanocidal drug candidate for Chagas disease therapy.
INTRODUCTION
Chagas disease (CD), a neglected lifelong malady caused by the intracellular protozoan parasite Trypanosoma cruzi, is a notorious health problem in Latin America and an emerging global health issue, mainly due to immigration of infected individuals to other continents (primarily to North America and Europe) but also because of broadening of the insect vector distribution areas (1). No vaccine is available, and the current therapy for the infection (nifurtimox [Nf] and benznidazole [Bz]) is largely insufficient, having undesirable side effects and limited efficacy especially in the later chronic stages (2). However, novel compounds for CD do not attract the investment of most pharmaceutical companies, especially due to large costs versus low economic profits, exposing the importance of academic research and technological development support for drug design and development of new leads and hits for this as well as other neglected parasitic illnesses that afflict millions of the poorest people worldwide. In this vein, for over 3 decades inhibitors of sterol biosynthesis have been considered potential antichagasic agents (3–6), because, similar to the case for pathogenic fungi, sterol biosynthesis in T. cruzi is vital. The pathway leads to formation of ergosterol-like products which are essential for parasite survival (viable membranes) and differentiation (cell growth and division). Like fungi, T. cruzi cannot replace these endogenous ergosterol derivatives with host cholesterol or with its metabolites, thus justifying the idea of repurposing antifungal agents for treating CD (6).
Currently, the vast majority of clinical and agricultural antifungals are represented by azole derivatives (7). These drugs inhibit sterol 14α-demethylase (CYP51), a cytochrome P450 enzyme, which is required for the pathway to proceed beyond squalene cyclization. This prevents accumulation of toxic methylated sterol precursors and provides essential membrane components and regulatory molecules (8). The azole ring nitrogen coordinates to the P450 heme iron, making it more complicated for the enzyme to be activated by reduction, while the nonligated portion of the inhibitor molecule forms multiple contacts with the amino acid residues inside the CYP51 active site (9). Two antifungal azoles (posaconazole and ravuconazole) have recently entered clinical trials for CD (1), but the extremely high cost, adverse side effects (posaconazole [10]), and lack of a curative effect (ravuconazole [11]) are important concerns that must be faced. Moreover, fungal CYP51s share less than 30% amino acid sequence identity with the T. cruzi enzyme ortholog (12), which may be the reason why antifungal drug repurposing is often not efficient enough (13–15) and in turn suggests that novel, more potent, pathogen-specific agents discovered using direct screening for the protozoan CYP51 inhibition are highly desirable (16).
Recently, we have shown that one such inhibitor, VNI (17), cures, with 100% survival and no observable side effects, both acute and chronic models of T. cruzi infection (18). In these models, female BALB/c mice infected with the Tulahuen strain were treated with 25 mg/kg of VNI, administered 24 h after infection, twice a day (b.i.d.) by oral gavage for 30 daily consecutive doses. PCR analysis after immunosuppression showed 100% parasitological clearance in the blood and in all analyzed organs and tissues (18). Low acute toxicity in vivo was found to be another advantageous feature of VNI: after being given orally in dosages up to 400 mg/kg, there were no alterations either in mouse weight (body and organs) or in the biochemical blood analysis (19). These biological analyses together with the low cost of synthesis and favorable pharmacokinetics clearly suggest VNI as a highly promising drug candidate for CD.
However, T. cruzi is not a homogeneous population (20) but represents a pool of >70 different strains (www.dbbm.fiocruz.br/TcruziDB/strain.html 01/02/2013) which circulate in domestic, peridomiciliar, and sylvatic cycles involving humans, insect vectors, and animal reservoirs (>100 species) of the parasite. In the most recent classification (21), T. cruzi strains are distributed into 6 major phylogenetic groups (TcI to TcVI) on the basis of zymodemes and several genetic markers. Although no direct correlation between disease severity and parasite lineage has yet been established (22), it is well known that different T. cruzi strains vary broadly in drug sensitivity. Among them, CL-Brener (TcVI), whose complete genome sequencing was reported in 2005 (23), and Tulahuen (also TcVI), whose CYP51 gene we cloned and have been studying (24), are known to be drug sensitive, while there are other strains of the parasite that display medium [e.g., Y (TcII)] or high [e.g., Colombiana (TcI)] resistance to the current clinically available nitroderivative compounds (Bz and Nf) (20, 25, 26). Interestingly, this drug resistance appears to encompass antifungal azoles, including posaconazole (14, 27).
In the present study, we have investigated effects of VNI in highly stringent in vitro and in vivo protocols conducted with the T. cruzi Y and Colombiana strains. Although, as revealed by immunosuppression, no complete parasitological clearance was achieved under these conditions, our experimental data show that VNI and its derivative VNI/VNF are very active against various drug-resistant T. cruzi strains, justifying further preclinical studies that are under way.
MATERIALS AND METHODS
Compounds.
Synthesis of VNI and its derivative VNI/VNF was performed as reported previously (19). Benznidazole (Bz) (Laboratório Farmacêutico do Estado de Pernambuco [LAFEPE], Brazil) was used as a reference drug (28). For in vitro studies, stock solutions (5 mM) were prepared in dimethyl sulfoxide (DMSO), and the fresh final solvent concentration never exceeded 0.6%, which is not toxic for either parasites or mammalian cells. For in vivo studies, VNI was either (i) dissolved (as specified in the figure legends) in DMSO (never exceeding 10%, which does not cause any detectable mice toxicity [28]) and then diluted to the final dose using distilled sterile water or (ii) dissolved in 10% DMSO plus 5% arabic gum as reported previously (18).
Mammalian cell cultures.
For both drug toxicity and infection assays, primary cultures of cardiac cells (CC) were obtained as reported (29). The cultures were sustained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% horse serum, 5% fetal bovine serum (FBS), 2.5 mM CaCl2, 1 mM l-glutamine, and 2% chicken embryo extract. Cell cultures were maintained at 37°C in an atmosphere of 5% CO2 and air, and assays were run at least three times in duplicate.
Parasites.
The Y and Colombiana strains of T. cruzi were used throughout the experiments. Bloodstream trypomastigotes (BT) were harvested by heart puncture from T. cruzi-infected Swiss mice at the parasitemia peak day (29). Culture derived trypomastigotes (CT) were obtained from supernatant of infected murine fibrosarcoma L929 cell line cultures, as reported previously (30). Intracellular amastigotes were evaluated after the infection of primary cultures of cardiac cells (29) or L929 cells (31) using the BT and CT forms of the parasite, respectively.
In vitro cytotoxicity assays.
In order to rule out toxic effects of the compounds against mammalian host cells, uninfected cardiac cells (CC) were incubated for 24 and 48 h at 37°C in the presence or absence of each compound diluted in DMEM. The CC morphology and spontaneous contractibility were evaluated by light microscopy. The cell death rates were measured by the MTT [3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide] colorimetric assay (28). The absorbance was measured as previously reported (28) with a spectrophotometer (VersaMax tunable microplate reader; Molecular Devices), which allows for the determination of the compound concentration that reduces 50% of cellular viability (EC50).
Trypanocidal analysis.
BT and CT were incubated at 37°C for 24 h in the presence of increasing nontoxic concentrations of the tested compounds diluted in RPMI 1640 medium (Roswell Park Memorial Institute–Sigma-Aldrich) supplemented with 5% fetal bovine serum. Death rates were determined by light microscopy through direct quantification of the number of live parasites using a Neubauer chamber, and the drug concentration that reduces 50% of the number of the treated parasites (EC50) was calculated. For the analysis of the effect against intracellular amastigotes, after 24 h of parasite-host cell interaction (at a parasite/CC ratio of 10:1), the infected cultures were washed to remove unbound trypomastigotes and then incubated for another 48 h with increasing but nontoxic doses of the test compounds. CC were maintained at 37°C in an atmosphere of 5% CO2 and air and then the medium replaced every 24 h. Next, untreated and treated infected CC were fixed and stained with Giemsa solution, and the mean numbers of infected host cells and of parasites per infected cell were scored as reported previously (28). Only characteristic parasite nuclei and kinetoplasts were counted as surviving parasites, since irregular structures could indicate parasites undergoing death. The drug activity was estimated by calculating the infection index (II), which represents the mean value of the percentage of infected cells multiplied by the average number of intracellular amastigotes per infected host cell (32). Statistical analysis was performed by analysis of variance (ANOVA) as reported previously (28).
All in vitro assays were run at least twice in duplicate.
TEM.
Bloodstream trypomastigotes treated or not for 2 h at 37°C with the EC50 of the VNI (corresponding to 24 h of drug incubation) were fixed for 60 min at 4°C with 2.5% glutaraldehyde and 2.5 mM CaCl2 in 0.1 M cacodylate buffer (pH 7.2) and postfixed for 1 h at 4°C with 1% OsO4, 0.8% potassium ferricyanide, and 2.5 mM CaCl2 using this same buffer. The samples were then routinely processed for transmission electron microscopy (TEM) (33) and examined in a Jeol electron microscope from the Oswaldo Cruz Institute Electron Microscopy Platform Unit.
Mouse infection and treatment schemes.
Male Swiss mice were obtained from the Fundação Oswaldo Cruz (FIOCRUZ) animal facilities (CECAL/FIOCRUZ, Rio de Janeiro, Brazil). Mice were housed at a maximum of 8 per cage and kept in a conventional room at 20 to 24°C under a 12 h/12 h light/dark cycle. The animals were provided with sterilized water and chow ad libitum. Infection was performed by intraperitoneal (i.p.) injection of 104 and 5 × 103 bloodstream trypomastigotes for the Y and Colombiana strains, respectively. The animals (18 to 21 g) were divided into the following groups (at least six per group): uninfected (noninfected and nontreated), untreated (infected with T. cruzi but treated only with vehicle), and treated (infected and treated [receiving 0.2-ml oral daily doses] with 6 to 50 mg/kg b.i.d.). VNI was given twice a day due to its pharmacokinetics (the compound lifetime in mouse plasma is 16 h [18]). In parallel, infected mice were orally treated with 100 mg/kg/day benznidazole once a day. All treatments were started at the parasitemia onset (5 days postinfection [dpi] for Y and 11 to 12 dpi for Colombiana), followed by (i) 5, (ii) 20, or (iii) 26 consecutive daily doses.
Parasitemia, mortality rates, and ponderal curve analysis.
Parasitemia was individually checked by direct microscopic counting of parasites in 5 μl of blood, as described before (34). At different time points, body weight was evaluated. Mortality was checked daily until 130 days posttreatment and expressed as percent cumulative mortality (CM) (28).
Cure assessment.
Cure criteria were based on three parasitological methods: (i) negative parasitemia as observed by light microscopy, (ii) hemoculture assays, and (iii) PCR (31). Animals presenting negative results for all tests were considered cured. Parasitological cure assessment was performed at least 30 days after compound administration; surviving animals were subjected to immunosuppression with cyclophosphamide (Genuxal) administered in three cycles with doses of 50 mg/kg/day. Each cycle consisted of four consecutive days of cyclophosphamide administration, with 3-day intervals between cycles.
Parasitemia relapse was then followed for the next 3 weeks (35). Those mice that maintained negative parasitemia levels were then euthanized and about 1,000 μl of their blood collected for hemoculture and PCR analysis (29). The DNA extraction and PCR protocols were adapted and standardized for rodent samples as previously reported (28). Briefly, 500 μl blood was diluted 1:3 in guanidine solution (6 M guanidine HCl, 0.2 M EDTA) and heated for 90 s in boiling water in order to cleave the parasite kinetoplast DNA (kDNA) network. The PCR was performed using the primers: 121 (5′AAATAATGTACGGG[T/G]GAGATGCATGA3′) and 122 (5′GGTTCGATTGGGGTTGGTGTAATATA3′), which amplify a 330-bp sequence from the minicircle kinetoplast DNA (ca. 120,000 copies/parasite), as previously described (36). The PCR was carried out using a GeneAmp PCR System 9700 (Applied Biosystems) as follows: one step at 94°C for 3 min (to activate the Taq Platinum DNA polymerase), 2 cycles at 98°C for 1 min and 64°C for 2 min, 38 cycles at 94°C for 1 min and 64°C for 1 min, and a final extension at 72°C for 10 min. The amplification products were detected by 1.5% agarose gel electrophoresis following staining with ethidium bromide (5 mg/ml). For hemoculture, 200 μl of blood was added to 5 ml liver infusion tryptose (LIT) medium and incubated at 28°C for 30 days, with weekly examination by light microscopy to detect epimastigote forms (28). Only samples with negative parasitemia and hemoculture assay results were further screened by PCR analysis.
Ethics.
All procedures were carried out in accordance with the guidelines established by the FIOCRUZ Committee of Ethics for the Use of Animals (CEUA 0028/09).
CYP51 gene sequencing.
Colombiana genomic DNA was obtained from 109 epimastigote forms grown in liver infusion tryptose (LIT) medium supplemented with 10% fetal calf serum with weekly passages (37), using a commercial kit based on silica membrane technology (QIAamp DNA Mini Kit; Qiagen, Valencia, CA). The parasites were rinsed in saline buffer and then incubated in DNA lysis buffer. The protocol was carried out according to the manufacturer's instructions, and the DNA was eluted with 100 μl elution (AE) buffer. The DNA was stored at −20°C, and its purity and concentration were determined using a NanoDrop 2000c spectrophotometer (Thermo Scientific) at 260/280 and 260/320 nm. The length of genomic DNA was determined by electrophoresis through a 1% agarose gel.
Next, the CYP51 gene was PCR amplified using a FailSafe PCR Premix Selection Kit (Epicentre). The upstream primer 5′-CGCCATATGTTCATTGAAGCCATTGTATTGG-3′ contained a unique NdeI cloning site (underlined) and corresponded to the Tulahuen T. cruzi CYP51 cDNA from bp 1 to 25. The downstream primer 5′-CGCAAGCTTCAGTGATGGTGATGCGAGGGCAATTTCTTCTTGCG-3′ included a unique HindIII cloning site (underlined) followed by a stop codon (bold) and a C-terminal 4-histidine tag (italics) and was complementary to the T. cruzi CYP51 sequence from bp 1443 to 1423. The PCR mixture included 50 ng genomic DNA, 1 μM each forward and reverse primers, and 0.5 μl FailSafe PCR enzyme in a final volume of 25 μl. Twenty-five microliters of FailSafe PCR 2× Premix I was added, and amplification was carried out by denaturation at 95°C for 2 min and then 28 cycles of denaturation at 95°C for 60 s, annealing at 52°C for 30 s, and extension at 72°C for 140 s. Terminal extension for 2 min at 72°C completed the reaction. The PCR products were subcloned into pGEM-T Easy vector (Promega) and sequenced (T7 and SP6 primers) at Vanderbilt DNA Sequencing Core facility.
Salmonella enterica serovar Typhimurium reverse mutation test.
VNI was tested in reverse mutation tests (with and without metabolic activation with S9) following the protocol proposed by Maron and Ames (38), ICH Guidance for Industry S2 (R1) Genotoxicity Testing and Data Interpretation for Pharmaceuticals Intended for Human Use (June 2012) (U.S. Department of Health and Human Services, Food and Drug Administration [FDA], Center for Drug Evaluation and Research [CDER], and Center for Biologics Evaluation and Research [CBER]), and OECD test guideline 471 (39). Aliquots of 100 μl of S. Typhimurium strains TA97, TA98, TA100, and TA102 (2 ×108 cells/ml) were preincubated (20 min, 37°C) with 100 μl of VNI (from 0.06 nM up to 3,550 nM) and 500 μl of 0.2 M sodium phosphate buffer (pH 7.4). After 20 min, 2 ml top agar (0.7% agar, 0.6% NaCl, 50 μM l-histidine, 50 μl biotin, pH 7.4) at 45°C was added to the test tubes, and the final mixtures were poured into petri dishes with minimal agar (1.5% agar and Vogel-Bonner E medium containing 2% glucose). These final mixtures were incubated at 37°C for 72 h, and the His+ revertant colonies were counted. The mutagenic indexes (MI) were calculated as the mean value obtained from each concentration divided by the mean value of the negative control (DMSO, 5%). For determination of the cytotoxic effects of VNI, the preincubation assay mixture in the bacterial (reverse) mutation test was diluted in 0.9% (wt/vol) NaCl. This suspension contained 2 ×103 cells/ml. An aliquot of 100 μl of this suspension was plated on nutrient agar (0.8% Bacto nutrient broth [Difco], 0.5% NaCl, and 1.5% agar). The plates were then incubated at 37°C for 24 h, and survival fractions were calculated as percent survival compared to that of the negative group. All experiments were performed in triplicate and repeated twice. The tested compound was considered mutagenic when (i) the number of revertant colonies in the assay was at least twice the number of spontaneous revertants (MI ≥ 2), (ii) analysis of variance (ANOVA) revealed a significant response (P ≤ 0.05), and (iii) a reproducible concentration-response curve (≤0.01) was obtained.
Nucleotide sequence accession number.
The cDNA and protein sequences for the CYP51 A and B genes have been deposited into the NCBI GenBank database (http://www.ncbi.nih.gov/Genbank) under nucleotide accession numbers KC330216 and KC330217, respectively.
RESULTS
In vitro analysis of VNI and its derivative VNI/VNF (19) showed dose-dependent trypanocidal activities against bloodstream trypomastigotes (BT) from strain Y, reaching EC50s of 11 and 32 μM for VNI and VNI/VNF, respectively, after 24 h at 37°C (Table 1). VNI also proved to be very effective against culture-derived trypomastigotes (CT), exhibiting EC50s of ∼3.0 μM for both the Y and Colombiana strains (Table 1). The compounds were further screened on the intracellular forms of Y strain T. cruzi-infected cardiac cell cultures and demonstrated even higher activity against the multiplying intracellular forms of the parasite (EC50 = 0.9 ± 0.2 μM). Remarkably, against intracellular T. cruzi, VNI/VNF displayed a stronger effect than VNI, since it reduced 90% of cardiac cell parasitism when only 7 μM was used, while the EC90 for VNI was 38 μM (Table 1). Next, toxicity aspects of these inhibitors on mammalian cells were studied using cardiac cell cultures. Treatment at 37°C for 24 h resulted in loss of cellular viability only when high doses were employed, showing a 50% lethal concentration of 200 μM (Table 1). Incubation for 48 h with VNI and VNI/VNF resulted in EC50s of 50 and 150 μM, respectively, showing that besides being more effective against intracellular parasites, VNI/VNF is also less toxic to mammalian host cells, leading to a higher selectivity index (SI) of 170 (Table 1).
Table 1.
In vitro assays on T. cruzia
| Compound | EC50, μM (SI) |
Intracellular forms, Y |
|||
|---|---|---|---|---|---|
| BT, Y | CT |
||||
| Y | Colombiana | EC50, μM (SI) | EC90, μM | ||
| VNI | 11 ± 5 A (18) | 3.1 ± 0.8 (67) | 3.4 ± 0.5 (59) | 0.9 ± 0.2 C (56) | 38 ± 5 |
| VNI/VNF | 32 ± 15 B (6) | ND | ND | 0.9 ± 0.2 D (170) | 7 ± 5 |
| Bz | 13 ± 2 AB (>77) | ND | ND | 2.8 ± 1.9 CD (>360) | ND |
The trypanocidal effect (EC50 values and selectivity index [SI]) were evaluated for bloodstream (BT) (Y strain) and tissue culture-derived (CT) (Y and Colombiana strains) trypomastigotes incubated at 37°C for 24 h and on intracellular forms (Y strain, reduction of infection index) incubated for 48 h at 37°C. SIs were determined at 24 h of incubation for BT and CT (EC50 = 200 μM) and at 48 h of incubation for intracellular forms (EC50 = 50 and 150 μM for VNI and VNI/VNF, respectively) (see Materials and Methods). Values are means and standard deviations. The same letter is placed next to values for which statistical evaluation by ANOVA did not show statistical significance (P > 0.05). ND, not done.
Transmission electron microscopy (TEM) analysis was next performed, and as expected, untreated parasites presented typical morphology in regard to nuclei, endoplasmic reticulum, mitochondria, and kDNA (Fig. 1A to C). In contrast, after 2 h of incubation with VNI, bloodstream trypomastigotes of the Y (Fig. 1C to I) and Colombiana (Fig. 2A and B) strains displayed an altered morphology, and the most prominent features included well-developed endoplasmic reticulum profiles in very close proximity to and surrounding different parasite organelles such as nuclei (Fig. 1D [insets] and E, white arrows) and the Golgi apparatus (Fig. 1F and G, white arrows). Also, blebs of the plasma membrane could be seen (Fig. 1E, black arrow), in addition to an intense disorganization and detachment of the nuclear envelop (Fig. 2B) and dilated Golgi cisternae (Fig. 2A, black arrow), as well as swollen mitochondria (Fig. 2A [inset] and 1D [lower inset, asterisks]) with dilated and crista disorganization (Fig. 2A and B, insets). The presence of numerous cytosolic concentric membrane structures (Fig. 1D, H, and I, black arrows) was also frequently noticed in BT exposed to VNI.
Fig 1.
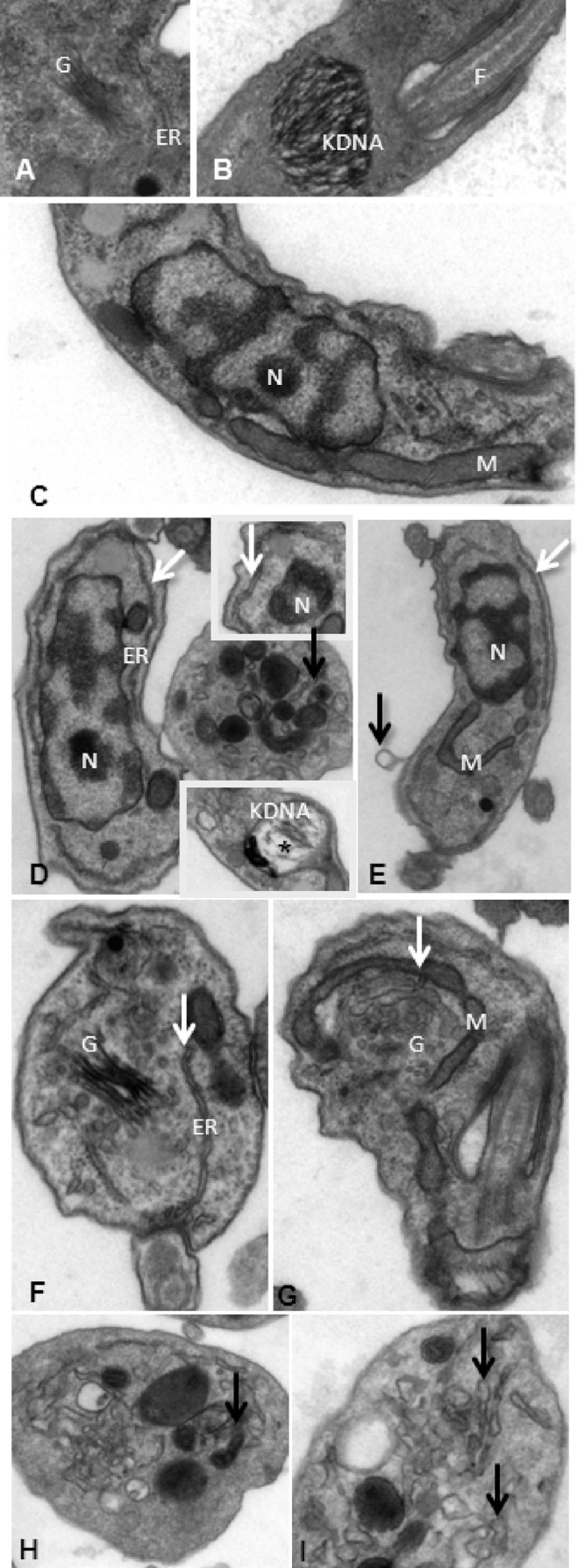
Ultrastructural aspects of bloodstream trypomastigotes of T. cruzi (Y strain) subjected (D to I) or not (A to C) to VNI exposure. N, nuclei; M, mitochondria, ER, endoplasmic reticulum, F, flagellum; G, Golgi apparatus.
Fig 2.

Ultrastructural aspects of bloodstream trypomastigotes of T. cruzi (Colombiana strain) subjected to VNI exposure. N, nuclei; M, mitochondria, ER, endoplasmic reticulum, F, flagellum; G, Golgi apparatus.
In order to explore the potential mutagenic aspects of VNI, the bacterial reverse mutation test (Ames test) was performed without and with metabolic activation (S9). These tests are commonly employed as an initial screen for genotoxic activity and, in particular, for point mutation-inducing activity. Since those point mutations are the cause of many human genetic diseases, the evidence for these mutations represents a useful tool to predict DNA damage in human cells. The strains used in the Ames test have several features that make them more sensitive for the detection of mutations, including responsive DNA sequences at the reversion sites, increased cell permeability to large molecules, and elimination of DNA repair systems or enhancement of error-prone DNA repair processes. Under the conditions used, VNI showed no mutagenic potential (MI < 2) (Table 2) at all tested doses.
Table 2.
Induction of His+ revertants in S. Typhimurium strains by VNI in a reverse mutation test without metabolic activation
| Strain | VNI (nM) | Without S9 |
With S9 |
||||
|---|---|---|---|---|---|---|---|
| MIa | No. of His+ revertants/plateb | Survival (%)c | MI | No. of His+ revertants/plate | Survival (%) | ||
| TA97 | 0 | 1.00 | 81.0 ± 4.4 | 100.0 | 1.00 | 192.0 ± 22.7 | 100.0 |
| 0.06 | 1.08 | 87.7 ± 3.8 | 100.0 | 1.09 | 208.7 ± 10.1 | 73.6 | |
| 0.12 | 1.10 | 89.0 ± 13.9 | 100.0 | 1.05 | 200.7 ± 9.9 | 100.0 | |
| 0.23 | 1.17 | 94.7 ± 0.6 | 100.0 | 1.02 | 196.0 ± 19.7 | 100.0 | |
| 0.45 | 1.12 | 90.3 ± 16.8 | 100.0 | 1.11 | 212.7 ± 32.9 | 100.0 | |
| 0.89 | 1.23 | 100.0 ± 5.2 | 100.0 | 1.10 | 210.7 ± 43.1 | 100.0 | |
| TA98 | 0 | 1.00 | 24.0 ± 7.1 | 100.0 | 1.00 | 45.0 ± 15.6 | 100.0 |
| 0.06 | 1.85 | 44.3 ± 3.1 | 83.2 | 0.99 | 44.7 ± 17.2 | 100.0 | |
| 0.12 | 1.14 | 27.3 ± 7.2 | 100.0 | 0.84 | 38.0 ± 4.0 | 97.1 | |
| 0.23 | 1.19 | 28.7 ± 9.1 | 70.9 | 0.87 | 39.3 ± 6.1 | 100.0 | |
| 0.45 | 1.22 | 29.3 ± 4.0 | 74.4 | 1.04 | 46.7 ± 6.1 | 99.6 | |
| 0.89 | 1.63 | 39.0 ± 4.4 | 80.3 | 1.02 | 46.0 ± 12.5 | 99.3 | |
| TA100 | 0 | 1.00 | 178.0 ± 28.6 | 100.0 | 1.00 | 84.0 ± 5.3 | 100.0 |
| 0.06 | 0.88 | 156.3 ± 9.1 | 100.0 | 1.09 | 91.3 ± 11.0 | 56.4 | |
| 0.12 | 0.81 | 144.3 ± 9.8 | 93.8 | 1.03 | 86.7 ± 6.4 | 70.5 | |
| 0.23 | 0.86 | 153.3 ± 14.2 | 100.0 | 1.18 | 99.3 ± 4.6 | 79.9 | |
| 0.45 | 1.16 | 205.7 ± 11.0 | 100.0 | 1.28 | 107.3 ± 6.4 | 75.9 | |
| 0.89 | 0.99 | 177.0 ± 23.3 | 100.0 | 0.96 | 80.7 ± 18.5 | 62.8 | |
| TA102 | 0 | 1.00 | 346.7 ± 40.1 | 100.0 | 1.00 | 405.0 ± 33.0 | 100.0 |
| 0.06 | 0.77 | 266.3 ± 18.0 | 100.0 | 1.00 | 403.3 ± 14.8 | 74.2 | |
| 0.12 | 1.20 | 414.7 ± 37.0 | 100.0 | 0.75 | 303.3 ± 33.3 | 90.3 | |
| 0.23 | 0.90 | 311.0 ± 28.6 | 100.0 | 0.73 | 294.0 ± 53.8 | 86.2 | |
| 0.45 | 1.11 | 386.0 ± 30.0 | 100.0 | 0.79 | 318.0 ± 11.4 | 95.7 | |
| 0.89 | 1.06 | 368.0 ± 5.7 | 100.0 | 0.85 | 344.7 ± 16.9 | 76.7 | |
Mutagenic index (MI), number of His+ revertants induced in the sample/number of spontaneous His+ revertants in the negative control (5% DMSO).
Mean value ± standard deviation from at least three experiments.
Calculated in relation to the negative control. Bold numbers are statistically significant (P ≤ 0.005).
Next, due to the low mutagenic potential of VNI and its previously reported low acute toxicity (NOAEL [no observed adverse effect level] values of 400 mg/kg as shown in reference 19) and excellent activity against BT, CT, and intracellular forms and different strains of T. cruzi, exhibiting a high SI and an effect superior to that of reference drug (Bz), this compound was subjected to in vivo analysis using highly stringent protocols. These in vivo assays were performed using Swiss male mice (which are less resistant to T. cruzi infection than female mice [40]) inoculated with BT forms from strains Y (104) and Colombiana (5 × 103) (29), employing doses equal to or lower than the dosage used in the previous study (25 mg/kg b.i.d.) (18) and shorter treatment periods (from 5 to 26 consecutive daily doses). In contrast to the previous VNI study (18) and other, less stringent protocols for evaluating drug efficacy in murine acute models of Chagas disease (14, 27) that start the compound administration as early as 24 h after T. cruzi inoculation, the present VNI treatment was started only at the onset of parasitemia (5 and 11 to 12 days after infection for the Y and Colombiana strains, respectively) to ensure that the parasite had the chance to actively start proliferation within the tissues (28, 29). Only those mice that presented positive parasitemia were used in the following studies.
In the first scheme of treatment employing Colombiana acute infection (scheme 1), VNI was administered for 5 daily consecutive doses using 6 to 25 mg/kg twice a day by the oral route (p.o.), totaling 12, 25, and 50 mg/kg/day (Fig. 3). Except for the lowest dose (6 mg/kg), VNI considerably reduced the parasitemia and protected (100%) against mortality induced by the parasite infection (Fig. 3A and B). As expected, 25 mg/kg VNI also reduced the parasitemia in the experimental mouse model of T. cruzi acute infection using strain Y: while infected but untreated mice presented high parasitemia levels, peaking at 8 dpi (Fig. 4A), VNI-treated mice displayed low circulating parasitism (>80% decreased levels), resulting in 100% mouse survival (Fig. 4A and B). Next, a second scheme of treatment was used, aiming to achieve a complete suppression of T. cruzi infection. Colombiana-infected animals were treated b.i.d. for 20 consecutive days using 25 and 50 mg/kg VNI (Fig. 5). Our data showed that both doses of VNI suppressed the parasitemia levels as seen with administration of 100 mg/kg Bz (Fig. 5). Both doses also induced 100% animal survival, as Bz also did, while animals untreated or treated only with the vehicle (DMSO plus arabic gum) displayed similar parasitemia levels, which caused >80% mortality (data not shown). Since no significant difference was found using 25- and 50-mg/kg doses, another set of analyses (scheme 3) was performed using 25 mg/kg VNI administered b.i.d. for 26 days, starting at parasitemia onset (Fig. 6). Under these conditions, VNI not only completely suppressed parasitemia but also protected against mouse ponderal weight loss and against mortality triggered by T. cruzi infection, leading to 100% survival and showing efficacy similar to that of 100 mg/kg Bz (Fig. 6A to C). However, the analysis of parasitological cure after immunosuppression demonstrated that no cure was achieved in either Bz- or VNI-treated mice, since all groups relapsed and most (5 out of 6) died during cyclophosphamide administration (Table 3).
Fig 3.

Parasitemia (A) and percent cumulative mortality (B) in Swiss Webster male mice infected with 5 × 103 trypomastigote forms of T. cruzi (Colombiana strain) and treated or not with VNI (p.o., b.i.d.) from dpi 12 to 16 at 6-, 12-, or 25-mg/kg doses or with 100 mg/kg Bz. VNI was diluted in 10% DMSO plus 5% arabic gum.
Fig 4.

Parasitemia (A) and percent cumulative mortality (B) in Swiss Webster male mice infected with 104 trypomastigote forms of T. cruzi (Y strain) and treated or not with VNI (via gavage, b.i.d.) from dpi 5 to 9 at 25 mg/kg or with Bz at 100 mg/kg as a reference drug. VNI was diluted in DMSO (10%).
Fig 5.

Parasitemia levels in Swiss Webster male mice infected with 5 × 103 trypomastigote forms of T. cruzi (Colombiana strain) and treated (p.o.) or not from dpi 11 to 30 with 10% DMSO, VNI (b.i.d. at 25-mg/kg and 50-mg/kg doses), or Bz at 100 mg/kg as a reference drug. Mortality rates until 81 dpi were 83% in untreated mice and mice treated with 10% DMSO plus 5% arabic gum (5 out of 6) and 0% in VNI- and Bz-treated mice (0 out of 6). VNI was diluted in 10% DMSO plus 5% arabic gum.
Fig 6.
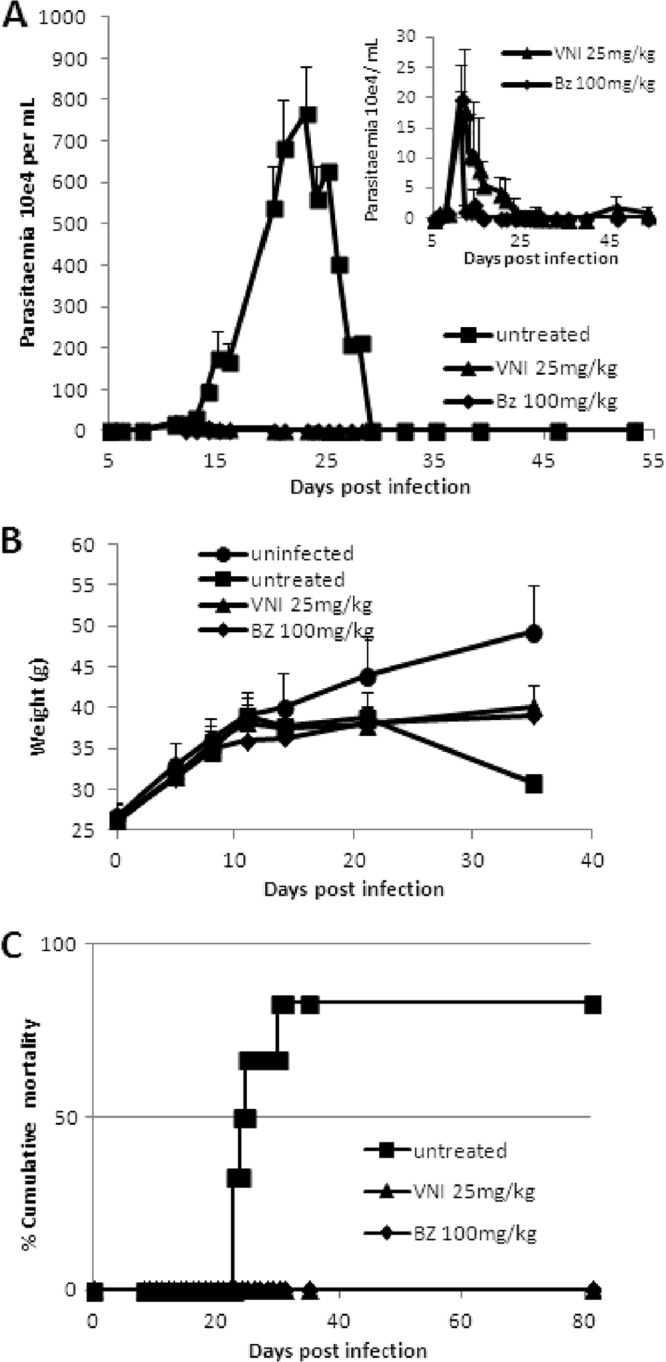
Parasitemia (A), ponderal curve (B), and percent cumulative mortality (C) in Swiss Webster male mice infected by 5 × 103 trypomastigote forms of T. cruzi (Colombiana strain) and treated or not with VNI (p.o., b.i.d.) from dpi 11 to 36 at 25 mg/kg and with Bz at 100 mg/kg as a reference drug. VNI was diluted in 10% DMSO plus 5% arabic gum.
Table 3.
Parasitological cure analysis of VNI and Bz-treated micea
| Group | No. of doses | No. of surviving mice/no. of mice that relapsed after Cy administrationb | No. of tested samples/no. of negative hemocultures after Cy administrationc |
|---|---|---|---|
| Untreated | 0 | 1/1 | 0/0 |
| Bz, 100 mg/kg, p.o. | 26 | 6/5 | 1/0 |
| VNI, 25 mg/kg b.i.d., p.o. | 26 | 6/5 | 1/0 |
Swiss Webster male mice were infected by 5 × 103 trypomastigote forms of T. cruzi (Colombiana strain) and treated or not with VNI (via gavage, b.i.d.) from dpi 11 to 36 at 25 mg/kg and with Bz at 100 mg/kg as a reference drug. VNI was diluted in 10% DMSO plus 5% arabic gum.
After 45 days of treatment (at 81dpi), surviving mice (6/6 in both the VNI and Bz groups and 1/6 untreated animals) were subjected to 3 cycles of cyclophosphamide (Cy) administration, and then the parasitemia was followed up to 121 dpi.
Five animals each from the Bz and VNI groups died due to parasitemia relapse (up to 130 dpi).
DISCUSSION
VNI (Fig. 7) was originally obtained from Novartis, from a 400-compound library of potential inhibitors for vitamin D-metabolizing cytochromes P450 (41). Two compounds with very similar structure (both carboxamide-containing β-phenyl-imidazoles), SDZ-284692 (current name, VNI [17]) and SDZ-285604 (current name, VNF [42]), have been identified as the most potent T. cruzi CYP51 inhibitors upon testing of this library in the reconstituted enzyme reaction against protozoan, fungal, and human CYP51 orthologs. The inhibitory effects of the compounds on the protozoan CYP51 activity correlated well with their antiparasitic activity in T. cruzi cells (43), but no in vivo experiments were done at that time due to insufficient amounts of the compounds provided by Novartis. Subsequently, costructures of the protozoan CYP51s in complexes with VNI (17) and VNF (42) have been determined, providing an explanation both for the high potency of this novel inhibitory scaffold and for the CYP51 enzyme susceptibility to inhibition (12, 16). Surprisingly, since they are structurally highly similar and both interact with the CYP51 heme iron via the imidazole ring nitrogen (N3), inside the active site of the enzyme VNI and VNF were found to acquire the opposite orientations (Fig. 7). While the long three-ring arm of VNI protrudes toward the substrate access channel entrance, the two-ring arm of VNF occupies the deepest portion of the substrate binding cavity. This finding served as the basis for the synthesis of VNI/VNF, the derivative which was designed to fill the majority of the CYP51 substrate binding cavity. This structural modification was proven to strengthen the inhibitory effect of this derivative on the CYP51 activity (19), and the analytical use of VNI/VNF in the current study (Table 1) supports the possibility of the compound having higher antiparasitic efficiency as well.
Fig 7.

Chemical structures of VNI and VNI/VNF. The VNI/VNF molecule was obtained by adding a second aromatic ring to VNI and replacing two Cl atoms with F (in order to maintain lower lipophilicity [log P = 5.4, versus 6.2 for the chlorinated analog]). The design was based on the costructures of CYP51 with VNI (dark) and VNF (light), which both coordinate to the heme iron but have the opposite orientation inside the CYP51 active site, with the goal of filling the majority of the CYP51 binding cavity (sphere).
Overall, cellular experiments revealed excellent antiparasitic effects of these CYP51 inhibitors against both forms of T. cruzi that are relevant for human infection (trypomastigotes and amastigotes) and high selectivity indexes. Transmission electron microscopy (TEM) confirmed that VNI targets membrane elements. Interestingly, in addition to the previously described altered plasma membrane with blebs (44), we also observed altered Golgi apparatus and endoplasmic reticulum elements and nuclear envelope detachment. Altogether, these alterations may be indicative of a morphological phenotype of autophagy as demonstrated by reticulum and mitochondrion profiles surrounding typical parasite organelles as well as the occurrence of cytosolic concentric membrane structures similar to those reported for other natural and synthetic anti-T. cruzi compounds (45, 46). The TEM data suggest that the endoplasmic reticulum may be the source of membranes for the formation of preautophagosomal structures as reported previously (45). The autophagic process and respective specific inhibitors will be further studied in more detail in order to explore and better understand the cell death mechanisms and intracellular components involved in the VNI trypanocidal effect.
Another aspect investigated was the potential of VNI to induce mutagenicity as assayed by Ames tests. The bacterial reverse mutation test detects point mutations restoring the functional capability of the bacteria to synthesize the essential amino acid histidine. The revertant bacteria are detected by their ability to grow in the absence of the amino acid, which is required by the parent test strain (39). Our present data did not show a mutagenicity profile of VNI until the concentration of 3,550 nM. However, although in all tested bacterial assays no mutagenic aspects could be seen, the potential of mutagenicity of VNI cannot be completely discarded, since its high in vitro toxicity against these strains can mask mutagenicity, which was not presently observed in any strain or at any concentration tested. Once registration of pharmaceuticals requires a comprehensive assessment of their genotoxic potential, extensive follow-up testing to assess the in vivo mutagenic and carcinogenic potentials would be warranted to assess the potential risk for treatment of patients, unless justified by appropriate risk-benefit analysis. Further studies are under way to explore this aspect in more detail.
Our recent study of VNI activity in mice infected with the Tulahuen strain confirmed the efficiency of the compound in vivo, using both acute and chronic models of T. cruzi infection (18). In the present work we have further demonstrated that VNI is also highly active against a variety of T. cruzi isolates, including not only the drug-sensitive Tulahuen strain but also the medium and highly drug-resistant Y and Colombiana strains. Even when used in the highly stringent acute protocols, VNI was able to suppress the parasitemia and protect against mortality (100% animal survival) in both the Y- and Colombiana-infected mice. The fact that we did not reach the sterile parasitological cure in these experiments does not diminish the VNI scaffold value but supports the existence of differences in susceptibilities to CYP51 inhibitors in various T. cruzi strains. In fact, amplification of the CYP51 gene from the Colombiana genomic DNA revealed the presence of two genes, with 8 and 7 amino acid differences from the CYP51 of the Tulahuen strain (Table 4). Although none of the side chains of these residues appears to be oriented inside the CYP51 substrate binding cavity (Fig. 8), this finding suggests that Colombiana can potentially have a CYP51-related lower susceptibility to azoles due to higher CYP51 protein abundance (two genes) and/or indirect involvement of some of the altered residues in CYP51 function, such as modulation of catalytic activity via interaction with the electron donor partner, efficiency of reduction, or flexibility. This will be tested by expression of Colombiana CYP51s and structure/function characterization in vitro. A deeper investigation regarding possible enzyme specificities according to parasite stage (trypomastigotes and epimastigotes) may also be considered. Concerning the lack of parasitological cure noticed with the Colombiana strain, our next step will be to use longer treatment schemes of VNI administration (60 days b.i.d.), as currently have been used for most azoles (6) and suggested as a target product profile of novel trypanocidal agents for CD (http://www.dndi.org/diseases-projects/diseases/chagas/target-product-profile.html). Alternative drug formulations (particularly cyclodextrin solutions, which are generally used to increase bioavailability of lipophilic compounds, including posaconazole [14, 27]) as well as VNI-Bz combinatorial therapy and comparative testing of the efficiencies of VNI and VNI/VNF in vivo are also forthcoming, and we believe that CYP51 structure-based VNI scaffold optimization will produce a drug candidate(s) sufficiently active against the entire diverse T. cruzi population.
Table 4.
Sequence variations in CYP51s from different T. cruzi strainsa
| T. cruzi strain | CYP51 (protein accession no., database) | Amino acids |
DNA |
||
|---|---|---|---|---|---|
| % Identity | No. of differences (amino acid substitutions) | % Identity | No. of differences (no. silent) | ||
| CL-Brener | A (XP_820210.1, GB) | 100 | 0 (none) | >99 | 2 (2) |
| B (XP_821219.1, GB) | 99 | 4 (G9A, D62E, A117S, E160K) | 97 | 35 (31) | |
| Colombiana | A (KC330216, GB; CCP42464.1, EMBL) | 98 | 8 (A117S, E160K, H196R, K203Q,b V245I, K314E,b D405E, P480Sb) | 98 | 19 (11) |
| B (KC330217, GB; CCP42465.1, EMBL) | 98 | 7 (G9A, L13P,b P32S,b D62E, A117S, E160K A288Sb) | 97 | 39 (32) | |
| Sylvio | Single, A-like (EKG07251.1, GB) | 98 | 6 (G9C, A117S, E160K, H196R, V245I, D405E) | ||
| Marinkellei | Single, B-like (EKF39305.1, GB) | 95 | 24 | ||
CYP51 from the Tulahuen strain (GenBank [GB] protein accession number AAW47718.1) is used as a reference.
Amino acid difference not found in other T. cruzi strains.
Fig 8.

Mapping of amino acid substitutions in the CYP51 genes (A and B) from the Colombiana strain of T. cruzi. G9 and L13 are not present in the structure [3k1o] because the N-terminal transmembrane peptide (31 amino acid residues in T. cruzi CYP51) was truncated for crystallization purposes; P480 is not seen due to flexibility of the protein C terminus.
ACKNOWLEDGMENTS
This study was supported by Fiocruz, Fundação Carlos Chagas Filho de Amparo a Pesquisa do Estado do Rio de Janeiro, Conselho Nacional Desenvolvimento Científico e Tecnológico (CNPq), PDTIS/Fiocruz, and PROEP/CNPq. The work was also supported by National Institutes of Health (NIH) grant GM067871 (G.I.L.).
We thank the Program for Technological Development in Tools for Health-PDTIS-FIOCRUZ for use of its facilities. VNI and VNI/VNF were synthesized by the Vanderbilt Institute of Chemical Biology Synthesis Core.
Footnotes
Published ahead of print 17 June 2013
REFERENCES
- 1.Leslie M. 2011. Drug developers finally take aim at a neglected disease. Science 333:933–935 [DOI] [PubMed] [Google Scholar]
- 2.Soeiro MN, Dantas AP, Daliry A, Silva CF, Batista DG, de Souza EM, Oliveira GM, Salomão K, Batista MM, Pacheco MG, Silva PB, Santa-Rita RM, Barreto RF, Boykin DW, Castro SL. 2009. Experimental chemotherapy for Chagas disease: 15 years of research contributions from in vivo and in vitro studies. Mem. Inst. Oswaldo Cruz 104(Suppl 1):301–310 [DOI] [PubMed] [Google Scholar]
- 3.Docampo R, Moreno SN, Turrens JF, Katzin AM, Gonzalez-Cappa SM, Stoppani AO. 1981. Biochemical and ultrastructural alterations produced by miconazole and econazole in Trypanosoma cruzi. Mol. Biochem. Parasitol. 3:169–180 [DOI] [PubMed] [Google Scholar]
- 4.Beach DH, Goad LJ, Holz GG. 1986. Effects of ketoconazole on sterol biosynthesis by Trypanosoma cruzi epimastigotes. Biochem. Biophys. Res. Commun. 136:851–856 [DOI] [PubMed] [Google Scholar]
- 5.Urbina JA, Lazardi K, Aguirre T, Piras MM, Piras R. 1988. Antiproliferative synergism of the allylamine SF 86-327 and ketoconazole on epimastigotes and amastigotes of Trypanosoma (Schizotrypanum) cruzi. Antimicrob. Agents Chemother. 32:1237–1242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Urbina JA. 2009. Ergosterol biosynthesis and drug development for Chagas disease. Mem. Inst. Oswaldo Cruz 104(Suppl 1):311–318 [DOI] [PubMed] [Google Scholar]
- 7.Maertens JA. 2004. History of the development of azole derivatives. Clin. Microbiol. Infect. 10:1–10 [DOI] [PubMed] [Google Scholar]
- 8.Roberts CW, McLeod R, Rice DW, Ginger M, Chance ML, Goad LJ. 2003. Fatty acid and sterol metabolism: potential antimicrobial targets in apicomplexan and trypanosomatid parasitic protozoa. Mol. Biochem. Parasitol. 126:129–142 [DOI] [PubMed] [Google Scholar]
- 9.Lepesheva GI, Waterman MR. 2011. Sterol 14alpha-demethylase (CYP51) as a therapeutic target for human trypanosomiasis and leishmaniasis. Curr. Top. Med. Chem. 11:2060–2071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pinazo MJ, Espinosa G, Gállego M, López-Chejade PL, Urbina JA, Gásgon J. 2010. Successful treatment with posaconazole of a patient with chronic Chagas disease and systemic lupus erythematosus. Am. J. Trop. Med. Hyg. 82:583–587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Diniz LF, Caldas IS, Guedes PM, Crepalde G, de Lana M, Carneiro CM, Talvani A, Urbina JA, Bahia MT. 2010. Effects of ravuconazole treatment on parasite load and immune response in dogs experimentally infected with Trypanosoma cruzi. Antimicrob. Agents Chemother. 54:2979–2986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lepesheva GI, Waterman MR. 2011. Structural basis for conservation in the CYP51 family. Biochim. Biophys. Acta 1814:88–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Apt W, Aguilera X, Arribada A, Pérez C, Miranda C, Sánchez G, Zulantay I, Cortés P, Rodriguez J, Juri D. 1998. Treatment of chronic Chagas' disease with itraconazole and allopurinol. Am. J. Trop. Med. Hyg. 59:133–138 [DOI] [PubMed] [Google Scholar]
- 14.Molina J, Martins-Filho O, Brener Z, Romanha AJ, Loebenberg D, Urbina JA. 2000. Activities of the triazole derivative SCH 56592 (posaconazole) against drug-resistant strains of the protozoan parasite Trypanosoma (Schizotrypanum) cruzi in immunocompetent and immunosuppressed murine hosts. Antimicrob. Agents Chemother. 44:150–155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Urbina JA, Payares G, Sanoja C, Lira R, Romanha AJ. 2003. In vitro and in vivo activities of ravuconazole on Trypanosoma cruzi, the causative agent of Chagas disease. Int. J. Antimicrob. Agents. 21:27–38 [DOI] [PubMed] [Google Scholar]
- 16.Lepesheva GI, Villalta F, Waterman MR. 2011. Targeting Trypanosoma cruzi sterol 14α-demethylase (CYP51). Adv. Parasitol. 75:65–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lepesheva GI, Park HW, Hargrove TY, Vanhollebeke B, Wawrzak Z, Harp JM, Sundaramoorthy M, Nes WD, Pays E, Chaudhuri M, Villalta F, Waterman MR. 2010. Crystal structures of Trypanosoma brucei sterol 14 alpha-demethylase and implications for selective treatment of human infections. J. Biol. Chem. 285:1773–1780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Villalta F, Dobish MC, Nde P, Kleshchenko Y, Hargrove TY, Johnson CA, Waterman MR, Johnston JN, Lepesheva GI. 4 March 2013. VNI cures the acute and chronic experimental Chagas disease. J. Infect. Dis. [Epub ahead of print.] 10.1093/infdis/jit042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hargrove TY, Kim K, Soeiro MNC, da Silva CF, Batista DGJ, Batista MM, Yazlovitskaya EM, Waterman MR, Sulikowski GA, Lepesheva GI. 2012. CYP51 structures and structure-based development of novel, pathogen-specific inhibitory scaffolds. Int. J. Parasitol. Drugs Drug Resist. 2:178–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Filardi LS, Brener Z. 1987. Susceptibility and natural resistance of Trypanosoma cruzi strains to drugs used clinically in Chagas disease. Trans. R. Soc. Trop. Med. Hyg. 81:755–759 [DOI] [PubMed] [Google Scholar]
- 21.Zingales B, Andrade S, Briones M, Campbell D, Chiari E, Fernandes O, Guhl F, Lages-Silva E, Macedo A, Machado C, Miles MA, Romanha AJ, Sturm NR, Tibayrenc M, Schijman AG. 2009. A new consensus for Trypanosoma cruzi intraspecific nomenclature: second revision meeting recommends TcI to TcVI. Mem. Inst. Oswaldo Cruz 104:1051–1054 [DOI] [PubMed] [Google Scholar]
- 22.Vázquez-Chagoyán JC, Gupta S, Garg NJ. 2011. Vaccine development against Trypanosoma cruzi and Chagas disease. Adv. Parasitol. 75:121–146 [DOI] [PubMed] [Google Scholar]
- 23.El-Sayed NM, Myler PJ, Bartholomeu DC, Nilsson D, Aggarwal G, Tran AN, Ghedin E, Worthey EA, Delcher AL, Blandin G, Westenberger SJ, Caler E, Cerqueira GC, Branche C, Haas B, Anupama A, Arner E, Aslund L, Attipoe P, Bontempi E, Bringaud F, Burton P, Cadag E, Campbell DA, Carrington M, Crabtree J, Darban H, da Silveira JF, de Jong P, Edwards K, Englund PT, Fazelina G, Feldblyum T, Ferella M, Frasch AC, Gull K, Horn D, Hou L, Huang Y, Kindlund E, Klingbeil M, Kluge S, Koo H, Lacerda D, Levin MJ, Lorenzi H, Louie T, Machado CR, McCulloch R, McKenna A, Mizuno Y, Mottram JC, Nelson S, Ochaya S, Osoegawa K, Pai G, Parsons M, Pentony M, Pettersson U, Pop M, Ramirez JL, Rinta J, Robertson L, Salzberg SL, Sanchez DO, Seyler A, Sharma R, Shetty J, Simpson AJ, Sisk E, Tammi MT, Tarleton R, Teixeira S, Van Aken S, Vogt C, Ward PN, Wickstead B, Wortman J, White O, Fraser CM, Stuart KD, Andersson B. 2005. The genome sequence of Trypanosoma cruzi, etiologic agent of Chagas disease. Science 309:409–415 [DOI] [PubMed] [Google Scholar]
- 24.Lepesheva GI, Zaitseva NG, Nes WD, Zhou W, Arase M, Liu J, Hill GC, Waterman MR. 2006. CYP51 from Trypanosoma cruzi: a phyla-specific residue in the B′ helix defines substrate preferences of sterol 14 alpha-demethylase. J. Biol. Chem. 281:3577–3585 [DOI] [PubMed] [Google Scholar]
- 25.Murta SMF, Gazzinelli RT, Brener Z, Romanha AJ. 1998. Molecular characterization of susceptible and naturally resistant strains of Trypanosoma cruzi to benznidazole and nifurtimox. Mol. Biochem. Parasitol. 93:203–214 [DOI] [PubMed] [Google Scholar]
- 26.Andrade SG, Magalhães JB, Pontes AL. 1985. Evaluation of chemotherapy with benznidazole and nifurtimox in mice infected with Trypanosoma cruzi strains of different types. Bull. World Health Organ. 63:721–726 [PMC free article] [PubMed] [Google Scholar]
- 27.Buckner F, Bahia MT, Suryadevara PK, White KL, Shackleford DM, Chennamaneni NK, Hulverson MA, Laydbak JU, Chatelain E, Scandale I, Verlinde CL, Charman SA, Lepesheva GI, Gelb MH. 2012a. Pharmacological characterization, structural studies, and in vivo activity of anti-Chagas disease lead compounds derived from tipifarnib. Antimicrob. Agents Chemother. 56:4914–4921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.da Silva CF, Batista DG, Oliveira GM, de Souza EM, Hammer ER, da Silva PB, Daliry A, Araujo JS, Britto C, Rodrigues AC, Liu Z, Farahat AA, Kumar A, Boykin DW, Soeiro MN. 2012. In vitro and in vivo investigation of the efficacy of arylimidamide DB1831 and its mesylated salt form—DB1965—against Trypanosoma cruzi infection. PLoS One 7:e30356. 10.1371/journal.pone.0030356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Batista DG, Batista MM, de Oliveira GM, do Amaral PB, Lannes-Vieira J, Britto CC, Junqueira A, Lima MM, Romanha AJ, Sales Junior PA, Stephens CE, Boykin DW, Soeiro MN. 2010. Arylimidamide DB766, a potential chemotherapeutic candidate for Chagas' disease treatment. Antimicrob. Agents Chemother. 54:2940–2952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.De Souza EM, Menna-Barreto R, Araújo-Jorge TC, Kumar A, Hu Q, Boykin DW, Soeiro MN. 2006. Antiparasitic activity of aromatic diamidines is related to apoptosis-like death in Trypanosoma cruzi. Parasitology 133:75–79 [DOI] [PubMed] [Google Scholar]
- 31.Romanha AJ, Castro SL, Soeiro Mde N, Lannes-Vieira J, Ribeiro I, Talvani A, Bourdin B, Blum B, Olivieri B, Zani C, Spadafora C, Chiari E, Chatelain E, Chaves G, Calzada JE, Bustamante JM, Freitas-Junior LH, Romero LI, Bahia MT, Lotrowska M, Soares M, Andrade SG, Armstrong T, Degrave W, Andrade Z A. 2010. In vitro and in vivo experimental models for drug screening and development for Chagas disease. Mem. Inst. Oswaldo Cruz 105:233–238 [DOI] [PubMed] [Google Scholar]
- 32.Silva CF, Batista MM, Mota RA, de Souza EM, Stephens CE, Som P, Boykin DW, Soeiro MN. 2007. 2007. Activity of “reversed” diamidines against Trypanosoma cruzi “in vitro”. Biochem. Pharmacol. 73:1939–1946 [DOI] [PubMed] [Google Scholar]
- 33.De Souza EM, Lansiaux A, Bailly C, Wilson WD, Hu Q, Boykin DW, Batista MM, Araújo-Jorge TC, Soeiro MN. 2004. Phenyl substitution of furamidine markedly potentiates its anti-parasitic activity against Trypanosoma cruzi and Leishmania amazonensis. Biochem. Pharmacol. 68:593–600 [DOI] [PubMed] [Google Scholar]
- 34.da Silva CF, da Silva PB, Batista MM, Daliry A, Tidwell RR, Soeiro MN. 2010. The biological in vitro effect and selectivity of aromatic dicationic compounds on Trypanosoma cruzi. Mem. Inst. Oswaldo Cruz 105:239–245 [DOI] [PubMed] [Google Scholar]
- 35.Caldas IS, Talvani A, Caldas S, Carneiro CM, de Lana M, da Matta Guedes PM, Bahia MT. 2008. Benznidazole therapy during acute phase of Chagas disease reduces parasite load but does not prevent chronic cardiac lesions. Parasitol. Res. 103:413–421 [DOI] [PubMed] [Google Scholar]
- 36.Wincker P, Britto C, Pereira JB, Cardoso MA, Oelemann W, Morel CM. 1994. Use of a simplified polymerase chain reaction procedure to detect Trypanosoma cruzi in blood samples from chronic chagasic patients in a rural endemic area. Am. J. Trop. Med. Hyg. 51:771–777 [DOI] [PubMed] [Google Scholar]
- 37.Camargo EP. 1964. Growth and differentiation in Trypanosoma cruzi. I. Origin of metacyclic trypanosomes in liquid media. Rev. Inst. Med. Trop. São Paulo 6:93–100 [PubMed] [Google Scholar]
- 38.Maron DM, Ames BN. 1983. Revised methods for the Salmonella mutagenicity test. Mutat. Res. 113:173–215 [DOI] [PubMed] [Google Scholar]
- 39.OECD 1997. Test guideline 471. Bacterial reverse mutation test. In OECD guideline for testing of chemicals. Organization for Economic Cooperation and Development, Paris, France [Google Scholar]
- 40.de Souza EM, Rivera MT, Araújo-Jorge TC, de Castro SL. 2001. Modulation induced by estradiol in the acute phase of Trypanosoma cruzi infection in mice. Parasitol. Res. 87:513–520 [DOI] [PubMed] [Google Scholar]
- 41.Schuster I, Egger H, Nussbaumer P, Kroemer RT. 2003. Inhibitors of vitamin D hydroxylases: structure-activity relationships. J. Cell Biochem. 88:372–380 [DOI] [PubMed] [Google Scholar]
- 42.Lepesheva GI, Hargrove TY, Anderson S, Kleshchenko Y, Furtak V, Wawrzak Z, Villalta F, Waterman MR. 2010. Structural insights into inhibition of sterol 14 alpha-demethylase in the human pathogen Trypanosoma cruzi. J. Biol. Chem. 285:25582–25590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lepesheva GI, Ott RD, Hargrove TY, Kleshchenko YY, Schuster I, Nes WD, Hill GC, Villalta F, Waterman MR. 2007. Sterol 14 alpha-demethylase as a potential target for antitrypanosomal therapy: enzyme inhibition and parasite cell growth. Chem. Biol. 14:1283–1293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lepesheva G, Hargrove T, Kleshchenko Y, Nes W, Villalta F, Waterman M. 2008. CYP51: a major drug target in the cytochrome P450 superfamily. Lipids 43:1117–1125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fernandes MC, Da Silva EN, Pinto AV, De Castro SL, Menna-Barreto RF. 2012. A novel triazolic naphthofuranquinone induces autophagy in reservosomes and impairment of mitosis in Trypanosoma cruzi. Parasitology. 139:26–36 [DOI] [PubMed] [Google Scholar]
- 46.Soeiro Mde N, de Castro SL. 2011. Screening of potential anti-Trypanosoma cruzi candidates: in vitro and in vivo Studies. Open Med. Chem. J. 5:21–30 [DOI] [PMC free article] [PubMed] [Google Scholar]