

Abstract
Despite recent advances in atomic level understanding of drug and inhibitor interactions with human cytochromes P450, the decades-old questions of chemical and structural determinants of hydrocarbon binding are still unanswered. (+)-α-Pinene is a monoterpene hydrocarbon that is widely distributed in the environment and a potent P450 2B inhibitor. Therefore, a combined biophysical and structural analysis of human P450 2B6 interactions with (+)-α-pinene was undertaken to elucidate the basis of the very high affinity binding. Binding of (+)-α-pinene to the P450 active site was demonstrated by a Type I spectral shift. Thermodynamics of ligand binding were explored using isothermal titration calorimetry and compared to those of P450 2A6, which is much less flexible than 2B6 based on comparison of multiple X-ray crystal structures. Consistent with expectation, entropy is the major driving force for hydrocarbon binding to P450 2A6, as evidenced by the calorimetric results. However, formation of the 2B6-(+)-α-pinene complex has a significant enthalpic component. A 2.0 Å resolution crystal structure of this enzyme ligand complex reveals that the highly plastic 2B6 utilizes previously unrecognized rearrangements of protein motifs. The results indicate that the specific components of enthalpic contribution to ligand binding are closely tied to the degree of enzyme flexibility.
INTRODUCTION
Cytochromes P450 (P450, EC 1.14.14.1) comprise a superfamily of heme-thiolate enzymes that oxidize a diverse range of endogenous chemicals, pharmaceuticals, and xenobiotics.1 Utilizing molecular oxygen and NADPH in conjunction with an oxidoreductase, P450 enzymes catalyze a diverse variety of chemical reactions, the most common being monooxygenation.2 This reaction generally increases the water solubility of the target compound. Over the past decade, structural analysis of human and other mammalian P450s has provided crucial insights into the determinants of potent and selective binding of a variety of drugs.3-6 Despite the broad range of substrates, the P450 fold is highly conserved within the superfamily.3,7 Adaptation to molecules upon ligand binding reflects the notable plasticity of some of these enzymes.
Members of the P450 2B subfamily were among the first mammalian microsomal P450s isolated and characterized and have served as a model for investigation of mechanisms of gene activation mediated by drugs and other exogenous compounds and for exploration of the structural plasticity of mammalian drug metabolizing P450s.8,9 In humans, 2B6 is found in the liver, lung, kidney, and brain, and it prepares a chemically diverse set of compounds for clearance in addition to metabolizing many endogenous compounds.10,11 P450 2B6 also exhibits potent inhibition by a wide variety of compounds ranging from 4-(4-chlorophenyl)imidazole to itraconazole.12,13
Biochemical and biophysical investigations of this subfamily of enzymes have focused on protein-ligand and protein-protein interactions and the catalytic mechanisms of mammalian monooxygenases.9,14 In addition, the discrete amino acid substitutions responsible for marked species differences seen across the P450 2B subfamily have been established.14 Enzyme plasticity has also been demonstrated by X-ray crystallography, isothermal titration calorimetry (ITC), and hydrogen-deuterium (H-D) exchange coupled to mass spectrometry (DXMS).9,14 Structural studies of P450s 2B have focused on either imidazole inhibitors or tightly binding drugs. Furthermore, a recent report described a possible mechanism by which the structurally plastic P450 2B enzymes are able to bind large molecules.15 However, it remains unclear how a single enzyme can bind molecules across a wide range of sizes (Mr roughly 80 – 800) with similar high affinity.
At present, the factors governing how individual human P450s bind and oxidize environmentally important small molecules, such as organic solvents or certain natural products, are poorly understood. Previous reports indicated that cyclohexane, n-hexanes, and several monoterpenes were metabolized by P450 2B enzymes.16-18 Monoterpenes and their oxygenated derivatives are found in numerous plant oils. These hydrocarbons are used as odorants, solvents, and as treatment components in alternative medicine, and monoterpene emissions from terrestrial plants play a major role in atmospheric chemistry.19,20 Additionally, monoterpenes may be prototypical plant toxins that were among the major driving forces of the evolution of multiple P450s in mammals, including those involved in drug metabolism.21
With this in mind, we initiated structural and biophysical studies of the interaction of (+)-α-pinene (Figure 1) with 2B6. Spectral binding titrations and ITC demonstrated the high affinity of (+)-α-pinene for 2B6, and the thermodynamic behavior was compared with that of the more rigid P450 2A6. A 2.0 Å resolution X-ray crystal structure of 2B6 in complex with (+)-α-pinene provides insight into structural adaptations required for 2B6 to bind small hydrocarbons. After three decades of investigation, the first structure of a human drug metabolizing P450 complexed with a pure hydrocarbon is presented.
Figure 1.
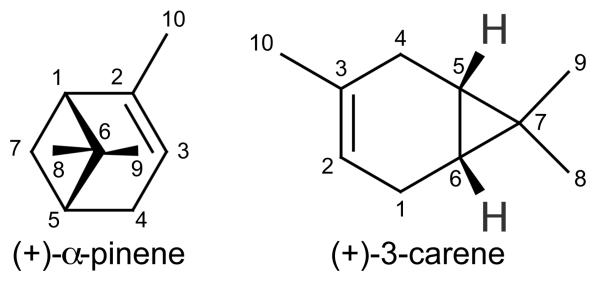
Molecular representations of (+)-α-pinene and (+)-3-carene.
EXPERIMENTAL SECTION
Chemical Reagents
(+)-α-Pinene and (+)-3-carene were obtained from Sigma-Aldrich (St. Louis, MO). CHAPS (3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate) was purchased from Calbiochem (EMD Chemicals, San Diego, CA). CYMAL-5 (5-cyclohexyl-1-pentyl-ß-D-maltoside) was acquired from Anatrace (Maumee, OH). Nickel-nitrilotriacetic acid (Ni2+-NTA) affinity resin was from Thermo Scientific (Rockford, IL). Macroprep CM cation exchange resin was received from Bio-Rad Laboratories (Hercules, CA). Amicon ultrafiltration devices were from Millipore (Billerica, MA). The pGro7 plasmid was from Takara Bio (Shiba, Japan). Escherichia coli JM109 and TOPP3 cells were from Stratagene (La Jolla, CA). 3(R)-Hydroxy-7(R),12(R)-bis(ethyloxy)-cholane (234-chol) is a custom-made facial amphiphile.22 All protein figures were created using PyMOL.23 All chemical structures were created using MarvinSketch v. 5.10.3, 2012, ChemAxon (http://www.chemaxon.com).
Protein Expression and Purification
Cytochrome P450 2B6 was expressed in E. coli JM109 cells using the pKK2B6dH (Y226H/K262R) plasmid and the pGro7 plasmid, containing the GroEL/ES chaperone pair, as previously described.15,24,25 For purification, the pellet was resuspended in 10% of the original culture volume in buffer containing 20 mM potassium phosphate (pH 7.4 at 4 °C), 20% (v/v) glycerol, 10 mM 2-mercaptoethanol (BME), and 0.5 mM phenylmethanesulfonyl fluoride (PMSF). The resuspended cells were further treated with lysozyme (0.2 mg/mL) and stirred for 120 min at 4 °C, followed by centrifugation for 30 min at 7500 × g in a JA-14 rotor in a Beckman Coulter Avanti J-26 XPI Centrifuge. After decanting the supernatant, spheroplasts were resuspended in 5% of the original culture volume in buffer containing 500 mM potassium phosphate (pH 7.4 at 4 °C), 20% (v/v) glycerol, 10 mM BME, and 0.5 mM PMSF and were sonicated for 3 × 45 s on ice. CHAPS was added to the sample at a final concentration of 0.8% (w/v), and the sample was allowed to stir for 90 min at 4 °C prior to ultracentrifugation for 45 min at 245,000 × g using a fixed-angle Ti 50.2 rotor in a Beckman Coulter Optima L-80 XP Ultracentrifuge. The P450 concentration was measured using the reduced CO difference spectra from the resulting supernatant.26
The supernatant was applied to a Ni2+-NTA column. The column was washed with buffer containing 100 mM potassium phosphate (pH 7.4 at 4 °C), 100 mM NaCl, 20% (v/v) glycerol, 10 mM BME, 0.5 mM PMSF, 0.5% (w/v) CHAPS and 5 mM histidine, and the protein was eluted using buffer containing 10 mM potassium phosphate (pH 7.4 at 4 °C), 100 mM NaCl, 20% (v/v) glycerol, 10 mM BME, 0.5 mM PMSF, 0.5% (w/v) CHAPS, and 50 mM histidine. The P450 containing fractions were pooled, diluted to reduce ionic strength with buffer containing 5 mM potassium phosphate, pH 7.4 at 4 °C, 20% (v/v) glycerol, 1 mM ethylenediaminetetraacetic acid (EDTA), 0.2 mM dithiothreitol (DTT), 0.5 mM PMSF and 0.5% (w/v) CHAPS, and loaded onto a Macroprep CM-Sepharose column. The cation exchange column was washed using low salt buffer containing 5 mM potassium phosphate (pH 7.4 at 4 °C), 20% (v/v) glycerol, 1 mM EDTA, and 0.2 mM DTT. The protein was eluted using the wash buffer with 500 mM NaCl. The P450 fractions were pooled, and the concentration was measured using the reduced CO-difference spectra.26 Cytochrome P450 2A6 was expressed in JM109 E. coli using the pCW2A6dH plasmid obtained from Dr. Eric Johnson at the Scripps Research Institute (La Jolla, CA) and as described above for P450 2B6.
Spectral Binding Titrations
The absorbance spectra were measured with an MC2000-2 multi-channel CCD rapid scanning spectrometer (Ocean Optics, Dunedin, FL, USA) equipped with one absorbance and one fluorescence channel, a pulsed Xe-lamp PX-2 light source, and a home-made thermostated cell chamber with a magnetic stirrer. A semi-micro quartz cell with a stirring compartment (10 × 4 mm light path) from Hellma GmbH (Müllheim, Germany) was used in the titration experiments. All titration experiments were carried out at 25 °C with continuous stirring in buffer containing 50 mM potassium phosphate (pH 7.4 at 4 °C), 500 mM NaCl, 1 mM EDTA, and 1 mM tris(2-carboxyethyl)phosphine (TCEP). Ligand was dissolved in acetone, and total solvent concentration at the end of each titration was less than 1%. A baseline was recorded between 350 and 700 nm using this buffer. A spectrum was recorded after the addition of protein to the buffer. Spectra were recorded after the addition of aliquots of ligand to the sample cuvette.
Data Analysis
Principal component analysis (PCA) combined with least-squares approximation of the spectra of principal components with a linear combination of appropriate prototype spectral standards was used to interpret changes in absorbance spectra in spectral titration experiments, as previously described.27,28 The spectral standards used in the titrations included the spectra of ferric high-spin, ferric low-spin, and ferric P450 states obtained for full length P450 2B4.28 The spectral dissociation constants (KS) were generally obtained by fitting the data to the equation for ligand binding for high affinity ligands 2ΔA = (ΔAmax/[E0])((KD + [I0] + [E0]) − (KD + [I0] + [E0])2 − 4[E0][I0])1/2) where ΔAmax is the maximum change in the fraction of high-spin P450, E0 is total enzyme concentration, and I0 is total inhibitor concentration when data are normalized to total P450 concentration using SPECTRALAB.27 Data for (+)-3-carene binding to 2B6 were fit to the Hill equation.
Dynamic Light Scattering
Prior to ITC experiments, the aggregation state of P450 2A6 and 2B6 in solution was measured by Dynamic Light Scattering (DLS), as previously described.29 The buffer contained 50 mM potassium phosphate (pH 7.4 at 4 °C), 500 mM NaCl, 1 mM EDTA, and 1 mM TCEP. Positive controls were bacterial cytochrome P450 eryF (P450eryF), since it is known to be monomeric in solution, and rabbit P450 2B4, since it is known to be monomeric in this buffer.30 Experiments were performed on a Zetasizer Nano-ZS (Malvern Instruments, Worcestershire, UK) instrument. The results were analyzed using the Zetasizer software, assuming a viscosity and refractive index of water to approximate the solution conditions. Due to the estimations made to calculate the hydrodynamic radii and the molecular weight of the aggregate, they should be considered approximate.
ITC Experiments and Data Analysis
ITC experiments were performed on a VP-ITC calorimeter (MicroCal, Inc.). The volume of the calorimetric cell is 1.4 mL, and the titrations in this paper were conducted by adding the titrant in steps of 10 μL. Experiments were performed at 25 °C with a 180 s initial delay. All solutions were thoroughly degassed to prevent bubble formation in the cell from stirring. Freshly prepared 2B6 was dialyzed extensively against degassed buffer containing 50 mM potassium phosphate (pH 7.4 at 4 °C), 500 mM NaCl, 1 mM EDTA, and 1 mM tris(2-carboxyethyl)phosphine (TCEP), as previously reported for rabbit 2B4.30 The protein concentration was 10 μM, and the ligand concentration was 25 μM. Cold methanol was added to a final concentration of 2 % immediately prior to temperature equilibration. Ligand stock solutions were prepared in 100 % methanol. Care was taken to prevent methanol evaporation from each of the solutions. The stability and CO binding properties of the protein were not altered in the buffer containing 2% methanol for the duration of the ITC experiments. Protein and ligand samples were quickly preincubated to the required temperature using a ThermoVac (MicroCal, Inc.) and loaded into the calorimetric cell and titration syringe, respectively. The titration cell was stirred continuously at 305 rpm. Reference titrations were carried out by injecting each ligand into buffer alone in the calorimetric cell, and heat of dilution was subtracted from the ligand-protein titration data. The binding isotherms were best fit to a one-set binding site model by Marquardt nonlinear least-squares analysis to obtain the binding stoichiometry (N), association constant (KA), and thermodynamic parameters of the interaction using ORIGIN, Version 7. Fitting of the data to a two-site model or sequential binding model did not improve the fit and yielded unreasonable error values. When total heme protein concentration was used to calculate enthalpic changes, thermodynamic parameters remained unchanged, but the stoichiometric ratio decreased ~20% for 2B6 and ~10% for 2A6. This observation is consistent with the proposal of Muralidhara et al. that the 1:1 stoichiometric binding ratio represents the active pool of P450 determined by reduced CO difference spectra, as shown in the binding of imidazole ligands to rabbit P450 2B4.30
Protein Crystallization and Data Collection
Following purification, the pooled protein was diluted to a final concentration of 18 μM in 50 mM potassium phosphate (pH 7.4 at 4 °C), 500 mM sucrose, 500 mM NaCl, 1 mM EDTA, and 0.2 mM DTT. The ligand (dissolved in acetone) was added to the protein solution to a final concentration of 180 μM. In order to prevent the evaporation of the volatile monoterpene, the solution was confined in a 50 ml plastic centrifuge tube with minimum void space above the surface of the liquid prior to incubating overnight at 4 °C on ice. This 2B6 (+)-α-pinene complex was concentrated until the protein concentration reached 280 μM and was supplemented with 4.8 mM Cymal-5, 1 mM (+)-α-pinene, and 0.028% (w/v) 234-chol. The sitting-drop vapor diffusion method was used to screen crystallization conditions using the Hampton Research Crystal Screen HR2-110. Crystals of 2B6 with (+)-α-pinene were obtained at 18 °C after incubating the protein in a 1:1 ratio with the precipitant solution containing 0.1 M HEPES sodium pH 7.5 and 1.4 M sodium citrate tribasic dihydrate for 3-4 days. Crystals were soaked in the above screen solution containing 20% (v/v) sucrose and 0.1 mM (+)-α-pinene for 30 seconds before flash freezing them in liquid nitrogen. Crystallographic data were collected remotely at Stanford Synchrotron Radiation Lightsource (SSRL) beam line 7-131 using 1° oscillations over 240 frames and 10 s exposures using a Quantum 315 CCD detector at 100 K. Crystals of 2B6 (+)-α-pinene diffracted to 2.0 Å resolution and data were integrated using iMOSFLM 32 and scaled using SCALA in CCP4 33.
Structure Determination and Refinement
The structure of 2B6 in complex with (+)-α-pinene was determined using the coordinates of the 2B6-4CPI structure (PDB ID: 3IBD) as a starting model in the molecular replacement program Phaser from the CCP4 software suite. The solvent content, as determined by the Matthews coefficient analysis, was 61.7% assuming the presence of one molecule in the asymmetric unit. The output model from Phaser, with the space group P3221, was subjected to rigid body and restrained refinement in REFMAC. The model was built manually in COOT 34 using Fo-Fc and 2Fo-Fc electron density maps contoured at 3σ and 1σ, respectively. PRODRG server was used to make the library description for (+)-α-pinene.35 Iterative refinement was continued until the R-factor and Rfree stopped improving. The model was validated by MOLPROBITY36, which ranked the structure in the 98th percentile among structures of comparable resolution. There were no Ramachandran outliers or bad bond lengths or angles in the final model. The crystal structure contained a total of 375 molecules of water, three molecules of CYMAL-5 detergent, one molecule of sucrose, and protein residues from 28 to 492 with the terminal residue being the first histidine of the C-terminal 4-His-tag. Coordinates and structure factors were deposited in the Protein Data Bank (PDB ID: 4I91), and the refinement statistics for the above structure are summarized in Table S1.
RESULTS
Binding of (+)-α-pinene to 2B6 produced a pronounced Type I spectral shift, with an increase at 390 nm and a decrease at 417 nm, indicating an increase in the high-spin fraction of the enzyme (Figure S1). The spectral dissociation constant (KS) of (+)-α-pinene binding to 2B6 was 0.38 ± 0.23 μM with a maximal increase in the fraction of high-spin P450 of 49.3 ± 11.0 % (Table 1). While the binding affinity of this small hydrocarbon for 2B6 is similar to previously reported values for 4-CPI (0.19 μM), ticlopidine (0.30 μM), and clopidogrel (0.10 μM), (+)-α-pinene produces a much greater change in the fraction of high-spin P450 than the latter two compounds (>49 % vs. 2 – 4 %).37,38 The very high affinity binding of (+)-α-pinene is remarkable for a compound containing no apparent functional groups to interact with the enzyme. Furthermore, the maximal binding affinity at 298 K of (+)-α-pinene for 2B6 predicted from its experimentally determined octanol/water partitioning coefficient (logP) of 4.4439 and based solely on enthalpy of desolvation would be −1.364×logP (~ −6 kcal/mol, ~39 μM). This calculated value differs from the experimentally derived spectral binding affinity by about one hundred-fold.
Table I. Thermodynamic Parameters of Interaction Derived from ITC and Spectral Titrations.
| Enzyme | ΔH (kcal/mol) |
ΔS (cal/mol/K) |
ΔG (kcal/mol) |
N (unitless) |
KD1 (10−6 M) |
Ks (10−6 M) |
ΔFh (%) |
|---|---|---|---|---|---|---|---|
| 2A62 | −3.8 (2.3)4 | 18.2 (7.3) | −9.3 (0.2) | 1.0 (0.0) | 0.17 (0.05) | 34.20 (7.21) | 78.5 (2.1) |
| 2B62 | −13.1 (2.3) | −12.1 (9.4) | −9.4 (1.0) | 0.9 (0.2) | 0.22 (0.17) | 0.38 (0.23) | 49.3 (11.0) |
| 2B63 | −6.3 (2.2) | 3.2 (2.3) | −7.5 (1.4) | 1.2 (0.3) | 1.82 (0.61) | 1.60 (0.14)* | 42.5 (3.4) |
KA−1 from ITC
(+)-α-pinene
(+)-3-carene
Values represent mean of at least three unique experiments. The standard deviation is displayed in parentheses.
Measured using the Hill equation giving the S50 with n=2.
These results suggested that hydrophobicity (and hence entropy) alone cannot account for the binding affinity. Therefore, thermodynamic contributions of (+)-α-pinene binding to 2B6 were explored by ITC. ITC provides direct measurement of heat changes in a system, and analysis of binding isotherms provides the values of thermodynamic parameters. This technique has previously been used to analyze ligand binding to P450s eryF, 2B4 and 3A4.40,41 A representative binding isotherm of 2B6 titrated with (+)-α-pinene (Figure 2A) shows the decrease in exothermic heat of binding (upper panel) and the integrated enthalpic changes for each injection fit to a one-site binding model (lower panel). Thermodynamic constants are reported in Table 1. The stoichiometric binding ratio was 1:1; the KD value was ~0.22 μM. Dynamic light scattering (DLS) experiments revealed no significant effect of the ligand on 2B6 in the conditions utilized for the ITC experiments (Table S1).
Figure 2.

Typical calorimetric titrations (upper panels) and resulting integrated binding isotherms with the best fit to the single site binding model (lower panels) of (+)-α-pinene (A) or (+)-3-carene (B) binding to 2B6 or (+)-α-pinene binding to 2A6 (C) at 25 °C are shown.
Binding of a second hydrocarbon, (+)-3-carene (Fig. 1), to 2B6 was investigated by spectral titration and ITC to verify ligand binding and the contributions of enthalpy to ligand binding, respectively (Table 1). This monoterpene produces a Type I spectral shift upon binding to 2B6 with an S50 of 1.60 ± 0.14 μM and 42.5 ± 3.4 % maximal increase in high-spin P450 content. A representative ITC binding isotherm is shown in Figure 2B, and the thermodynamic constants are contained in Table 1. The stoichiometric binding ratio was 1:1, and the KD was 1.74 ± 0.72 μM. As with (+)-α-pinene, binding of (+)-3-carene to 2B6 is driven primarily by enthalpy.
The results of hydrocarbon binding to 2B6 indicate that enthalpy drives formation of these complexes, in contrast to the prevailing opinion in the P450 field that favorable entropy is the critical factor. However, binding behavior with a more rigid P450, namely P450 2A6 (2A6), was explored to assess the role of enzyme flexibility in the thermodynamics of binding. While substrates of 2A6 or 2B6 are weakly basic or neutral in nature42 and have overlapping ranges of relative molecular masses (2A6 Mr = 136 - 265; 2B6 Mr = 86 - 450), 2A6 substrates tend to be more hydrophilic than those of 2B6 (average logP of 1.50 versus 2.70) and usually contain two hydrogen bond acceptors versus a single acceptor.42 Furthermore, QSAR studies indicate that the inhibition potency of 2A6 substrates and inhibitors is governed by hydrogen bond formation with the active site residue N297 and lipophilicity of the active site.43 Conversely, P450 2B ligand potency has been correlated to compound lipophilicity, molecular size, and relative molecular mass.43 As for monoterpenoids, some substrate overlap is seen. 2A6 is reported to metabolize alcohols, such as (+)-fenchol, and ketones, such as (−)-verbenone and (−)-fenchone, whereas 2B6 metabolizes ketones, including (−)-verbenone and (−)-fenchone, and hydrocarbons, including (−)-α-pinene.17,44-46
Spectral titrations of 2A6 with (+)-α-pinene yielded a KS of 34.20 ± 7.21 μM and a 78.5 ± 2.1 % maximal increase in high-spin P450 content (Table 1). A representative ITC binding isotherm is shown in Figure 2C, and the thermodynamic constants are contained in Table 1. The stoichiometric binding ratio was 1:1, and the KD was ~0.17 μM. DLS confirmed that the aggregation state of 2A6 was unchanged by the addition of (+)-α-pinene in the conditions used for ITC (Table S1). As expected, binding of (+)-α-pinene to 2A6 is entropy driven, demonstrating a striking difference between hydrocarbon binding to the more rigid 2A6 and more flexible 2B6.
Because of the high affinity of (+)-α-pinene for 2B6, the significant change in heme iron spin state, and the intriguing results of initial thermodynamic measurements, we solved an X-ray crystal structure of a (+)-α-pinene 2B6 complex (PDB ID: 4I91) (See Table S2 for collection and refinement data). An unbiased electron density for the ligand was clearly observed and subsequently modeled in the active site near the heme (Figure 3A). Residues within a 5 Å radius of the ligand include I101, I114, F115, F206, S294, F297, A298, T302, L363, V367, and V477 (Figure 3B); these amino acids were previously identified in the active site of several complexes of 2B6 or 2B4.9
Figure 3.

Unbiased electron density map and the active site residues of P450 2B6 in complex with (+)-α-pinene. (A) An unbiased Fo-Fc omit map in brown contoured at 3σ surrounds the (+)-α-pinene molecule (blue). (B) Active site residues located within 5 Å from (+)-α-pinene (blue sticks) are shown in green sticks in divergent stereo view.
While 2B6 metabolism of (+)-α-pinene has not yet been reported, metabolism of its enantiomer, (−)-α-pinene, yields (−)-trans-verbenol via hydroxylation at C4 and (−)-myrtenol by hydroxylation at C10, but no ratio of products was indicated.17 The closest atom of (+)-α-pinene (C10 in Figure 1) to the heme iron was located at a distance of 4.8 Å, which would be conducive to production of myrtenol. Initial experiments demonstrated that (+)-α-pinene tripled NADPH consumption by a reconstituted system containing P450 2B6 from 16 to 52 nmol/min/nmol with no change in hydrogen peroxide formation (7 nmol/min/nmol). These data clearly demonstrate rapid formation of the reactive oxygen complex of P450 2B6 in the presence of (+)-α-pinene. However, the distribution between product formation and uncoupling to water remains to be determined.
The 2B6 (+)-α-pinene structure was nearly identical to two previously solved 2B6 complexes (4-benzylpyridine (4-BP) and 4-(4-nitrobenzyl)pyridine (4-NBP)),47 similar to one (4-(4-chlorophenyl)imidazole),37 and significantly different from the fourth (amlodipine).15 These complexes, in decreasing extent of similarity based on the RMSD of a Cα overlay with the (+)-α-pinene are: 4-BP (0. 17 Å) > 4-NBP (0.28 Å) > 4-CPI (0.63 Å) > amlodipine (1.05 Å). Marked differences were noted upon comparison of the (+)-α-pinene and the amlodipine complexes (Figure 4A). Specifically, locations of the H-helix and the H-I loop were as much as 6 Å apart, and the F-G cassette shifted up to ~3.5 Å. Furthermore, the A-, B′-, and C-helices and the β4-loop shifted by 1.5-2.5 Å. Rearrangements of two phenylalanine residues (F206 and F297) also contributed to a more compact active site in the (+)-α-pinene complex, with F206 moving in and F297 moved out. The organization of these side chains is similar to the arrangement seen in the complexes of 2B6 with 4-BP and with 4-NBP.47 The combined effect of these shifts was to close the active site around the relatively small monoterpene compared to the space needed for the much larger amlodipine.
Figure 4.

Structural differences revealed upon comparison of 2B6 (+)-α-pinene complex and amlodipine complex. A) Active site of 2B6 (+)-α-pinene (orange) and 2B6 amlodipine (green) complex structures. B) Rearrangement of the I-helix among the 2B6 amlodipine (3UA5, orange), 4-CPI (3IBD, blue), and (+)-α-pinene (4I91, green) complexes.
DISCUSSION
Precedent for the apparent enthalpically driven binding of (+)-α-pinene to 2B6 can be found in studies of other proteins with occluded hydrophobic pockets, in which favorable enthalpy from van der Waals interactions between protein and bound ligand is a major contributor to binding free energy.48-50 In the case of 2B6, the ability of the protein to maximize such interactions by changing its conformation to conform closely to the (+)-α-pinene molecule likely contributes to the favorable enthalpy. An alternate explanation is that the favorable entropy does not reflect the binding event per se. Rather, as found in a recent report by Gilson and coworkers51 of long time scale molecular dynamics simulation of protein-ligand interactions, the thermodynamics of global protein conformational changes can mask the thermodynamics of local phenomena, including protein-ligand binding. These conformational changes could release water from one or more small hydrophobic pockets where the water is confined and unable to make its full complement of hydrogen bonds. One such pocket seen in multiple X-ray crystal structures of 2B6 is that occupied by CYMAL-5 in the (+)-α-pinene complex (4I91) and comprising residues A176, C180, F188, F195, M198, L199, F202, I241, Y244, I245, F296, and T300 within 5 Å of the CYMAL-5 molecule. Release of water from such a hydrophobic pocket could produce a significant enthalpic boost to binding.
Smaller contributions to differences from the expected thermodynamics of ligand binding could also be explained by alterations in the disruption of ordered waters in the active site. In mouse major urinary protein, which binds a broad array of small hydrophobic ligands, gains in solute-solute dispersion energy are not offset by interactions between solvent and binding-site residues prior to association.48 In the case of 2B6, favorable solute-solute enthalpy is what appears in the enthalpy-driven thermodynamic signature of monoterpene binding. It was also of interest to compare our ITC results with a previous proposal of Backes et al. that aromatic hydrocarbon binding to P450s is a combination of the ligand being forced from an aqueous environment (“push”) and being drawn into the hydrophobic active site of the enzyme (“pull”).52 With 2B6, the hydrophobic nature of the monoterpene would provide the “push” from solution, and the sum of the protein dynamics, the exclusion of water from the active site of the enzyme upon (+)-α-pinene binding, and the hydrophobic nature of the active site could be responsible for the “pull” phenomenon.
Comparison of X-ray crystal structures of 2B6 in complex with ligands of varying sizes shows significant conformational flexibility, with up to 1.05 Å difference between the two most divergent structures and likely greater plasticity in solution. 2A6 is much more rigid, displaying ~0.34 Å RMSD difference between the two most different structures, those containing coumarin (1Z10)53 and pilocarpine (3T3R)54; this enzyme is also likely more rigid than 2B6 in solution. In the case of 2B6 and 2A6, the more plastic enzyme displays the unexpected driving force of enthalpy for hydrocarbon binding, while the less plastic protein exhibits the expected entropy-driven binding profile. This observation would support the inference that conformational changes following (+)-α-pinene binding to 2B6 are probably drowning out the contributions of binding per se. In addition, the free energy of (+)-α-pinene binding and resulting association/dissociation constants are similar in the case of 2B6 and 2A6 (Table 1). The ΔH and TΔS for 2B6 are both −9 kcal/mol relative to 2A6. A crude interpretation of this information would suppose that the “direct” interaction of (+)-α-pinene with 2B6 is about the same as that with 2A6, and the difference in binding thermodynamics reflects the differences in conformational adjustment upon ligand binding between the two enzymes.
For hydrocarbon binding to 2B6, dissociation constants measured by spectral transition are similar to those from ITC; however, the KS for (+)-α-pinene binding to 2A6 is three orders of magnitude higher than the KD from ITC. The low to high spin transition in P450s usually occurs when the sixth coordination site of the heme is opened by loss of water.55 Protein rearrangement restricts solvent access and organizes the heme iron, molecular oxygen, and the substrate in close proximity to each other, facilitating catalysis and minimizing side reactions, such as peroxide or superoxide production. In this light, (+)-α-pinene or (+)-3-carene binding to 2B6 probably leads to a compaction of the active site that causes the dissociation of water from the heme due to ligand proximity. Since 2A6 has demonstrated less conformational flexibility, the compression of the active site is likely not as great upon (+)-α-pinene binding. In addition, (+)-α-pinene is unable to interact with N297 in 2A6, leading to a less restricted orientation of the ligand in the active site and making dissociation of the heme water ligand less likely. Moreover, interaction of water with N297, T305 and the heme iron could create a more ordered lattice in 2A6 than found in the interaction of water with T302 and the heme iron in 2B6. Based on all these considerations, the tight binding of (+)-α-pinene indicated by ITC may not lead to a spin shift, which is observed instead at much higher concentrations.
Differences in secondary structure arrangement were seen among the 2B6 complexes with (+)-α-pinene (4I91), 4-CPI (3IBD), and amlodipine (3UA5). Major differences in the overall conformation of two of these structures are highlighted by overlays (Figure S2). The distance of residues 296 - 306 from the heme differs significantly among the three 2B6 structures, in decreasing order: 4I91 > 3IBD > 3UA5 (Figure 4B). In order to analyze the differences in secondary structure arrangement in the 2B6 complexes, we mapped onto the 2B6 (+)-α-pinene complex the difference in H-D exchange rate between ligand-free 2B4 and its tBPA complex.29 We used 2B4 as a surrogate because of the overall similarity between the structures of 2B6 and 2B4 when complexed with the same ligand and availability of the H-D exchange data with 2B4.56 Furthermore, the 2B6 (+)-α-pinene complex and the 2B4-tBPA structure are fairly similar (0.64 Å RMSD, Figure 5A). Differences in placement of secondary structure ele-ments in 2B6 structures (Figure S2) map nicely to the DXMS data from 2B4 (Figure 5B). Specifically, slowing of the H-D exchange rate, shown in shades of blue, is found in the same parts of 2B4 that show the greatest differences between the amlodipine and (+)-α-pinene complexes of 2B6. Changes along the I-helix are likely due to the compact structure of the 2B4-tBPA complex.
Figure 5.

Areas of P450 2B4 showing significant changes in H-D exchange rate are also those showing significant structural movement among conformations represented in the crystal structures of 2B6 as seen from the side (left) and from above the heme (right). A) Overlay of 2B6 complexed with (+)-α-pinene (green) and 2B4 labeled with t-BPA (3R1A, blue); and B) Data for differences in the H-D exchange rate between 2B4 without ligand and covalently labeled with tert-butylphenylacetylene (t-BPA) at the 10000s time point are mapped onto the 2B6 (+)-α-pinene complex.56
In summary, the widely distributed monoterpene (+)-α-pinene was shown to bind tightly to both 2B6 and 2A6, and (+)-3-carene was shown to bind tightly to 2B6. However, using ITC as a probe of the thermodynamic profile of ligand binding revealed the strong contribution of enthalpy in the case of 2B6 and entropy with 2A6. This difference in thermodynamic contributions of entropy and enthalpy is likely due in part to the difference in enzyme plasticity. Concurrently, an X-ray crystal structure of a 2B6 (+)-α-pinene complex highlighted the structural plasticity of 2B6. Furthermore, changes in H-D exchange rates in 2B4 and observed structural rearrangements in 2B6 correspond to the same regions of the protein, reinforcing the validity of conclusions made from comparison among X-ray crystal structures of 2B6. Due to the hydrophobic nature of many P450 substrates and of many P450 active sites, the fact that many hydrophobic associations in solution do not have an entropy-driven thermodynamic signature should also extend this possibility to these enzymes.57 Determination of thermodynamic contributions of ligand binding to P450 enzymes appears to depend not only upon ligand and active site hydrophobicity but also upon how the architecture of the active site lends itself to solvation. Apparently, 2A6 is more solvated than 2B6, where Van der Waals interactions dominate small hydrocarbon binding.
Supplementary Material
ACKNOWLEDGMENT
This work is supported by grant ES003619 (JRH) from the National Institutes of Health. We thank Dr. Michael Gilson from the Skaggs School of Pharmacy and Pharmaceutical Sciences at UCSD for critical reading of the manuscript. We also thank the staff at the Stanford Synchrotron Radiation Lightsource (SSRL), operated by Stanford University on behalf of the United States Department of Energy, Office of Basic Energy Sciences, for assistance with data collection. The Stanford Synchrotron Radiation Lightsource is supported by the National Institute of Health, the National Center for Research Resources, the Biomedical Technology Program and the United States Department of Energy of Biological and Environmental Research.
ABBREVIATIONS
- P450
cytochrome P450
- ITC
isothermal titration calorimetry
- H-D
hydrogen-deuterium
- DXMS
hydrogen-deuterium exchange coupled to mass spectrometry
- CHAPS
3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate
- CYMAL-5
5-cyclohexyl-1-pentyl-ß-D-maltoside
- Ni2+-NTA
nickel-nitrilotriacetic acid
- 234-chol
3(R)-Hydroxy-7(R),12(R)-bis(ethyloxy)-cholane
- BME
2-mercaptoethanol
- PMSF
phenylmethanesulfonyl fluoride
- EDTA
ethylenediaminetetraacetic acid
- DTT
dithiothreitol
- TCEP
tris(2-carboxyethyl)phosphine
- PCA
principal component analysis
- DLS
dynamic light scattering
- SSRL
Stanford Synchrotron Radiation Lightsource
- PDB
Protein Data Bank
Footnotes
The authors declare no competing financial interest.
Accession Code
Atomic coordinates and structure factors for the P450 2B6-(+)-α-pinene complex is deposited in the Protein Data Bank as entries 4I91.
Tables and figures as described in the text. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- (1).Johnson EF, Stout CD. Biochem. Biophys. Res. Co. 2005;338:331. doi: 10.1016/j.bbrc.2005.08.190. [DOI] [PubMed] [Google Scholar]
- (2).Guengerich FP. Chem. Res. Toxicol. 2001;14:611. doi: 10.1021/tx0002583. [DOI] [PubMed] [Google Scholar]
- (3).Otyepka M, Skopalik J, Anzenbacherova E, Anzenbacher P. BBA - Gen. Subjects. 2007;1770:376. doi: 10.1016/j.bbagen.2006.09.013. [DOI] [PubMed] [Google Scholar]
- (4).Gay SC, Roberts AG, Halpert JR. Future Med. Chem. 2010;2:1451. doi: 10.4155/fmc.10.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Otyepka M, Berka K, Anzenbacher P. Curr. Drug. Metab. 2012;13:130. doi: 10.2174/138920012798918372. [DOI] [PubMed] [Google Scholar]
- (6).Dong D, Wu B, Chow D, Hu M. Drug Metab. Rev. 2012;44:192. doi: 10.3109/03602532.2011.645580. [DOI] [PubMed] [Google Scholar]
- (7).Poulos TL. Drug Metab. Dispos. 2005;33:10. doi: 10.1124/dmd.104.002071. [DOI] [PubMed] [Google Scholar]
- (8).Wang H, Negishi M. Curr. Drug Metab. 2003;4:515. doi: 10.2174/1389200033489262. [DOI] [PubMed] [Google Scholar]
- (9).Wilderman PR, Halpert JR. Curr. Drug Metab. 2012;13:167. doi: 10.2174/138920012798918417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Ekins S, Iyer M, Krasowski MD, Kharasch ED. Curr. Drug Metab. 2008;9:363. doi: 10.2174/138920008784746346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Gervot L, Rochat B, Gautier JC, Bohnenstengel F, Kroemer H, de Berardinis V, Martin H, Beaune P, de Waziers I. Pharmacogenetics. 1999;9:295. [PubMed] [Google Scholar]
- (12).Walsky RL, Astuccio AV, Obach RS. J. Clin. Pharmacol. 2006;46:1426. doi: 10.1177/0091270006293753. [DOI] [PubMed] [Google Scholar]
- (13).Wang HB, Tompkins LM. Curr. Drug Metab. 2008;9:598. doi: 10.2174/138920008785821710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Zhao Y, Halpert JR. BBA-Gen. Subjects. 2007;1770:402. doi: 10.1016/j.bbagen.2006.07.006. [DOI] [PubMed] [Google Scholar]
- (15).Shah MB, Wilderman PR, Pascual J, Zhang Q, Stout CD, Halpert JR. Biochemistry. 2012;51:7225. doi: 10.1021/bi300894z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Guengerich FP, Ballou DP, Coon MJ. Biochem. Bioph. Res. Co. 1976;70:951. doi: 10.1016/0006-291x(76)90684-7. [DOI] [PubMed] [Google Scholar]
- (17).Sugie A, Miyazawa M. Proc. 47th TEAC. 2003:159. [Google Scholar]
- (18).Shimada T, Shindo M, Miyazawa M. Drug Metab. Pharmacok. 2002;17:507. doi: 10.2133/dmpk.17.507. [DOI] [PubMed] [Google Scholar]
- (19).Mercier B, Prost J, Prost M. Int. J. Occup. Med. Environ. Health. 2009;22:331. doi: 10.2478/v10001-009-0032-5. [DOI] [PubMed] [Google Scholar]
- (20).Harrison SP, Morfopoulos C, Dani KGS, Prentice IC, Arneth A, Atwell BJ, Barkley MP, Leishman MR, Loreto F, Medlyn BE, Niinemets Ü, Possell M, Peñuelas J, Wright IJ. New Phytol. 2013;197:49. doi: 10.1111/nph.12021. [DOI] [PubMed] [Google Scholar]
- (21).Gonzalez FJ, Nebert DW. Trends Genet. 1990;6:182. doi: 10.1016/0168-9525(90)90174-5. [DOI] [PubMed] [Google Scholar]
- (22).Zhang QH, Ma XQ, Ward A, Hong WX, Jaakola VP, Stevens RC, Finn MG, Chang G. Angew. Chem.-Int. Edit. 2007;46:7023. doi: 10.1002/anie.200701556. [DOI] [PubMed] [Google Scholar]
- (23).Schrodinger L. 1.3 ed. Schrodinger, LLC; 2010. [Google Scholar]
- (24).Mitsuda M, Iwasaki M. Protein Expres. Purif. 2006;46:401. doi: 10.1016/j.pep.2005.10.017. [DOI] [PubMed] [Google Scholar]
- (25).Scott EE, Spatzenegger M, Halpert JR. Arch. Biochem. Biophys. 2001;395:57. doi: 10.1006/abbi.2001.2574. [DOI] [PubMed] [Google Scholar]
- (26).Omura T, Sato R. J. Biol. Chem. 1964;239:2370. [PubMed] [Google Scholar]
- (27).Davydov DR, Deprez E, Hui Bon Hoa G, Knyushko TV, Kuznetsova GP, Koen YM, Archakov AI. Arch. Biochem. Biophys. 1995;320:330. doi: 10.1016/0003-9861(95)90017-9. [DOI] [PubMed] [Google Scholar]
- (28).Renaud JP, Davydov DR, Heirwegh KPM, Mansuy D, Hoa GHB. Biochem. J. 1996;319:675. doi: 10.1042/bj3190675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Gay SC, Zhang H, Wilderman PR, Roberts AG, Liu T, Li S, Lin HL, Zhang Q, Woods VL, Stout CD, Hollenberg PF, Halpert JR. Biochemistry. 2011;50:4903. doi: 10.1021/bi200482g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Muralidhara BK, Negi S, Chin CC, Braun W, Halpert JR. J. Biol. Chem. 2006;281:8051. doi: 10.1074/jbc.M509696200. [DOI] [PubMed] [Google Scholar]
- (31).Soltis SM, Cohen AE, Deacon A, Eriksson T, Gonzalez A, McPhillips S, Chui H, Dunten P, Hollenbeck M, Mathews I, Miller M, Moorhead P, Phizackerley RP, Smith C, Song J, van dem Bedem H, Ellis P, Kuhn P, McPhillips T, Sauter N, Sharp K, Tsyba I, Wolf G. Acta Crystallogr. D. 2008;64:1210. doi: 10.1107/S0907444908030564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Leslie AGW. Acta Crystallogr. D. 1999;55:1696. doi: 10.1107/s090744499900846x. [DOI] [PubMed] [Google Scholar]
- (33).Bailey S. Acta Crystallogr. D. 1994;50:760. [Google Scholar]
- (34).Emsley P, Cowtan K. Acta Crystallogr. D. 2004;60:2126. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- (35).van Aalten DMF, Bywater R, Findlay JBC, Hendlich M, Hooft RWW, Vriend G. J. Comput. Aid. Mol. Des. 1996;10:255. doi: 10.1007/BF00355047. [DOI] [PubMed] [Google Scholar]
- (36).Chen VB, Arendall WB, 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. Acta Crystallogr. D. 2010;66:12. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Gay SC, Shah MB, Talakad JC, Maekawa K, Roberts AG, Wilderman PR, Sun L, Yang JY, Huelga SC, Hong W-X, Zhang Q, Stout CD, Halpert JR. Mol. Pharmacol. 2010;77:529. doi: 10.1124/mol.109.062570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Talakad JC, Kumar S, Halpert JR. Drug Metab. Dispos. 2009;37:644. doi: 10.1124/dmd.108.023655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Griffin S, Wyllie SG, Markham J. J. Chromatogr. A. 1999;864:221. doi: 10.1016/s0021-9673(99)01009-2. [DOI] [PubMed] [Google Scholar]
- (40).Isin EM, Guengerich FP. J. Biol. Chem. 2006;281:9127. doi: 10.1074/jbc.M511375200. [DOI] [PubMed] [Google Scholar]
- (41).Muralidhara BK, Halpert JR. Drug Metab. Rev. 2007;39:539. doi: 10.1080/03602530701498182. [DOI] [PubMed] [Google Scholar]
- (42).Lewis DFV. Curr. Med. Chem. 2003;10:1955. doi: 10.2174/0929867033456855. [DOI] [PubMed] [Google Scholar]
- (43).Sridhar J, Liu JW, Foroozesh M, Stevens CLK. Molecules. 2012;17:9283. doi: 10.3390/molecules17089283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Miyazawa M, Nakanishi K. Biosci. Biotech. Bioch. 2006;70:1259. doi: 10.1271/bbb.70.1259. [DOI] [PubMed] [Google Scholar]
- (45).Miyazawa M, Sugie A, Shimada T. Drug Metab. Dispos. 2003;31:1049. doi: 10.1124/dmd.31.8.1049. [DOI] [PubMed] [Google Scholar]
- (46).Miyazawa M, Sugie A, Shindo M. Biosci. Biotech. Bioch. 2002;66:2458. doi: 10.1271/bbb.66.2458. [DOI] [PubMed] [Google Scholar]
- (47).Shah MB, Pascual J, Zhang Q, Stout CD, Halpert JR. Mol. Pharmacol. 2011;80:1047. doi: 10.1124/mol.111.074427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Homans SW. In: Bioactive Conformation I. Peters T, editor. Vol. 272. Springer-Verlag Berlin; Berlin: 2007. p. 51. [Google Scholar]
- (49).Syme NR, Dennis C, Bronowska A, Paesen GC, Homans SW. J. Am. Chem. Soc. 2010;132:8682. doi: 10.1021/ja101362u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Young T, Abel R, Kim B, Berne BJ, Friesner RA. Proc. Natl. Acad. Sci. U.S.A. 2007;104:808. doi: 10.1073/pnas.0610202104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Fenley AT, Muddana HS, Gilson MK. Proc. Natl. Acad. Sci. U.S.A. 2012;109:20006. doi: 10.1073/pnas.1213180109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Backes WL, Cawley G, Eyer CS, Means M, Causey KM, Canady WJ. Arch. Biochem. Biophys. 1993;304:27. doi: 10.1006/abbi.1993.1317. [DOI] [PubMed] [Google Scholar]
- (53).Yano JK, Hsu M-H, Griffin KJ, Stout CD, Johnson EF. Nat. Struct. Mol. Biol. 2005;12:822. doi: 10.1038/nsmb971. [DOI] [PubMed] [Google Scholar]
- (54).DeVore NM, Meneely KM, Bart AG, Stephens ES, Battaile KP, Scott EE. FEBS J. 2012;279:1621. doi: 10.1111/j.1742-4658.2011.08412.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Haines DC, Tomchick DR, Machius M, Peterson JA. Biochemistry. 2001;40:13456. doi: 10.1021/bi011197q. [DOI] [PubMed] [Google Scholar]
- (56).Wilderman PR, Shah MB, Liu T, Li S, Hsu S, Roberts AG, Goodlett DR, Zhang Q, Woods VL, Jr., Stout CD, Halpert JR. J. Biol. Chem. 2010;285:38602. doi: 10.1074/jbc.M110.180646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Barratt E, Bingham RJ, Warner DJ, Laughton CA, Phillips SEV, Homans SW. J. Am. Chem. Soc. 2005;127:11827. doi: 10.1021/ja0527525. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.