

Abstract
Burst suppression is an electroencephalogram pattern that consists of a quasi-periodic alternation between isoelectric ‘suppressions’ lasting seconds or minutes, and high-voltage ‘bursts’. It is characteristic of a profoundly inactivated brain, occurring in conditions including hypothermia, deep general anaesthesia, infant encephalopathy and coma. It is also used in neurology as an electrophysiological endpoint in pharmacologically induced coma for brain protection after traumatic injury and during status epilepticus. Classically, burst suppression has been regarded as a ‘global’ state with synchronous activity throughout cortex. This assumption has influenced the clinical use of burst suppression as a way to broadly reduce neural activity. However, the extent of spatial homogeneity has not been fully explored due to the challenges in recording from multiple cortical sites simultaneously. The neurophysiological dynamics of large-scale cortical circuits during burst suppression are therefore not well understood. To address this question, we recorded intracranial electrocorticograms from patients who entered burst suppression while receiving propofol general anaesthesia. The electrodes were broadly distributed across cortex, enabling us to examine both the dynamics of burst suppression within local cortical regions and larger-scale network interactions. We found that in contrast to previous characterizations, bursts could be substantially asynchronous across the cortex. Furthermore, the state of burst suppression itself could occur in a limited cortical region while other areas exhibited ongoing continuous activity. In addition, we found a complex temporal structure within bursts, which recapitulated the spectral dynamics of the state preceding burst suppression, and evolved throughout the course of a single burst. Our observations imply that local cortical dynamics are not homogeneous, even during significant brain inactivation. Instead, cortical and, implicitly, subcortical circuits express seemingly different sensitivities to high doses of anaesthetics that suggest a hierarchy governing how the brain enters burst suppression, and emphasize the role of local dynamics in what has previously been regarded as a global state. These findings suggest a conceptual shift in how neurologists could assess the brain function of patients undergoing burst suppression. First, analysing spatial variation in burst suppression could provide insight into the circuit dysfunction underlying a given pathology, and could improve monitoring of medically-induced coma. Second, analysing the temporal dynamics within a burst could help assess the underlying brain state. This approach could be explored as a prognostic tool for recovery from coma, and for guiding treatment of status epilepticus. Overall, these results suggest new research directions and methods that could improve patient monitoring in clinical practice.
Keywords: intracranial electroencephalogram, human, neurophysiology, general anaesthesia
Introduction
The anaesthetized brain, though profoundly inactivated, is nevertheless characterized by rich electrophysiological dynamics. At deep levels of general anaesthesia, the brain reaches a state of burst suppression, in which periods of high voltage brain activity (bursts) alternate with periods of isoelectric quiescence (suppressions). This alternation is quasi-periodic and the suppression periods become longer in duration as the brain becomes more inactivated, lasting for seconds or even minutes (Amzica, 2009; Brown et al., 2010). Burst suppression seems to be a fundamental characteristic of the deeply inactivated brain, and can occur in a range of conditions including hypothermia (Stecker et al., 2001), deep levels of general anaesthesia (Akrawi et al., 1996), certain infant encephalopathies (Ohtahara and Yamatogi, 2003) and coma (Young, 2000). Clinically, it is commonly used as an electrophysiological endpoint in neurological intensive care after traumatic injury (Hall and Murdoch, 1990) and in the treatment of status epilepticus (Claassen et al., 2002; Rossetti et al., 2004). However, despite the presence of burst suppression in this broad range of inactivated brain states, its biophysical mechanisms are poorly understood.
Burst suppression has classically been viewed as a homogeneous brain state. This perspective has been derived from EEG studies in which bursts and suppressions have been shown to occur concurrently across the scalp (Clark and Rosner, 1973). However, because scalp EEG is spatially blurred, the underlying dynamics are not fully understood. In vivo studies in anaesthetized animals have helped to identify the potential cellular correlates of burst suppression, showing that although nearly all cortical neurons are inhibited during suppression periods, a subset of thalamocortical neurons can continue firing at delta frequencies (Steriade et al., 1994). This work established the involvement of different cortical and subcortical cell-types within both burst and suppression. However, a broader mechanism for burst suppression has not been determined, and the notion of burst suppression as a global state remains.
In search of a more detailed and complete mechanistic understanding, studies have suggested that burst suppression is associated with enhanced excitability in cortical networks (Kroeger and Amzica, 2007; Ferron et al., 2009). These studies implicate extracellular calcium as a correlate for the switches between burst and suppression. Ching et al. (2012) have proposed an alternative mechanism, using computational methods, where burst suppression manifests in a state of reduced neuronal activity and cerebral metabolism. In such a state, insufficient production of ATP in local cortical networks can gate neuronal potassium channels, leading to suppression of action potentials. Such a mechanism accounts for the general features of burst suppression previously observed, as well as its occurrence under multiple aetiologies and also predicts a specific frequency structure for the neuronal activity within each burst.
A crucial element that has yet to be studied is how large-scale cortical networks function during burst suppression. Little is known about these systems-level circuits in burst suppression, partly due to the technical challenges of sampling multiple cortical sites simultaneously. Furthermore, it is not clear if burst suppression in the human brain matches what is described in animal models. Animal studies have typically been done over millimetre-scale areas of cortex, and it is possible that larger-scale recordings in human cortex could reveal spatial differences in burst suppression across cortex. Such spatial differences could identify certain brain regions to be differentially sensitive during burst suppression and pharmacologically induced coma, impacting strategies for clinical monitoring and patient care.
Here, we examine the cortical dynamics underlying burst suppression, and test whether their spatiotemporal properties are consistent with predictions from previous animal and computational studies. Specifically, we test: (i) whether the spatial distribution of burst suppression is homogeneous across cortex; and (ii) whether the temporal structure of the state preceding burst suppression is replicated within bursts and decelerates throughout bursts, as suggested by a computational model (Ching et al., 2012). We tested these hypotheses by recording intracranial electrocorticograms from five patients with intractable epilepsy who entered a state of burst suppression while undergoing general anaesthesia for clinical treatment. We recorded from subdural grid, strip and depth electrodes distributed throughout temporal, parietal and frontal cortex. We investigated spatial dynamics in the four patients with grid electrodes, and temporal dynamics in all five patients. This combination of broad spatial coverage and excellent temporal resolution enabled an in-depth examination of the spatiotemporal dynamics of burst suppression.
Materials and methods
Data acquisition
We enrolled five patients with epilepsy intractable to medication, who were implanted with intracranial electrocorticography electrodes for standard clinical monitoring (AdTech Inc). Informed consent was obtained from all patients in accordance with the local institutional review board. Electrode placement was determined solely by clinical criteria. One patient was implanted only with depth electrodes and the other four had a combination of depth electrodes and subdural grid and strip electrodes, with 1 cm spacing between electrode contacts. Recordings were collected throughout induction of general anaesthesia using propofol, at the beginning of a surgery to explant the electrodes. Relevant clinical information, including anaesthetic protocol and recording sites, can be found in Tables 1 and 2. A portion of one recording from one patient was previously reported in a separate analysis of slow oscillations (Lewis et al., 2012). Electrocorticography data were recorded with a sampling rate of 2000 Hz, low pass filtered at 100 Hz and resampled to 250 Hz. For all analyses of spatial dynamics (Figs 1–4), grid channels were referenced with a Laplacian montage in which the average of all available neighbouring channels (up to four nearest neighbours) was subtracted, in order to minimize spatial spread of the signals. When analysing temporal structure across all channels (grid, strip and depth electrodes; Figs 5 and 6), they were referenced in a bipolar scheme as the depth and strip electrodes were positioned in one-dimensional arrays. Channels with large artefacts or with periods of signal saturation were excluded from the analysis. All data were exported to Matlab (Mathworks) for further analysis with custom software.
Table 1.
Clinical information for each study participant
| Patient | Age | Gender | Weight (kg) | Location of epileptogenic area | Aetiology | Electrodes |
|---|---|---|---|---|---|---|
| A | 52 | F | 65 | Temporal lobe | Cortical dysplasia | 64-contact grid covering left temporal cortex, extending partially over frontal and parietal regions; 4-contact strip on left occipital cortex; three 4-contact strips spanning left anterior, middle, and posterior subtemporal cortex; two 8-contact depth electrodes targeting anterior and posterior left hippocampus. |
| B | 32 | M | 67 | Frontal lobe | Unknown | 64-contact grid over temporal and parietal cortex; 4-contact strip extending to frontal cortex; 8-contact strip along parietal cortex; 4-contact strip on subtemporal cortex; two 8-contact depths into medial temporal cortex; one occipital 8-contact depth. |
| C | 23 | F | 55 | Frontal lobe | Unknown | 64-contact grid over frontal, temporal, and parietal cortex; two 4-contact subfrontal strips; two 4-contact interhemispheric frontal strips; 8-contact medial temporal depth; 8-contact cingulate depth; 8-contact prefrontal depth |
| D | 34 | F | 112 | Frontal lobe | Cortical dysplasia | 64-contact grid over frontal, temporal, and parietal cortex; 4-contact frontopolar strip; 4-contact subfrontal strip; 4-contact subtemporal strip; two 8-contact medial temporal depths; two 8-contact frontal depths |
| E | 29 | M | 75 | Temporal lobe | Post-traumatic epilepsy | 4 8-contact depth electrodes targeting left and right temporal and hippocampal cortex; 6 6-contact depth electrodes in subfrontal, cingulate, and posterior frontal cortex |
Table 2.
Anaesthetic protocol for each study participant
| Patient | Propofol bolus | Propofol infusion | Co-medication | Mean arterial pressure | CO2 |
|---|---|---|---|---|---|
| A | 130 mg (2 mg/kg) | None | 150 µg fentanyl ∼1 min post-induction; 2 mg midazolam ∼10 mins pre-induction | 88 | 35 |
| B | Total of 350 mg (5.2 mg/kg) over four boluses | None | 0.05 µg/kg/min remifentanil throughout induction | 131 | 49 |
| C | Total of 200 mg (3.64 mg/kg) over two boluses | None | 250 µg fentanyl simultaneously with propofol | 83 | 21 |
| D | None | Gradual infusion with varying rate, for a total of 3.3 mg/kg over 14 min | None | 81 | 31 |
| E | Total of 400 mg (5.3 mg/kg) over four boluses | None | None | 70 | 29 |
Figure 1.

The state of burst suppression can be limited to a local cortical region. (A) Reconstructed MRI for Patient A, showing grid electrode locations. Arrows mark the channels that are displayed in panels B and C. (B) Example time-series in different cortical regions where all channels are in burst suppression, but bursts are asynchronous in different regions. (C) Example from later in recording in the same regions as A: channel 55 is in burst suppression, whereas channels 17 and 12 are not. The state of burst suppression is therefore not necessarily cortex-wide. (D) Zoomed-in example from Patient B of the burst suppression probability (BSP) changing over time: initially most channels have a high burst suppression probability, but then a subset of channels exit burst suppression (e.g. channel 44) whereas other channels maintain high burst suppression probabilities (e.g. channel 5 remains in burst suppression with a burst suppression probability >0.5). (E) Standard deviation of the burst suppression probability across all channels from panel B: the increasing standard deviation demonstrates that the burst suppression probabilities in different cortical regions are becoming uncoupled as they diverge into different states.
Figure 2.
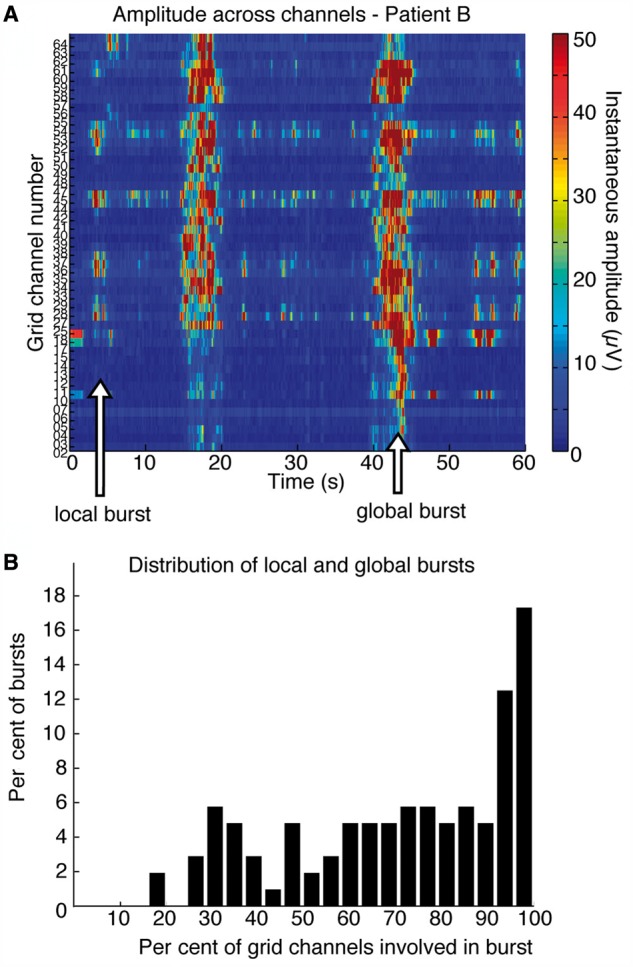
Bursts can occur in limited areas of cortex. (A) Instantaneous amplitude across all grid channels. The burst at 42 s involves all channels, but the burst at 5 s occurs in only a small subset of electrodes, indicating that a limited cortical region is bursting. (B) Histogram shows the number of grid channels participating in each burst across four patients: many bursts are global, but there is a long leftward tail to the distribution, demonstrating the frequent occurrence of local bursts.
Figure 3.

Burst timing is heterogeneous across cortex. (A) Example trace from Patient C: the burst in channel 32 starts hundreds of milliseconds before the bursts in channels 36 and 34. (B) Plot of mean difference in burst onset times between electrodes shows that there are substantial timing differences in burst onsets between distant electrodes, with distant electrodes showing larger gaps in burst timing. Blue lines are mean and standard error, red stars mark distances that are significantly different than pairs 1 cm apart (n = 233 electrodes in four patients). (C) Probability that two electrodes are both bursting (total time where both electrodes burst, normalized by total time that either electrode has a burst.) Probability decreases with distance, demonstrating that distant electrodes are less likely to be simultaneously in a burst state.
Figure 4.

Principal components analysis demonstrates that bursts are spatially clustered. (A) Reconstruction of the grid electrode placement in Patient D. (B) Each panel shows one of the first four principal components from Patient D, and the colour variation across the grid demonstrates how burst probability is locally differentiated. Each of the components is significantly spatially clustered (P < 0.05), demonstrating that burst properties are anatomically clustered and differ in distant cortical regions.
Figure 5.
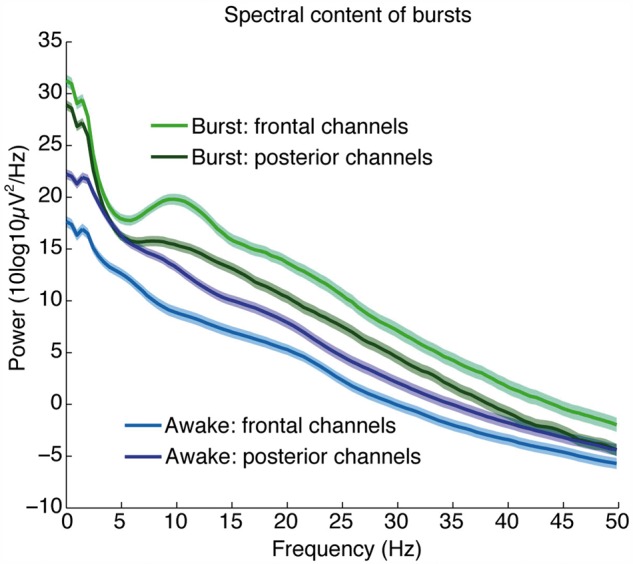
Bursts recover the spectral dynamics of propofol general anaesthesia. Average spectra (±standard error) within a burst, across all channels (n = 374 electrodes in five patients), categorized by anatomical location. Plot shows that the bursts contain increased slow power relative to the awake state, and a frontal alpha oscillation.
Figure 6.
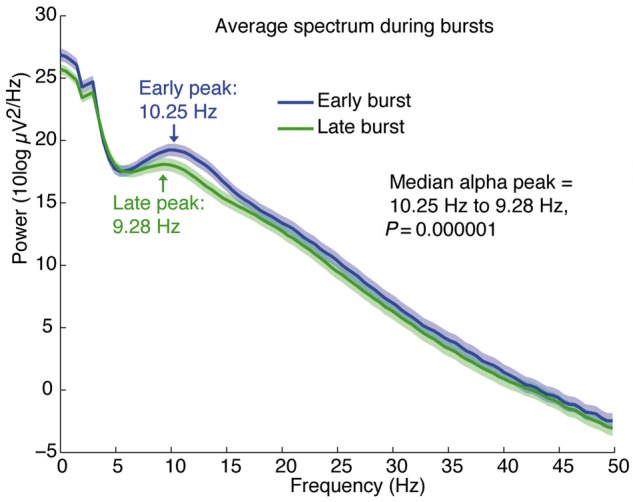
The alpha rhythm decelerates over the course of the burst. Average spectra across all channels with an alpha oscillation (n = 160 electrodes in five patients) show that there is a significant decrease in peak frequency between the early (0–1.5 s) and late (1.5–3 s) portions of a burst.
Segmentation of burst suppression
In each patient, a period of burst suppression was manually identified and extracted for further analysis. We used an automated method to segment bursts and suppressions (Supplementary Fig. 1). The method first required manual labelling of unambiguous suppression periods in the first 60 s of the recording. The data were then transformed in three steps: (i) signals were high pass filtered with a finite impulse response filter of length 2206, with a gain of 0 from 0–2.55 Hz and a gain of 1 from 3–125 Hz; (ii) the Hilbert transform of the transformed signal was used to calculate the instantaneous amplitude; and (iii) the instantaneous amplitude was smoothed with a moving average filter with a span of 50 samples (200 ms). These transformations yielded a continuous measure approximating high-frequency power. The value of this measure during the manually labelled suppression periods was used to set a threshold for burst detection [mean plus 4 standard deviations (SD) of the value during manually-labelled suppressions]. Threshold crossings lasting >500 ms were labelled as bursts, and burst terminations were labelled when the measure returned below threshold for 500 ms. We used 500 ms as a computational requirement for threshold crossings but manually confirmed that our method successfully detected the slow timescale shifts characteristic of burst suppression. In particular, we noted that the median duration of suppressions was 4.76 s, with an interquartile range of 3.76–7.31 s. To ensure that our results on burst timing were not an artefact of the burst detection algorithm, we implemented an alternative variance-based method. In this method, the variance of the raw signal was computed in 100 ms sliding windows and this measure replaced the instantaneous amplitude as the segmentation threshold.
Comparisons of burst timing
The difference in burst onset times was taken between every pair of electrodes in the grid. For each burst onset in a given electrode, the burst occurring closest in time in every other electrode was selected if it occurred within 1 s of the first burst. The absolute value of this timing difference was then calculated, and averaged across all pairs of electrodes in the grid. Timing differences were statistically compared across different distances of electrode separation using the Wilcoxon ranksum test.
The joint probability of bursting in two electrodes was computed for each pair of electrodes by calculating the amount of time that both electrodes were simultaneously in a burst state, and then normalizing by the total amount of time that either electrode was in a burst state. As above, significant changes in joint bursting probability at different distances were calculated using the Wilcoxon ranksum test.
Identification of local bursts
We plotted burst onsets across all channels and found that burst onsets were visibly clustered across channels (Supplementary Fig. 3), enabling an automated selection of multi-channel bursts using a simple threshold. Clusters of burst onsets were identified when at least five channels had a burst onset within a 200 ms bin. The number of channels involved in each burst was then computed by counting the number of channels that demonstrated a burst onset within 1.5 s of the main cluster, to ensure that all channels were counted even if burst onset was substantially delayed.
Spectral analysis of bursts
The spectral content of bursts was analysed using multitaper spectral estimation, computed with the Chronux toolbox (Bokil et al., 2010). Within-burst dynamics were analysed by selecting bursts lasting at least 3 s, and running a triggered spectral analysis at the onset of those bursts. Spectra were estimated with a T = 2-s window, a time-bandwidth product of TW = 3, and five tapers, yielding a spectral resolution of 1.5 Hz. An analogous calculation was performed on the baseline awake period by taking a triggered spectrum of an equal number of windows spaced 4 s apart. Error bars were computed as the standard error of the spectra across channels.
Comparisons of early and late portions of the burst were performed by selecting bursts lasting at least 3 s, and dividing them into two 1.5-s windows, marked ‘early’ and ‘late’. The analysis was restricted to channels with an alpha peak, defined as channels where the maximum power in the 8–14 Hz range was higher than the maximum power in the 4–7 Hz range. For each portion, the spectrum was calculated in a T = 1.5 s window, with time-bandwidth product TW = 4, with seven tapers, yielding a spectral resolution of 2.67 Hz. The peak alpha power was then identified as the frequency with the highest power lying between 8 and 14 Hz. Statistical testing was performed by identifying the difference between the early and late peak in each channel, and then performing a Wilcoxon signed-rank test on the difference across all channels. Plots show the average power across channels and error bars show the standard error.
Results
Spatially isolated states of burst suppression
We first examined the spatial dynamics of burst suppression in four subjects who each had a subdural grid of electrodes spaced 1 cm apart and spanning up to 11 cm of cortex (233 electrodes across four patients). This broad spatial sampling allowed us to examine how burst suppression dynamics varied across cortex. In contrast with the prevailing assumptions, we found clear cases in which the burst suppression pattern was localized. We computed the burst suppression probability across all grid electrodes, which ranges from 0 to 1 and indicates the probability of a suppression period at a given point in time (Chemali et al., 2011). We found that the standard deviation of the burst suppression probability across the recording sites ranged from 0.04 to as high as 0.2. Across the entire post-induction period in these four patients, the mean range of the burst suppression probability across electrodes at a given point in time was 0.46, demonstrating a large average difference in burst dynamics across channels. These measures indicated that burst suppression dynamics could often diverge substantially across different cortical areas, with different cortical regions exhibiting different propensities for suppression.
Given that the burst suppression probability could vary widely across cortex, we investigated whether the state of burst suppression itself could be limited to a restricted cortical region. We identified periods in which a subset of channels exited burst suppression, defined as an interval of at least 30 s in which the channel did not undergo a suppression. We found that in three of the four subdural grid patients, a subset of channels exited burst suppression while others remained in deep burst suppression, with burst suppression probabilities >0.5 (Fig. 1B and C, and Supplementary Fig. 2). To quantify the amount of time spent in spatially isolated burst suppression, we identified the total time over all patients in which some channels were deeply suppressed, whereas other channels remained nearly continuous. The epochs of deep suppression, defined as possessing any three channels with a burst suppression probability >0.5, amounted to 15.9 min. Of this time, 4.6 min (28.7%) included at least three other channels that had a burst suppression probability <0.2, that is, at least three channels that remained in a lighter state of suppression.
These findings demonstrated that one region of cortex can be in a state of burst suppression, while neighbouring cortical regions exhibit continuous activity characteristic of a lighter stage of anaesthesia. Burst suppression can therefore occur in limited cortical regions, and does not necessarily reflect a cortex-wide phenomenon.
Asynchronous bursts across cortex
We next asked whether there could be spatially isolated burst dynamics even when all of cortex is in burst suppression. Specifically, we examined the spatial distribution of individual bursts in order to test whether bursts were sometimes constrained to a limited cortical region, as was suggested by observation of raw traces (Fig. 1B). We analysed each burst individually and identified which grid channels participated in the burst by selecting those with nearby burst onset times (Supplementary Fig. 3). We found that bursts frequently occurred locally; bursts were observed in only a subset of the grid channels whereas other regions remained suppressed (Fig. 2A). These local bursts were interspersed with global bursts that occurred across all channels, demonstrating that the state of burst suppression was present across cortex but that individual bursts could occur asynchronously (Fig. 2A). The median percentage of channels involved in a single burst was only 76% (quartiles: 52–94%, Fig. 2B), demonstrating that local bursts made up a substantial portion of total bursting. We therefore concluded that bursts can be either global or local, and that local bursts reflect activation in a limited cortical area while other regions continue to be suppressed. The fact that suppressions can continue in one region despite high-amplitude bursts in a neighbouring region suggests that a profoundly inactivated state can be confined to specific cortical regions.
Timing of burst onsets varies across cortex
Despite the presence of spatially localized bursts, it is also clear that many bursts occur broadly across cortex, as nearly a third of bursts (31%) occurred in >90% of channels (Fig. 2B). We therefore tested whether these ‘global’ bursts across multiple channels began simultaneously in each channel, or whether there were consistent time lags between distant channels. We compared burst onset times between every pair of electrodes, and found that mean differences in burst onset time were significantly correlated with the distance between pairs of channels. Onset time differences were larger between more distant pairs of channels (Fig. 3B), increasing from 225 ± 83 ms (mean ± SD) in adjacent (1 cm) channels to 368 ± 107 ms (mean ± SD) in channels separated by >4 cm (difference = 143 ms, P < 10−5). Because the difference in burst times was correlated with distance (R = 0.30, P < 10−5), these timing differences could not be attributed simply to noise in the automated segmentation algorithm; even if the entire difference between adjacent channels was due to segmentation noise, an additional 143 ms would remain as the mean difference in burst timing between distant channels. To further ensure that these results were not an artefact of our burst detection algorithm, we repeated this analysis using the variance of the raw signal to detect burst onsets (see ‘Materials and methods’ section) (Löfhede et al., 2008), and replicated the correlation of timing differences with distance (difference = 153 ms, R = 0.27, P < 10−5). This result therefore demonstrated that even a single ‘global’ burst can be locally differentiated, and can begin hundreds of milliseconds apart in different cortical regions (Fig. 3A).
We further tested this spatial heterogeneity by computing the probability that two different electrodes would simultaneously be in a bursting state. We found that adjacent electrodes had a 78.1% probability of being in a burst state simultaneously, whereas more distant electrodes shared only a 62.0% chance of simultaneous bursting (Fig. 3C, P < 10−5). As with the differences in burst timing, this result demonstrated that burst onsets are asynchronous across cortex, with significant lags between distant cortical regions participating in a simultaneous burst.
Anatomically clustered burst dynamics
Taken together, these results demonstrate that there can be substantial heterogeneity in bursting dynamics across the cortex, and suggest that bursts are spatially clustered. To explicitly test for spatial clustering of bursts, we performed a principal components analysis on the burst state across all grid electrodes. We found that 78% of the variance could be explained by the first four components in each patient. To test whether spatial clustering was present in these first four principal components (across all four patients, a total of 16 components), we compared the spatial derivative of the estimated components to a randomly shuffled grid. This shuffling analysis demonstrated that 15 of the 16 components were significantly spatially clustered (P < 0.05, Fig. 4), supporting the hypothesis that clusters of anatomically close cortical areas tended to share burst properties. We therefore concluded that although burst suppression is sufficiently correlated across cortex to produce a seemingly synchronous pattern in scalp EEG recordings, the underlying dynamics exhibit substantial local heterogeneity.
Recovery of local dynamics within each burst
This demonstration of spatially differentiated dynamics suggested that bursts and suppressions depend on local cortical state. This finding could be compatible with a previously described model for the generation of burst suppression, which proposes that a depressed cerebral metabolism could lead to burst suppression by producing a slow cycle in ATP levels (Ching et al., 2012). This model makes specific predictions about the spectral content within individual bursts: first, that they will recover the dynamics of the state immediately preceding burst suppression, and second, that the recovered oscillatory features will decelerate through the course of each burst.
We therefore tested whether the spectral content of bursts returned to the pre-burst suppression state; in this case, a lighter stage of propofol general anaesthesia. Propofol general anaesthesia produces two striking features in the EEG: a large increase in low-frequency (0.1–4 Hz) power (Murphy et al., 2011; Lewis et al., 2012; Purdon et al., 2013) and an alpha (∼10 Hz) rhythm that occurs predominantly in frontal regions (Cimenser et al., 2011; Murphy et al., 2011; Supp et al., 2011; Purdon et al., 2013). We computed the within-burst spectrum across all electrocorticogram channels to test whether these features were present. As this analysis did not require spatial information, we expanded our data set to include an additional subject with implanted depth electrodes, as well as including all depth and strip electrodes from the first four patients (total of 374 electrodes in five patients). We classified each channel as ‘frontal’ if it was anterior to the central sulcus, as defined by visual inspection of reconstructed MRI images (Dykstra et al., 2012) and ‘posterior’ otherwise. Spectra were then calculated for the 2 s after burst onset, and averaged separately for frontal and posterior channels. The resulting spectra were highly consistent with previous reports of lighter stages of general anaesthesia: posterior channels had a strong power component at slow frequencies, and frontal channels had both increased slow power as well as a pronounced alpha oscillation (Fig. 5). Comparing these spectra to a baseline recording within the same patients before any anaesthesia was administered confirmed that these changes marked a departure from the awake state. Within-burst dynamics were therefore consistent with the dynamics observed during lighter stages of propofol general anaesthesia, in agreement with the prediction of the metabolism-based model.
Deceleration of frequency structure during bursts
Given that the propofol-induced alpha rhythm resumed during bursts, we next tested whether its frequency decelerated throughout a burst. We first selected all channels with a peak in power in the alpha band, defined as higher power in the alpha (8–14 Hz) band than in the theta (4–7 Hz) band. The time course of alpha dynamics was examined by comparing the early (0–1.5 s) and late (1.5–3 s) components of bursts, restricting the analysis to bursts lasting at least 3 s. The spectra showed that alpha rhythms did decelerate throughout a burst, with a peak frequency dropping from 10.25 Hz in the early period to 9.28 Hz in the late period of the burst (Fig. 6, P < 10−5). Burst dynamics were therefore not only variable across cortex, but also exhibited consistent patterns within a single burst, and these patterns aligned precisely with the predictions of the metabolism-based model.
Discussion
Local dynamics in burst suppression
In this paper we have shown local cortical dynamics in the state of burst suppression induced by propofol general anaesthesia. Specifically, our results establish that (i) bursts and suppressions can occur in a limited cortical region while continuous activity persists in other areas; (ii) even when all of cortex undergoes a ‘global’ burst, there are significant differences in the timing of onset of bursts between disparate cortical regions; (iii) within each burst, the frequency structure matches the brain state that was present before the onset of burst suppression; and (iv) this frequency structure decelerates through the course of each burst. Taken together, these findings suggest that burst suppression is highly dependent on local cortical dynamics, as the state evolves both across time and across different cortical areas.
Implications for neurological disease
Given that burst suppression is both a symptom of neurological conditions, for instance, in post-anoxic coma, and is induced as a treatment for conditions such as status epilepticus and traumatic brain injury, these findings could have a significant impact on clinical practice. In particular, detection of the spectral content within each burst could reveal the neural dynamics that remain intact when not interrupted by the suppression epochs. For instance, bursts may contain activity synonymous with general anaesthesia as observed here, or they could be morphologically similar to epileptiform patterns associated with seizure. Monitoring the transition between these two patterns could reveal the underlying brain state, and a shift in the spectral content of bursts could signal an opportunity to lift a pharmacologically induced coma. Similarly, tracking the complexity of burst dynamics could also aid in evaluation of coma recovery and of brain development in early neonates. Future clinical studies could examine in detail how the spectral content of the EEG during burst suppression may be useful for predicting patient outcomes.
In addition to monitoring spectral dynamics within a burst, the spatial heterogeneity of burst suppression has implications for our understanding of neurological disease, and could impact clinical treatment. First, these results indicate that patients exhibiting burst suppression may in fact have substantial local variation in brain function. Neurologists may therefore wish to examine spatial differences in burst suppression to ascertain whether specific cortical regions are more susceptible to circuit dysfunction, as inactivation in different brain structures may be a function of underlying pathology. Furthermore, these results suggest that medically-induced coma, as used for treatment of status epilepticus and traumatic brain injury, should be monitored across multiple cortical regions and the treatment adjusted accordingly, as dynamics in one brain region may not fully reflect the ongoing state. In addition, the ability to observe and characterize local expression of suppression epochs could allow for more precise tracking of anaesthetic induction and emergence, and of hypothermia induced during surgery (Michenfelder and Milde, 1991). Specialized monitoring systems could be designed that exploit EEG spatial patterns to enable superior control of drug dosages when inducing burst suppression to control status epilepticus or for treatment of traumatic brain injury, ensuring that a desired burst suppression ratio is achieved throughout the brain rather than at a single cortical site.
Balance between local and global dynamics
Burst suppression has previously been viewed as a global phenomenon, with synchronous bursts occurring simultaneously across cortical areas. In our studies, we indeed observed high correlation of bursts across cortex, demonstrating that on average bursts are broadly synchronous. However, we also identified substantial local variation in burst dynamics. Burst timing differed consistently across cortex, with larger timing offsets between bursts in distant regions. In addition, both bursts and suppressions frequently occurred locally, limited to a small cluster of electrodes while other cortical regions were in a different state. One possible explanation for this could be local variation in cerebral metabolism: when metabolism is globally depressed, bursts can spread across cortex, producing a gradient of timing differences; whereas when metabolic rates are more varied in different regions, they may enter dissociated states with different burst suppression probabilities and different refractory periods, leading to spatially isolated bursts and suppressions. This interaction could resolve the contrast inherent in these results, as this mechanism would produce dynamics in whereby bursts are often correlated but can nevertheless demonstrate substantial local variation.
Subcortical circuit mechanisms
Our data and results have centred on the cortical dynamics of burst suppression. However, subcortical structures are undoubtedly an important determinant in the expression of burst suppression in the brain. Indeed, the local differences we have shown in cortical measurements are suggestive of non-trivial subcortical participation in each burst and suppression. The state of burst suppression can be viewed as a severe reduction in the ability of cortical neurons to sustain continued processing. Whether the reason is protective, for instance by metabolic mechanisms, or otherwise, the neurons in question simply cannot fire for prolonged periods of time. In contrast, previous research on the cellular correlates of burst suppression has shown that certain subcortical populations, namely thalamic reticular and relay cells, may exhibit ongoing activity even during cortical suppressions (Steriade et al., 1994). The generation of individual bursts is thought to be caused by input from these relay neurons once cortical post-suppression refractory periods subside (Kroeger and Amzica, 2007). The extent to which burst suppression is expressed differentially in the cortex may thus be a reflection of the integrity of specific thalamocortical networks. In this scenario, the dynamic range in some subcortical loops—and the efficacy of ascending and descending excitation—can remain largely intact, despite existing in a significantly inactivated brain. These differences suggest that there are differential sensitivities of cortical regions and their associated functions to anaesthetic drugs at high concentrations, hypothermia and diffuse brain injury.
Relationship to neuronal and metabolic mechanisms
Our results are consistent with the neuronal and metabolic mechanisms proposed in recent computational work (Ching et al., 2012). In that model, it was suggested that lowered cerebral metabolism leads to periods of suppression, but that the activity within each burst recovers the oscillatory dynamics of the state preceding burst suppression. An alternative hypothesis is that bursts are due to cortical hyperexcitability (Amzica, 2009; Ferron et al., 2009). In the case of propofol general anaesthesia, the EEG before burst suppression contains two distinct rhythms: a slow (0.1–1 Hz) oscillation that is asynchronous across cortex, and an alpha (∼10 Hz) rhythm that is highly coherent across frontal electrodes (Supp et al., 2011; Lewis et al., 2012; Purdon et al., 2013). The slow oscillation contains EEG deflections that mark brief (<1 s) periods of local cortical neuron inactivation. These inactivated periods occur both during sleep (Cash et al., 2009; Nir et al., 2011) and general anaesthesia (Lewis et al., 2012), and correlate with loss of consciousness. In this study, we found that bursts indeed replicated the EEG signatures of lighter stages of general anaesthesia: they exhibited both a slow oscillation and a frontal alpha oscillation that decelerated throughout the burst, as predicted by the decreased cerebral metabolism model (Ching et al., 2012). The fact that slow oscillations were contained within bursts suggests that burst suppression may be due to prolonged epochs of suppression overriding the ongoing cortical state. Bursts would then reflect a transient recovery in which the oscillatory rhythms characteristic of the preceding state (i.e. the slow and alpha oscillations) resume. This theory is additionally consistent with the fact that patients remain anaesthetized during bursts, as their EEG continues to reflect the signatures of propofol general anaesthesia. Our analyses therefore suggest that the main emergent feature of burst suppression may in fact be the suppression, which acts as an intermittent but prolonged interruption of ongoing cortical activity. The burst content could then serve as a readout of the previous cortical state, which could provide useful clinical information when monitoring patients during burst suppression.
The spatial heterogeneity we observe here is also consistent with the metabolic model. In particular, it would follow from the model that bursts and suppressions may be shorter or longer in different brain regions depending on regional variations in perfusion, local network activity, ATP concentration, and metabolic state. These spatial results are also compatible with a calcium-based mechanism for burst suppression. Namely, it has been suggested that transient increases and decreases in extracellular calcium, leading to synaptic disfacilitation, are a key determinant in suppression duration (Amzica, 2009). Again, such a mechanism would naturally lead to local variability due to calcium distribution and expression. Taken together, our results support a model in which burst suppression is driven by local variations in cortical dynamics, and are consistent with the hypothesis that suppressions are caused by decreased cerebral metabolism (Ching et al., 2012). Nevertheless, our results do not definitively verify a mechanism for burst suppression, and future experiments combining cortical and subcortical recordings with direct metabolic measurements will be needed to determine the precise molecular mechanisms involved.
Future directions
There remain limitations in the current study that could be addressed by future work. In particular, the recordings were collected in patients with epilepsy, rather than in healthy subjects. However, the consistency of our results across patients, despite their heterogeneous clinical backgrounds, suggests that these findings are not an artefact of their clinical histories. In addition, our data replicate the burst suppression characteristics reported in previous studies, such as quasiperiodic bursts and increasing burst suppression probability with increasing anaesthetic dose, suggesting that the burst suppression dynamics in these patients do not substantially differ from those of the typical brain. Moreover, the majority of electrodes in this analysis were not immediately adjacent to the seizure focus, but rather in cortex that was presumed healthy and not resected during clinical treatment. A second consideration is that we have only analysed burst suppression induced by propofol general anaesthesia. Given the similarity of burst suppression across different aetiologies, we expect that these results would generalize to other neurological conditions that produce burst suppression, but future studies in patients with different aetiologies will be needed to verify this experimentally. In addition, future studies will be needed to investigate whether other causes of burst suppression (i.e. other anaesthetic drugs, coma and hypothermia) produce similar spatiotemporal dynamics to those observed here. A final limitation is the lack of scalp EEG recordings for comparison with the intracranial recordings. Future studies will be needed to determine how the spatial structure detected in intracranial electrodes translates to recordings at the scalp, which will be important for clinical applications.
Understanding brain function through burst suppression
Our findings provide new insight into the neurophysiology of the profoundly inactivated brain. Despite trends towards synchronous activity, local cortical dynamics vary across time and space, and can lead to uncoupled burst suppression states across cortex. These results demonstrate a previously unknown complexity in neural circuit dynamics during deep general anaesthesia, and suggest new roles for cortical and subcortical structures in producing neurophysiological diversity during profound neural inactivation. These findings indicate that burst suppression in neurological conditions could benefit from an examination of how cortical activity varies within bursts and across electrodes, as these dynamics may be highly variable. In addition, they suggest future clinical studies to investigate how analysis of the spatiotemporal structure of burst suppression patterns could improve patient monitoring and the effectiveness of clinical treatments.
Supplementary Material
Acknowledgements
We would like to thank A. Salazar-Gomez and A. Sampson for assisting with experiments, and A. Dykstra and A. Chan for help with electrode localization code.
Funding
This work was supported by a doctoral fellowship from the Canadian Institutes of Health Research to L.D.L., a Career Award at the Scientific Interface from the Burroughs-Wellcome Fund to S.C., and by the National Institutes of Health [Director’s Pioneer Award DP1OD003646 to E.N.B., NIH Transformative 1R01GM104948 to E.N.B., NIH New Innovator Award DP2-OD006454 to P.L.P.].
Supplementary material
Supplementary material is available at Brain online.
References
- Akrawi WP, Drummond JC, Kalkman CJ, Patel PM. A comparison of the electrophysiologic characteristics of EEG burst-suppression as produced by isoflurane, thiopental, etomidate, and propofol. J Neurosurg Anesthesiol. 1996;8:40–6. doi: 10.1097/00008506-199601000-00010. [DOI] [PubMed] [Google Scholar]
- Amzica F. Basic physiology of burst-suppression. Epilepsia. 2009;50:38–9. doi: 10.1111/j.1528-1167.2009.02345.x. [DOI] [PubMed] [Google Scholar]
- Bokil H, Andrews P, Kulkarni JE, Mehta S, Mitra PP. Chronux: a platform for analyzing neural signals. J Neurosci Methods. 2010;192:146–51. doi: 10.1016/j.jneumeth.2010.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown EN, Lydic R, Schiff ND. General anesthesia, sleep, and coma. N Engl J Med. 2010;363:2638–50. doi: 10.1056/NEJMra0808281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cash SS, Halgren E, Dehghani N, Rossetti AO, Thesen T, Wang C, et al. The human K-complex represents an isolated cortical down-state. Science. 2009;324:1084–87. doi: 10.1126/science.1169626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chemali JJ, Wong KF, Solt K, Brown EN. A state-space model of the burst suppression ratio. Conf Proc IEEE Eng Med Biol Soc. 2011;2011:1431–4. doi: 10.1109/IEMBS.2011.6090354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ching S, Purdon PL, Vijayan S, Kopell NJ, Brown EN. A neurophysiological-metabolic model for burst suppression. Proc Natl Acad Sci USA. 2012;109:3095–100. doi: 10.1073/pnas.1121461109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cimenser A, Purdon PL, Pierce ET, Walsh JL, Salazar-Gomez AF, Harrell PG, et al. Tracking brain states under general anesthesia by using global coherence analysis. Proc Natl Acad Sci USA. 2011;108:8832–7. doi: 10.1073/pnas.1017041108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claassen J, Hirsch LJ, Emerson RG, Mayer SA. Treatment of refractory status epilepticus with pentobarbital, propofol, or midazolam: a systematic review. Epilepsia. 2002;43:146–53. doi: 10.1046/j.1528-1157.2002.28501.x. [DOI] [PubMed] [Google Scholar]
- Clark DL, Rosner BS. Neurophysiologic effects of general anesthetics. I. The electroencephalogram and sensory evoked responses in man. Anesthesiology. 1973;38:564–82. [PubMed] [Google Scholar]
- Dykstra AR, Chan AM, Quinn BT, Zepeda R, Keller CJ, Cormier J, et al. Individualized localization and cortical surface-based registration of intracranial electrodes. Neuroimage. 2012;59:3563–70. doi: 10.1016/j.neuroimage.2011.11.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferron JF, Kroeger D, Chever O, Amzica F. Cortical inhibition during burst suppression induced with isoflurane anesthesia. J Neurosci. 2009;29:9850–60. doi: 10.1523/JNEUROSCI.5176-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall R, Murdoch J. Brain protection: physiological and pharmacological considerations. Part II: the pharmacology of brain protection. Can J Anaesth. 1990;37:762–77. doi: 10.1007/BF03006535. [DOI] [PubMed] [Google Scholar]
- Kroeger D, Amzica F. Hypersensitivity of the anesthesia-induced comatose brain. J Neurosci. 2007;27:10597–607. doi: 10.1523/JNEUROSCI.3440-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis LD, Weiner VS, Mukamel EA, Donoghue JA, Eskandar EN, Madsen JR, et al. Rapid fragmentation of neuronal networks at the onset of propofol-induced unconsciousness. Proc Natl Acad Sci USA. 2012;109:3377–86. doi: 10.1073/pnas.1210907109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Löfhede J, Löfgren N, Thordstein M, Flisberg A, Kjellmer I, Lindecrantz K. Classification of burst and suppression in the neonatal electroencephalogram. J Neural Eng. 2008;5:402–10. doi: 10.1088/1741-2560/5/4/005. [DOI] [PubMed] [Google Scholar]
- Michenfelder JD, Milde JH. The relationship among canine brain temperature, metabolism, and function during hypothermia. Anesthesiology. 1991;75:130–6. doi: 10.1097/00000542-199107000-00021. [DOI] [PubMed] [Google Scholar]
- Murphy M, Bruno M, Riedner B, Boveroux P, Noirhomme Q, Landsness E, et al. Propofol anesthesia and sleep: a high-density EEG study. Sleep. 2011;34:283–91. doi: 10.1093/sleep/34.3.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nir Y, Staba RJ, Andrillon T, Vyazovskiy VV, Cirelli C, Fried I, et al. Regional slow waves and spindles in human sleep. Neuron. 2011;70:153–69. doi: 10.1016/j.neuron.2011.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohtahara S, Yamatogi Y. Epileptic encephalopathies in early infancy with suppression-burst. J Clin Neurophysiol. 2003;20:398–407. doi: 10.1097/00004691-200311000-00003. [DOI] [PubMed] [Google Scholar]
- Purdon PL, Pierce ET, Mukamel EA, Prerau MJ, Walsh JL, Wong KF, et al. Electroencephalogram signatures of loss and recovery of consciousness from propofol. Proc Natl Acad Sci USA. 2013;110:E1142–51. doi: 10.1073/pnas.1221180110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossetti AO, Reichhart MD, Schaller P, Despland PA, Bogousslavsky J. Propofol treatment of refractory status epilepticus: a study of 31 episodes. Epilepsia. 2004;45:757–63. doi: 10.1111/j.0013-9580.2004.01904.x. [DOI] [PubMed] [Google Scholar]
- Stecker MM, Cheung AT, Pochettino A, Kent GP, Patterson T, Weiss SJ, et al. Deep hypothermic circulatory arrest: II. Changes in electroencephalogram and evoked potentials during rewarming. Ann Thorac Surg. 2001;71:22–8. doi: 10.1016/s0003-4975(00)02021-x. [DOI] [PubMed] [Google Scholar]
- Steriade M, Amzica F, Contreras D. Cortical and thalamic cellular correlates of electroencephalographic burst-suppression. Electroencephalogr Clin Neurophysiol. 1994;90:1–16. doi: 10.1016/0013-4694(94)90108-2. [DOI] [PubMed] [Google Scholar]
- Supp GG, Siegel M, Hipp JF, Engel AK. Cortical hypersynchrony predicts breakdown of sensory processing during loss of consciousness. Curr Biol. 2011;21:1988–93. doi: 10.1016/j.cub.2011.10.017. [DOI] [PubMed] [Google Scholar]
- Young GB. The EEG in coma. J Clin Neurophysiol. 2000;17:473–85. doi: 10.1097/00004691-200009000-00006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.