

This study identified a novel strategy to enhance cardiac differentiation of human induced pluripotent stem (iPS) cells by treating embryoid bodies with a histone deacetylase inhibitor, trichostatin A (TSA). It was found that cardiomyogenic differentiation of human iPS cells can be enhanced by TSA, and the cardiomyocytes so derived can be used to engineer vascularized, three-dimensional contracting tissue. This tissue could be used for studying the pathophysiology of cardiac disease, for drug discovery test beds, and potentially for generation of cardiac grafts to surgically replace damaged myocardium.
Keywords: Trichostatin A, Induced pluripotent stem cell, Cardiac differentiation, Cardiac tissue engineering
Abstract
Human induced pluripotent stem (iPS) cells are a promising source of autologous cardiomyocytes to repair and regenerate myocardium for treatment of heart disease. In this study, we have identified a novel strategy to enhance cardiac differentiation of human iPS cells by treating embryoid bodies (EBs) with a histone deacetylase inhibitor, trichostatin A (TSA), together with activin A and bone morphogenetic protein 4 (BMP4). Over a narrow window of concentrations, TSA (1 ng/ml) directed the differentiation of human iPS cells into a cardiomyocyte lineage. TSA also exerted an additive effect with activin A (100 ng/ml) and BMP4 (20 ng/ml). The resulting cardiomyocytes expressed several cardiac-specific transcription factors and contractile proteins at both gene and protein levels. Functionally, the contractile EBs displayed calcium cycling and were responsive to the chronotropic agents isoprenaline (0.1 μM) and carbachol (1 μM). Implanting microdissected beating areas of iPS cells into tissue engineering chambers in immunocompromised rats produced engineered constructs that supported their survival, and they maintained spontaneous contraction. Human cardiomyocytes were identified as compact patches of muscle tissue incorporated within a host fibrocellular stroma and were vascularized by host neovessels. In conclusion, human iPS cell-derived cardiomyocytes can be used to engineer functional cardiac muscle tissue for studying the pathophysiology of cardiac disease, for drug discovery test beds, and potentially for generation of cardiac grafts to surgically replace damaged myocardium.
Introduction
Ischemic heart disease is the leading cause of mortality and morbidity in the world [1]. Stem cell medicine has the potential to provide an alternative to conventional treatments for myocardial infarction and heart failure. The pluripotency of stem cells may be exploited for repair and regeneration of the injured heart. Induced pluripotent stem (iPS) cells are an ideal source for cardiomyocytes because they have the ability to self-renew while retaining the potential to differentiate into all cell types of the body, including cardiomyocytes [2, 3]. Furthermore, patient-specific iPS cells could offer an autologous source of pluripotent stem cells for personalized therapeutic strategies to circumvent immunological issues such as rejection of transplants.
Cardiomyogenesis is a highly organized process regulated by important signaling pathways such as transforming growth factor-β family and wingless-type mouse mammary tumor virus integration site (Wnt) proteins [4, 5]. It was also recently shown that small molecules can induce cardiac differentiation of human iPS cells [6]. In previous studies, it was reported that the treatment of murine and monkey embryonic stem cells [7, 8] and murine iPS cells [9] with trichostatin A (TSA), a histone deacetylase (HDAC) inhibitor, induces the acetylation of GATA4 and directs their differentiation into cardiomyocytes. However, the cardiogenic potential of TSA has not been tested in human pluripotent stem cells. Trichostatin A belongs to the hydroxamic acid class of HDAC inhibitors and was selected because of its potency and pan-HDAC selectivity [10]. Here we investigated the effect of TSA on cardiomyogenic differentiation of human iPS cells through the approach of embryoid body (EB) formation, and we showed that we can engineer vascularized human cardiac muscle tissue in vivo using cardiomyocytes derived from iPS cells.
Materials and Methods
Animals
Experimental procedures were approved by the Human Research Ethics Committee of the University of Melbourne (#829937 and #839910) and the Animal Ethics Committee of St. Vincent's Hospital (AEC 025/10; Melbourne, Victoria, Australia) and were conducted in accordance with the Australian National Health and Medical Research Council guidelines for the care and maintenance of animals. Human tissue sample collection was approved by the Human Research Ethics Committee-A of St. Vincent's Hospital (HREC-A 07/08). Male nude rats (CBH-rnu) were purchased from Animal Resources Centre (Perth, Western Australia, Australia, http://www.arc.wa.gov.au), maintained with a 12-hour dark/light cycle, and given water and chow ad libitum.
Human iPS Cell Culture
iPS(Foreskin)-1 and iPS(Foreskin)-2 cell lines were obtained from James A. Thomson (University of Wisconsin) [3]. The human Friedreich's ataxia iPS cell line FA3 was also used in this study [11]. iPS(Foreskin)-1 (passages 38–74), iPS(Foreskin)-2 (passages 23–60), and FA3 (passages 16–19) were maintained on a feeder layer of mitotically inactivated human foreskin fibroblasts (HFF:D551; American Type Culture Collection, Manassas, VA, http://www.atcc.org) in culture media containing Dulbecco's modified Eagle's medium (DMEM)/F-12 GlutaMAX medium supplemented with 20% knockout serum replacement, 0.1 mM 2-mercaptoethanol, 0.1 mM nonessential amino acids, 50 U/ml penicillin/streptomycin (all from Invitrogen, Carlsbad, CA, http://www.invitrogen.com), and 20 ng/ml recombinant human fibroblast growth factor-2 (Millipore, Billerica, MA, http://www.millipore.com).
Cardiac Differentiation
Spontaneous differentiation of iPS cells was induced through formation of EBs. EBs were formed by mechanically dissecting undifferentiated iPS cell colonies into approximately 0.2 mm2 pieces using the sharp edge of a flame-pulled capillary under the stereomicroscope. Pieces were transferred onto low-attachment plates and cultured in suspension in differentiation media containing DMEM/F-12 GlutaMAX medium supplemented with 20% fetal bovine serum, 0.1 mM 2-mercaptoethanol, 0.1 mM nonessential amino acids, and 50 U/ml penicillin/streptomycin, where they aggregated to form EBs over 6 days. On day 6 (day 0 post-plating), EBs were transferred to tissue culture plates precoated with 0.1% gelatin and 10 μg/ml fibronectin (both from Sigma-Aldrich, St. Louis, MO, http://www.sigmaaldrich.com), and cultured in differentiation media. The percentage of contractile EBs was measured as the number of EBs that showed spontaneous contraction divided by the total number of EBs plated.
Cardiac differentiation was directed by supplementation of media during EB formation with the following factors: (a) 0.5–50 ng/ml TSA (Sigma-Aldrich) for 6 days; (b) 100 ng/ml human recombinant activin A for 1 day, followed by 20 ng/ml human recombinant bone morphogenetic protein 4 (BMP4) (both from PeproTech, Rocky Hill, NJ, http://www.peprotech.com) for 5 days (AB); and (c) activin A, BMP4, and TSA (1 ng/ml, or 3.3 nM) (AB + TSA) (Fig. 1A). Media were changed every 2–3 days.
Figure 1.

TSA enhances cardiac differentiation and histone acetylation of human induced pluripotent stem (iPS) cells. (A): Treatment timeline of iPS cells for cardiac differentiation. (B): Concentration-dependent effect of TSA (n = 5–6). (C): Western blot analysis of histone acetylation induced by TSA (1 ng/ml for 6 days) (n = 3). *, p < .05; **, p < .01; ***, p < .001; ****, p < .0001 by two-way analysis of variance with the Bonferroni post hoc test. Abbreviations: BMP4, bone morphogenetic protein 4; D, day; EB, embryoid body; TSA, trichostatin A.
RNA Extraction and Quantitative Polymerase Chain Reaction
RNA was extracted from undifferentiated iPS cells, beating EBs, and nonbeating EBs using TriReagent (Invitrogen) followed by RNA precipitation with chloroform and isopropanol (both from Sigma-Aldrich). cDNA was synthesized using the high-capacity cDNA reverse transcription kit (Applied Biosystems, Foster City, CA, http://www.appliedbiosystems.com). A negative control with reverse transcriptase omitted was performed to check the absence of genomic DNA. A quantitative polymerase chain reaction (qPCR) was carried out using TaqMan Universal master mix, the 7900HT Fast Real-Time PCR system, and TaqMan gene expression assays (Applied Biosystems) for GAPDH (Hs03929097_g1), cardiac α-actin (ACTC1; Hs01109515_m1), troponin T (TNNT2; Hs01109515_m1), troponin I (TNNI3; Hs00165957_m1), myosin light chain atria isoform (MYL7; Hs01085598_g1), β-myosin heavy chain (MYH7; Hs00165276_m1), NKX2.5 (Hs00231763_m1), MEF2C (Hs00231149_m1), paired box protein 6 (PAX6, Hs_01088112_m1), and α-fetoprotein (AFP; Hs00173490_m1). All readings were performed in triplicate. The relative quantitation was achieved by applying the comparative CT method (ΔΔCt) whereby the mRNA expression levels were normalized against the level of GAPDH mRNA (ΔCt) with the level of candidate genes in undifferentiated iPS cell samples used as the reference genes (ΔΔCt). qPCR of non-reverse-transcribed sample was also conducted for each TaqMan probe listed above to check the genomic DNA contamination in the sample and the specificity of the probes. RNA extracted from human cardiac tissue served as a positive control.
Immunocytochemistry
For quantitative assessment of cardiomyocyte differentiation, spontaneously beating colonies at 7 days post-plating were trypsinized into a single cell suspension with 0.25% trypsin-EDTA and spun onto coated glass slides (4 minutes at 900 rpm; Shandon Cytospin 4; Thermo Fisher Scientific, Waltham, MA, http://www.thermofisher.com). For qualitative analysis, beating colonies at 7–10 days post-plating were dissociated into small clumps with 0.025% trypsin-EDTA (Invitrogen) for 10 minutes at 37°C followed by 0.25% trypsin-EDTA for 3–5 minutes at room temperature with constant agitation. Mixtures of single cells and clumps were seeded on coated eight-chamber slides for 5–7 days in differentiation media.
Cells were fixed with 4% paraformaldehyde (PFA) for 10 minutes and permeabilized with ice-cold ethanol for 10 minutes. Cells were then incubated with a serum-free blocking solution (Thermo Fisher Scientific) for 10 minutes prior to incubation with antibodies against cardiac troponin I (cTnI) (1 μg/ml, mouse monoclonal IgG; Millipore), cardiac troponin T (cTnT) (2 μg/ml, mouse monoclonal IgG; Abcam, Cambridge, MA, http://www.abcam.com), α-actinin (25 μg/ml, mouse monoclonal IgG, A7811; Sigma-Aldrich), or myosin heavy chain (2 μg/ml, mouse monoclonal IgG, ab15; Abcam) at 4°C overnight followed by Alexa Fluor 488 goat anti-mouse IgG (10 μg/ml; Invitrogen) for 60 minutes at room temperature. Cells were counterstained with 1 μg/ml 4′,6-diamidino-2-phenylindole (DAPI) (Invitrogen) for nuclear staining and mounted with fluorescence mounting agent (Dako, Campbellfield, Victoria, Australia, http://www.dako.com). Specificity of the staining was verified by the appropriate negative control immunoglobulin fraction. Images were taken using a fluorescence microscope (BX-61 microscope; Olympus, Tokyo, Japan, http://www.olympus-global.com), and at least 500 cells counted were from 10 areas in three to five independent experiments.
Video Microscopy and Calcium Imaging
Spontaneous contraction of cells was observed under phase contrast microscopy using an inverted microscope (IX71; Olympus) with heated stage (ThermoPlate; Tokai Hit, Fujinomiya-shi, Shizuoka-ken, Japan, http://www.tokaihit.com). Intracellular free calcium (Ca2+) imaging was performed in beating cells loaded with fluorescent Ca2+ indicator Fluo-4 AM (Invitrogen). Cells were incubated with 2 μg/ml Fluo-4 AM for 20 minutes in differentiation media at 37°C. Gray scale images were captured (15 frames per second) on an inverted microscope with a fluorescence source (X-Cite 120Q; Lumen Dynamics, Mississauga, Ontario, Canada, http://www.ldgi.com) and a digital camera (DP72; Olympus). Fluo-4 fluorescence was visualized with a 492 nm excitation beam (excitation filter BP492/18 with a xenon light source).
Microelectrode Array Recordings
The electrophysiological properties of the iPS cell-derived cardiomyocytes were assessed using a microelectrode array (MEA) recording system (Multichannel Systems, Reutlingen, Germany, http://www.multichannelsystems.com). With the contracting attached EBs, the beating cell clusters were microdissected at day 7 post-plating and transferred onto MEA plates coated with 0.1% gelatin and 10 μg/ml fibronectin. Responsiveness to pharmacological agents was determined 3–4 days later at 37°C in Krebs-Ringer buffer (composition in mM: 125 NaCl, 5 KCl, 1 Na2HPO4, 1 MgSO4, 20 HEPES, 5.5 glucose, 2 CaCl2; pH 7.4). The responsiveness of cells to the following agents was tested: isoproterenol hydrochloride (0.1 μM), carbamylcholine (1 μM), and nifedipine (0.1 μM) (all from Sigma-Aldrich). Each cell cluster was treated with all drugs in random order, and cells were allowed to recover to their baseline contraction in fresh Krebs-Ringer buffer between drug treatments. Extracellular field potentials were recorded at baseline and 2 minutes after the addition of the drugs. Data were analyzed offline with MC Rack software, version 4.3.5 (Multichannel Systems), for beating rate, RR interval, and extracellular field potential duration (FPD). RR interval was defined as the time elapsing between two consecutive beats, and FPD was defined as the time interval between the initial deflection of the field potential and the maximal local T wave. To avoid the influence of beat frequency on FPD, FPD measurements were normalized (corrected field potential duration [cFPD]) with the Bazzet correction formula: cFPD = FPD/√(RR interval) [12]. Data were expressed as percentage change from baseline, with the baseline set to 100%.
Detection of Histone Acetylation
Core histones of untreated or TSA (1 ng/ml for 6 days)-treated EBs were isolated and purified using a commercial histone purification kit (Active Motif, Carlsbad, CA, http://www.activemotif.com/) according to the manufacturer's instruction. Protein samples were mixed with Tris-glycine SDS sample buffer (Novex; Invitrogen) and boiled for 10 minutes. Five micrograms of histone proteins were fractionated using 12% Bis-Tris gel system (NuPAGE Novex; Invitrogen) by electrophoresis in 2-(N-morpholino)ethanesulfonic acid SDS running buffer (Invitrogen) at 180 V constant charge for 40 minutes and transferred onto a nitrocellulose membrane (Hybond-C Extra; Amersham Biosciences, Little Chalfont, Buckinghamshire, U.K., http://www.amersham.com) using a wet transfer method (Xcell II Blot Module; Invitrogen). The transfer of the protein was completed with 30 V constant charge for 60 minutes. The membrane was blocked in 5% skim milk in Tris-buffered saline containing 0.1% Tween-20 (TBS/T) for 1 hour at room temperature before overnight incubation with acetyl-histone H2A (Lys5), histone H2A, acetyl-histone H2B (Lys5), histone H2B, acetyl-histone H3 (Lys9), histone H3, acetyl-histone H4 (Lys8), or histone H4 antibodies (1:1,000; Cell Signaling Technology, Beverly, MA, http://www.cellsignal.com) in TBS/T with 3% bovine serum albumin (Sigma-Aldrich) at 4°C followed by Alexa Fluor 680 goat anti-rabbit or Alexa Fluor 680 goat anti-mouse antibodies (0.2 μg/ml; Invitrogen) for 1 hour at room temperature in the dark. The membrane was scanned using an Odyssey system (Li-Cor, Lincoln, NE, http://www.licor.com) at a 700-nm wavelength, and the relative protein level was quantified with densitometry using NIH ImageJ software.
In Vivo Vascularized Tissue Engineering Chamber
An in vivo tissue engineering chamber was prepared in the groin region of adult male nude rats weighing between 200 and 250 g as previously described [13]. Briefly, rats were anesthetized with isoflurane inhalation (4% induction and 2% maintenance). The femoral vessels were exposed through a longitudinal incision made on the medial thigh. The femoral artery and vein were separated from their surrounding tissue attachments, and small branches to the thigh musculature were cauterized to isolate the length of the vessels from the inguinal ligament to the branch point of the deep vein. Polyacrylic chambers (internal dimensions, 10 × 8 × 4 mm; Department of Chemical and Biomolecular Engineering, University of Melbourne, Melbourne, Victoria, Australia) were placed around the femoral vessels, by passing the intact vascular pedicle through slits on either end of the chamber, creating a flow-through model with intact femoral circulation. Lids were attached to the chamber base to create a protected volume for tissue growth. Skin wounds were then closed in two layers, and the animals were allowed to recover.
Beating and nonbeating cell clusters were microdissected at day 7 post-plating and separated into two wells of coated six-well plates. Cell clusters were suspended in 200 μl of rat plasma as the carrier matrix, and 30 μl of 2% calcium chloride was added to form a plasma clot in the sterilized chamber before implantation into the nude rat. Plasma clot was then folded over the femoral vessels to maximize the contact with the vessels. Each rat was implanted with two chambers (left and right femoral vessels) containing 40–80 either spontaneously beating or nonbeating clusters (approximately 50,000–100,000 cells) pooled together from two consecutive cultures.
Harvest of Chamber Tissue and Tissue Processing
Four weeks following implantation, rats were anesthetized and the chambers were opened for photography and video recording. Tissue constructs formed in the chambers were fixed in 4% PFA, divided into three equal transverse sections (distal, middle, and proximal), and paraffin embedded into the same block.
Immunohistochemistry
Paraffin-embedded 5-μm-thick sections were stained with hematoxylin and eosin for morphological assessment. Immunohistochemistry was performed to detect cardiac tissue with cTnT (4 μg/ml), α-actinin (125 μg/ml), and myosin heavy chain (4 μg/ml); gap junction with connexin 43 (7.5 μg/ml, mouse monoclonal IgM, ab11369; Abcam); endoderm lineage with human-specific α-fetoprotein (4.4 μg/ml, rabbit polyclonal IgG, A0008; Dako); ectoderm lineage with human-specific nestin (0.5 μg/ml, rabbit monoclonal IgG, ab105389; Abcam); human-derived cells with human mitochondria (2 μg/ml, mouse monoclonal IgG, E5204; Spring Bioscience, Pleasanton, CA, http://www.springbio.com); and blood vessels with human-specific CD31 (2.12 μg/ml, mouse monoclonal IgG, M0823; Dako) or biotinylated Griffonia simplicifolia isolectin B4 (1.67 μg/ml, B1205; Vector Laboratories, Burlingame, CA, http://www.vectorlabs.com). Sections were then incubated with the appropriate biotinylated secondary antibodies: rabbit anti-mouse (2.9 μg/ml, E0464; Dako) or goat anti-rabbit (7.5 μg/ml, BA-1000; Vector Laboratories), followed by streptavidin-biotinylated-peroxidase complex (Vectastain Elite ABC Standard, PK6100; Vector Laboratories). Peroxidase activity was visualized with diaminobenzidine (Thermo Scientific, Waltham, MA, http://www.thermoscientific.com), and sections were counterstained with hematoxylin before mounting. Streptavidin alkaline phosphatase (2 μg/ml; Millipore) and fast red (Sigma-Aldrich) were used to detect isolectin B4 staining. For immunofluorescence, sections were costained with cTnT and fluorescein-conjugated G. simplicifolia lectin (8 μg/ml, FL1101; Vector Laboratories), followed by a rabbit anti-mouse Dylight 549-streptavidin secondary antibody (1 μg/ml, 016-500-084; Jackson Immunoresearch Laboratories, West Grove, PA, http://www.jacksonimmuno.com) and then DAPI counterstaining. z-projections of high-resolution images at maximum intensity were taken with a laser scanning confocal microscope (A1R; Nikon, Tokyo, Japan, http://www.nikon.com). The appropriate IgG antibodies (rabbit or mouse) were used as negative controls. Rat and human heart tissues were used as positive controls.
Statistics
All sets of experiments were performed at least three times (n refers to the number of independent experiments unless specified). Data sets were expressed as mean ± SEM. The significance of the differences was evaluated using the unpaired Student's t test, and one-way or two-way analysis of variance for multiple comparisons with either Dunnett's or Bonferroni post hoc analyses where appropriate. p < .05 was considered statistically significant.
Results
Directed Cardiac Differentiation of Human iPS Cells With TSA
We used the EB formation method to induce spontaneous differentiation of iPS cells [14, 15]. In the iPS(Foreskin)-2 cell line, spontaneous myogenic contractions appeared as early as 1 day post-plating, yet spontaneous differentiation into beating cardiomyocytes was low in efficiency (3.8 ± 2.2% of EBs at 14 days; Fig. 1B). Treatment of iPS cells with TSA (1 ng/ml) for 6 days during EB formation significantly increased the percentage of beating colonies at day 14 post-plating (15.8 ± 3.5%, p < .05; Fig. 1B). TSA treatment at 1 ng/ml significantly increased the acetylation of histone-2B, histone-3, and histone-4 but not histone-2A (Fig. 1C). TSA at lower or higher concentrations failed to promote cardiac differentiation of iPS cells and was cytotoxic at 50 ng/ml. The percentage of beating EBs in all groups peaked around day 7 post-plating (Fig. 1B). The cardiogenic effects of TSA were confirmed in two other human iPS cell lines, iPS(Foreskin)-1 and FA3 (supplemental online Fig. 1).
Additive Effects of TSA and Growth Factors on Cardiac Differentiation
The optimal dose of TSA (1 ng/ml) was combined with established mesodermal differentiation factors, activin A (100 ng/ml) and BMP4 (20 ng/ml) (AB), to determine their combined effects on cardiac differentiation. In the iPS(Foreskin)-2 cell line, AB-directed cardiac differentiation resulted in 21.3 ± 5.8% of colonies beating spontaneously after 7 days (compared with 10.3 ± 2.7% in control), hence showing an efficiency similar to that produced by TSA treatment alone (Fig. 2A). Importantly, the percentage of beating EBs was increased additively in all three cell lines examined when TSA was combined with AB (48.4 ± 7.4% in iPS(Foreskin)-2, p < .0001; Fig. 2A; supplemental online Fig. 1). Cardiomyocytes derived from AB + TSA-treated EBs were positive for troponin I, troponin T, cardiac α-actin, and myosin heavy chain (Fig. 2B).
Figure 2.
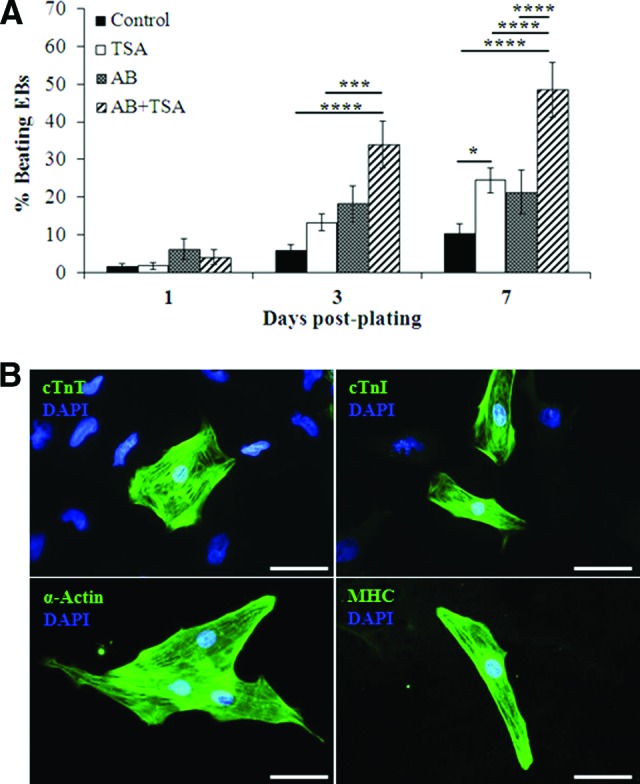
TSA enhances the cardiogenic effect of activin-A and bone morphogenetic protein 4 (BMP4). (A): Additive effect of TSA, and activin-A and BMP4 on directing cardiac differentiation (n = 8–11). (B): Immunocytochemistry for cTnI, cTnT, α-actinin, and myosin heavy chain (magnification, ×400). Scale bars = 50 μm. ***, p < .001; ****, p < .0001 by two-way analysis of variance with the Bonferroni post hoc test. Abbreviations: AB, activin A and bone morphogenetic protein 4; cTnI, cardiac troponin I; cTnT, cardiac troponin T; DAPI, 4′,6-diamidino-2-phenylindole; EB, embryoid body; MHC, myosin heavy chain; TSA, trichostatin A.
The percentage of cardiomyocytes within beating patches was similar among all treatment groups (Fig. 3A) and similar in the iPS(Foreskin)-1 and FA3 cell lines (supplemental online Fig. 2). Cotreatment with AB and TSA resulted in 9.1 ± 1.0% and 9.1 ± 1.7% of cTnT- and cTnI-positive cardiomyocytes, respectively, in contracting EBs at 7 days post-plating. When compared with undifferentiated iPS cells, qPCR analysis confirmed directed cardiac differentiation of iPS(Foreskin)-2 cells by the combination of AB and TSA, where expression of genes encoding the cardiac-restricted transcription factors MEF2C (Fig. 3B), GATA4 (Fig. 3C), and NKX2.5 (Fig. 3D) as well as the cardiac-specific structural and contractile proteins α-actin ACTC1 (Fig. 3E), TNNT2 (Fig. 3F), TNNI3 (Fig. 3G), MYL7 (Fig. 3H), and MYH7 (Fig. 3I) was significantly upregulated in beating EBs. This increase in cardiac-specific genes was absent in nonbeating EBs. The expression levels of genes coding for the cardiac progenitors MEF2C (Fig. 2B), GATA4 (Fig. 3C), and NKX2.5 (Fig. 3D) as well as contractile filament proteins ACTC1 (Fig. 2E), TNNT2 (Fig. 3F), MYL7 (Fig. 3H), and MYH7 (Fig. 3I) were also significantly higher in beating EBs derived from the AB + TSA group compared with control-beating EBs.
Figure 3.

Quantitative analysis of cardiomyogenic specification of human induced pluripotent stem (iPS) cells. (A): Percentage of cardiac troponin-positive cells in beating EBs at day 7 post-plating (n = 6–8 from three to five independent experiments). Right: Representative photomicrograph (magnification, ×200) of cells stained with cTnT, cTnI, and DAPI for nuclei. Scale bars = 100 μm. (B–I): Reverse transcription-polymerase chain reaction analysis of cardiac-specific gene expression markers at day 7 post-plating (n = 3–4, triplicate readings). *, p < .05; **, p < .01; ***, p < .001; ****, p < .0001 versus control. #, p < .05; ##, p < .01; ###, p < .001; ####, p < .0001 versus undifferentiated iPS cells by one-way analysis of variance with the Bonferroni post hoc test. Abbreviations: AB, activin A and bone morphogenetic protein 4; cTnI, cardiac troponin I; cTnT, cardiac troponin T; DAPI, 4′,6-diamidino-2-phenylindole; TSA, trichostatin A; Un, undifferentiated iPS cells.
Electrophysiological Responsiveness of Human iPS Cell-Derived Cardiomyocytes
Beating cells derived from iPS cells were cycling calcium (supplemental online Video 1). To determine whether human iPS(Foreskin)-2 cell-derived cardiomyocytes exhibit functional changes in membrane potential, microdissected beating areas from control and AB + TSA treated iPS(Foreskin)-2 were plated onto MEA. Extracellular field potentials measured by MEA were produced by action potential activities in cardiomyocytes [16]. The extracellular field potentials recorded were reproducible with different beating pieces but with variable signal amplitude, possibly because of impedance of the contact between cells and electrodes (Fig. 4A). Nonetheless, there was little heterogeneity in beating frequency across different locations within a single EB and even between different beating pieces on the same MEA (Fig. 4A).
Figure 4.

Electrophysiological properties of cardiomyocytes derived from human induced pluripotent stem cells treated with TSA and AB. (A): Microelectrode array with beating pieces and the extracellular field potential recorded. (B, C): The percentage change in beating rate (B) and cFPD (C) of cardiomyocytes derived from control or AB + TSA groups treated with isoproterenol hydrochloride (isoprenaline, 0.1 μM), carbamylcholine (carbachol, 1 μM), and nifedipine (0.1 μM). n = 4–6. *, p < .05 by paired t test. Abbreviations: AB, activin A and bone morphogenetic protein 4; cFPD, corrected field potential duration; TSA, trichostatin A.
The average contraction rate of the derived cardiomyocytes was 50 ± 4 bpm in AB + TSA groups and 59 ± 3 bpm in control (p > .05). In the AB + TSA group, the β-adrenoreceptor agonist isoproterenol (0.1 μM) increased the spontaneous beating frequency by 40%, and the muscarinic agonist carbamylcholine (1 μM) reduced the spontaneous beating rate of iPS cell-derived cardiomyocytes without affecting the field potential duration (corresponding to the action potential duration and QT interval of an electrocardiogram in vivo) of the cardiomyocytes (Fig. 4B, 4C). In contrast, cardiomyocytes derived from the control group did not exhibit chronotropic response to isoproterenol (0.1 μM) and carbamylcholine (1 μM). Nifedipine (an L-type calcium channel inhibitor) at 0.1 μM significantly reduced the beating frequency and field potential duration in control group, whereas in the AB + TSA group the field potential duration but not the beating frequency was significantly reduced by nifedipine (Fig. 4B, 4C).
Engineered Vascularized Human Cardiac Tissue
An in vivo tissue engineering approach was used to determine whether functional cardiac tissues could be generated from EBs containing human iPS cell-derived cardiomyocytes: these were enriched as microdissected, spontaneously beating clusters derived from iPS(Foreskin)-2 cells differentiated with the combined TSA and AB protocol (supplemental online Video 2). iPS(Foreskin)-2 cell line was selected for this study because of its higher potential to differentiate into cardiomyocytes than iPS(Foreskin)-1 or FA3 cell lines (Fig. 2; supplemental online Fig. 1). Microdissected, beating iPS cell clusters showed higher mRNA expression of cardiac-specific transcription factors and proteins than whole beating EBs (supplemental online Fig. 3). Four weeks postimplantation, tissue constructs generated from beating iPS cell clusters showed spontaneous focal beating independently of the host heart rate (two of three constructs) (Fig. 5C; supplemental online Video 3), whereas implanting nonbeating iPS cell clusters produced constructs devoid of spontaneous beating.
Figure 5.

Cardiac tissues engineered from human induced pluripotent stem (iPS) cell-derived cardiomyocytes. (A): The polyacrylic tissue engineering chamber placed around the femoral artery and vein in the groin region of a nude rat. A plasma clot containing human iPS cells (arrow) was implanted at the base of the chamber and folded over the femoral vessels (B). (C): Engineered tissue construct at 4 weeks postimplantation. (D–G): Cardiac tissue constructs were stained with cardiac troponin T (D, E), α-actinin (F), and myosin heavy chain (G). (H, I): Serial sections stained with cTnT (H) and connexin 43 (I). Magnification, ×400 (D, E, H) and ×200 (F, G). Scale bars = 5 mm (A–C) and 50 μm (D–I). Abbreviations: a, femoral artery; cTnT, cardiac troponin T; v, femoral vein.
Immunostaining with cardiac troponin T (Fig. 5D, 5E), α-actinin (Fig. 5F), and cardiac myosin heavy chain (Fig. 5G) showed implanted human cardiomyocytes not only survived but had engrafted into the constructs generated from beating iPS cell clusters (Fig. 5D–5I). Staining with anti-human mitochondria antibody confirmed the human origin of the cardiomyocytes (data not shown). Cardiac muscle cells with sarcomeric striations were either densely packed with an immature rounded morphology (Fig. 5D) or were elongated and aligned in parallel with each other (Fig. 5E–5G). Positive staining for gap junction connexin-43 was observed between human iPS cell-derived cardiomyocytes (Fig. 5H, 5I). Furthermore, staining with the vascular marker lectin indicated that the cardiac tissues were vascularized by host-derived neovessels, which were negative for human-specific CD31 staining (Fig. 6).
Figure 6.

Vascularization of the engineered human cardiac tissues. (A): Serial sections stained with cTnT, lectin (arrows indicate blood vessels), and human-specific CD31 (magnification, ×400). (B): z-projection of confocal image (magnification, ×200) of a section stained with cTnT, lectin, and DAPI. Scale bars = 50 μm. Abbreviations: cTnT, cardiac troponin T; DAPI, 4′,6-diamidino-2-phenylindole.
Teratomas were present in all constructs generated with nonbeating iPS cell clusters (n = 3) (supplemental online Fig. 4). Human cells representing all three germ layers (mesoderm, endoderm, and ectoderm) were also found in engineered constructs generated with beating iPS cell clusters. These data are proof of the pluripotency of human iPS cells used in the present study, but despite this, implantation of beating iPS cell clusters in vivo resulted in vascularized, contracting three-dimensional constructs that clearly contained fully differentiated cardiomyocytes.
Discussion
Here we have shown that cardiac differentiation of human iPS cells can be enhanced by judicious use of the HDAC inhibitor TSA. TSA also enhanced the cardiomyogenic potential of growth factors activin A and BMP4. The derived human cardiomyocytes expressed cardiac-specific transcription factors and contractile structural proteins, and were responsive to the chronotropic agents isoprenaline and carbachol. Using the in vivo vascularized tissue engineering chamber [13], human iPS cell-derived cardiomyocytes survived implantation in vivo and assembled into compact patches of vascularized, contractile cardiac muscle.
TSA is a selective and reversible inhibitor of class I and II HDAC [10, 17]. Acute treatment with TSA promoted differentiation of mouse embryonic stem cells [18], whereas chronic treatment reduced their differentiation [19]. In terms of cardiomyogenesis, TSA has previously been shown to induce cardiomyocyte differentiation in mouse pluripotent stem cells [7, 9] and mouse embryonic carcinoma cells P19 [20]. Here, we showed that TSA-induced histone acetylation is associated with cardiomyocyte differentiation of human iPS cells when supplemented during EB formation. TSA also promoted cardiomyogenesis in human iPS cell lines that have lower spontaneous cardiac differentiation efficiencies (iPS(Foreskin)-1 and FA3; supplemental online Fig. 1). Variation in differentiation efficiency could be due to differences in pluripotency reprogramming protocols or to different epigenetic backgrounds. The optimal concentration of TSA that we found to promote cardiomyogenic differentiation of human iPS cells was lower (1 ng/ml) than that previously reported in murine pluripotent stem cells (10–50 ng/ml) [7, 9]. Small changes in the concentration of TSA resulted in loss of cardiomyogenic effect: at a high concentration (>10 ng/ml), TSA killed cells. This discrepancy between effective concentrations could be due to the differences in species, treatment duration (6 days vs. 1 day), or differentiation protocol (EB vs. monolayer).
Mechanistically, TSA is thought to induce translocation of HDAC4, a class II HDAC, from the nucleus to the cytoplasm [9], removing a repressor of cardiac-specific transcription factors GATA4, MEF2c, and Nkx2.5 [7, 20] and providing additive effects to the activin A and BMP4 method. Conversely, a class I HDAC (HDAC1) is essential for cardiac differentiation of mouse pluripotent stem cells by regulating BMP2 and Sox17 [21, 22]. In the present study, cardiomyocytes derived from the TSA + AB group appeared to have a more mature electrophysiological phenotype, being more responsive to β-adrenergic and muscarinic agonists and exhibiting better calcium handling compared with cardiomyocytes derived from the control group. This finding is in agreement with a previous study in which TSA was shown to reversibly enhance electrophysiological maturation (shift from hypersensitivity toward more homogeneous and prolonged field potential duration in response to inhibitors of rapid delayed potassium rectifier current, IKr) of cardiomyocytes derived from pluripotent stem cells through an increase in histone H3 acetylation [23]. In light of these findings, TSA appears to promote cardiac differentiation of pluripotent stem cells through regulation of cardiac transcription factors, and the optimal treatment protocol needs to be tailored to the cell lines and species involved.
Transplantation of stem cell-derived cardiomyocytes represents a potential approach for treating ischemic heart disease. However, the success of direct cell transplantation has been limited by inadequate cell engraftment, cell loss along the needle track, and high cell death due to lack of anchorage matrix and the ischemic microenvironment [24]. Tissue engineering offers an attractive strategy to overcome these limitations [25]. With a vascularized chamber model, we engineered cardiac tissue by implanting human cell clusters containing iPS cell-derived cardiomyocytes. At 4 weeks postimplantation, the implanted cells had survived and engrafted into the surrounding tissue. Surprisingly, the cardiac tissue constructs exhibited spontaneous focal contraction even with a low number of implanted cells (less than 100,000). Histological analysis showed gap junction connexin 43 and sarcomeric striation patterns in elongated, rod-shaped cells organized in parallel arrays to resemble the morphology of adult myocardium. This may indicate that the three-dimensional in vivo environment of the chamber can provide the necessary cues, such as mechanical stimulation, blood flow, and other in vivo dynamic conditions, to support cardiomyocyte maturation from iPS-derived cardiomyocytes, similar to our previous studies with neonatal rat cardiomyocytes [26–28]. Further studies evaluating the maturation of human iPS cell-derived cardiac tissue after implantation are warranted, by evaluating the β- and α-myosin heavy chain transcript isoform ratio [26] and their electrophysiological properties [29, 30] in engineered constructs.
Vascularization is a key factor limiting the thickness of engineered cardiac tissues to within approximately 100 μm of capillary beds [31]. In the present study, the human cardiac tissues were vascularized by neovessels sprouting from the host femoral vessels. The functional vascular network allows engineering of a clinically relevant, thick cardiac tissue, as demonstrated previously with neonatal rat cardiomyocytes [27, 32], and supports implanted iPS cells and their differentiation [15]. What limits the clinical application of this strategy is the risk of teratoma formation from pluripotent stem cells coimplanted inadvertently. In the present study, the presence of all three germ layers in engineered cardiac tissues indicates the lack of purity in iPS cell-derived cardiomyocytes that were enriched by microdissecting the beating areas. The beating clusters may contain remnant pluripotent stem cells at implantation or predifferentiated cells of endoderm and ectoderm cell lineages, as shown here by endoderm (α-fetoprotein) and ectoderm (Pax6) lineage-specific gene expression in the microdissected beating clusters (supplemental online Fig. 5). It will also be important to evaluate the survival and engraftment of iPS cell-derived cardiomyocytes in the myocardium, especially in the infarcted myocardium, where the hostile ischemic microenvironment may significantly affect the survival and function of transplanted cells. Nevertheless, this tissue engineering model provides proof of principle for generating myocardium-like tissue from human pluripotent stem cell-derived cardiomyocytes, and it provides a model for testing the safety of constructs generated from mixed cell populations, which might prove useful as a prelude to safe clinical translation.
Conclusion
Cardiomyogenic differentiation of human iPS cells can be enhanced by the histone deacetylase inhibitor TSA, and the cardiomyocytes so derived can be used to engineer vascularized, three-dimensional contracting tissue. These engineered cardiac tissues allow more authentic recapitulation of the structural and functional complexity of the heart compared with two-dimensional cultures of cardiomyocytes, and thus offer a more physiologically relevant model. The constructs might also provide appropriate physiology and disease phenotypes to develop new treatment modalities. Moreover, engineered vascularized cardiac tissue might eventually be developed to replace and support damaged myocardium through surgical transplantation, especially if autologous cells are used to derive the cardiomyocytes.
Supplementary Material
Acknowledgments
These studies were supported by grants from National Heart Foundation and National Health and Medical Research Council of Australia (NHMRC 509271, 1024817). D.E.C. is supported by an NHMRC Gustav Nossal Scholarship, G.J.D. is a Principal Research Fellow of the NHMRC; and A.P. is a Career Development Award Fellow of the NHMRC. Support is also provided by the J.R. and J.O. Wicking Trust, Friedreich's Ataxia Research Alliance (research grant and 2012 Keith Michael Andrus Cardiac Research Award), the Tony and Gwyneth Lennon Foundation, and the Victorian State Government's Department of Innovation, Industry and Regional Development's Operational Infrastructure Support Program. We thank John Slavin, Laita Bokhari, and Zerina Lokmic for advising us in teratoma analysis, confocal imaging, and histology, respectively. We thank James A. Thomson (University of Wisconsin) for providing the iPS(Foreskin) cell lines.
Author Contributions
S.Y.L.: financial support, conception and design, collection and assembly of data, data analysis and interpretation, manuscript writing, final approval of manuscript; P.S.: administrative support, collection and assembly of data, data analysis and interpretation, final approval of manuscript; D.E.C.: provision of study material, data analysis and interpretation, final approval of manuscript; G.J.D. and R.J.D.: financial support, conception and design, data analysis and interpretation, manuscript writing, final approval of manuscript; A.P.: financial support, provision of study material, conception and design, data analysis and interpretation, manuscript writing, final approval of manuscript.
Disclosure of Potential Conflicts of Interest
The authors indicate no potential conflicts of interest.
References
- 1.Lloyd-Jones D, Adams R, Carnethon M, et al. Heart disease and stroke statistics—2009 update: A report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation. 2009;119:e21–e181. doi: 10.1161/CIRCULATIONAHA.108.191261. [DOI] [PubMed] [Google Scholar]
- 2.Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 3.Yu J, Vodyanik MA, Smuga-Otto K, et al. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007;318:1917–1920. doi: 10.1126/science.1151526. [DOI] [PubMed] [Google Scholar]
- 4.Burridge PW, Keller G, Gold JD, et al. Production of de novo cardiomyocytes: Human pluripotent stem cell differentiation and direct reprogramming. Cell Stem Cell. 2012;10:16–28. doi: 10.1016/j.stem.2011.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mummery CL, Zhang J, Ng ES, et al. Differentiation of human embryonic stem cells and induced pluripotent stem cells to cardiomyocytes: A methods overview. Circ Res. 2012;111:344–358. doi: 10.1161/CIRCRESAHA.110.227512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Minami I, Yamada K, Otsuji TG, et al. A small molecule that promotes cardiac differentiation of human pluripotent stem cells under defined, cytokine- and xeno-free conditions. Cell Rep. 2012;2:1448–1460. doi: 10.1016/j.celrep.2012.09.015. [DOI] [PubMed] [Google Scholar]
- 7.Kawamura T, Ono K, Morimoto T, et al. Acetylation of GATA-4 is involved in the differentiation of embryonic stem cells into cardiac myocytes. J Biol Chem. 2005;280:19682–19688. doi: 10.1074/jbc.M412428200. [DOI] [PubMed] [Google Scholar]
- 8.Hosseinkhani M, Hasegawa K, Ono K, et al. Trichostatin A induces myocardial differentiation of monkey ES cells. Biochem Biophys Res Commun. 2007;356:386–391. doi: 10.1016/j.bbrc.2007.02.151. [DOI] [PubMed] [Google Scholar]
- 9.Kaichi S, Hasegawa K, Takaya T, et al. Cell line-dependent differentiation of induced pluripotent stem cells into cardiomyocytes in mice. Cardiovasc Res. 2010;88:314–323. doi: 10.1093/cvr/cvq189. [DOI] [PubMed] [Google Scholar]
- 10.Vanhaecke T, Papeleu P, Elaut G, et al. Trichostatin A-like hydroxamate histone deacetylase inhibitors as therapeutic agents: Toxicological point of view. Curr Med Chem. 2004;11:1629–1643. doi: 10.2174/0929867043365099. [DOI] [PubMed] [Google Scholar]
- 11.Liu J, Verma PJ, Evans-Galea MV, et al. Generation of induced pluripotent stem cell lines from Friedreich ataxia patients. Stem Cell Rev. 2011;7:703–713. doi: 10.1007/s12015-010-9210-x. [DOI] [PubMed] [Google Scholar]
- 12.Zwi L, Caspi O, Arbel G, et al. Cardiomyocyte differentiation of human induced pluripotent stem cells. Circulation. 2009;120:1513–1523. doi: 10.1161/CIRCULATIONAHA.109.868885. [DOI] [PubMed] [Google Scholar]
- 13.Lim SY, Hsiao ST, Lokmic Z, et al. Ischemic preconditioning promotes intrinsic vascularization and enhances survival of implanted cells in an in vivo tissue engineering model. Tissue Eng Part A. 2012;18:2210–2219. doi: 10.1089/ten.tea.2011.0719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kehat I, Kenyagin-Karsenti D, Snir M, et al. Human embryonic stem cells can differentiate into myocytes with structural and functional properties of cardiomyocytes. J Clin Invest. 2001;108:407–414. doi: 10.1172/JCI12131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lim SY, Lee DG, Sivakumaran P, et al. In vivo tissue engineering chamber supports human induced pluripotent stem cell survival and rapid differentiation. Biochem Biophys Res Commun. 2012;422:75–79. doi: 10.1016/j.bbrc.2012.04.108. [DOI] [PubMed] [Google Scholar]
- 16.Halbach M, Egert U, Hescheler J, et al. Estimation of action potential changes from field potential recordings in multicellular mouse cardiac myocyte cultures. Cell Physiol Biochem. 2003;13:271–284. doi: 10.1159/000074542. [DOI] [PubMed] [Google Scholar]
- 17.Yoshida M, Kijima M, Akita M, et al. Potent and specific inhibition of mammalian histone deacetylase both in vivo and in vitro by trichostatin A. J Biol Chem. 1990;265:17174–17179. [PubMed] [Google Scholar]
- 18.Karantzali E, Schulz H, Hummel O, et al. Histone deacetylase inhibition accelerates the early events of stem cell differentiation: Transcriptomic and epigenetic analysis. Genome Biol. 2008;9:R65. doi: 10.1186/gb-2008-9-4-r65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee JH, Hart SR, Skalnik DG. Histone deacetylase activity is required for embryonic stem cell differentiation. Genesis. 2004;38:32–38. doi: 10.1002/gene.10250. [DOI] [PubMed] [Google Scholar]
- 20.Karamboulas C, Swedani A, Ward C, et al. HDAC activity regulates entry of mesoderm cells into the cardiac muscle lineage. J Cell Sci. 2006;119:4305–4314. doi: 10.1242/jcs.03185. [DOI] [PubMed] [Google Scholar]
- 21.Hoxha E, Lambers E, Wasserstrom JA, et al. Elucidation of a novel pathway through which HDAC1 controls cardiomyocyte differentiation through expression of SOX-17 and BMP2. PLoS One. 2012;7:e45046. doi: 10.1371/journal.pone.0045046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hoxha E, Lambers E, Xie H, et al. Histone deacetylase 1 deficiency impairs differentiation and electrophysiological properties of cardiomyocytes derived from induced pluripotent cells. Stem Cells. 2012;30:2412–2422. doi: 10.1002/stem.1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Otsuji TG, Kurose Y, Suemori H, et al. Dynamic link between histone H3 acetylation and an increase in the functional characteristics of human ESC/iPSC-derived cardiomyocytes. PLoS One. 2012;7:e45010. doi: 10.1371/journal.pone.0045010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Laflamme MA, Murry CE. Regenerating the heart. Nat Biotechnol. 2005;23:845–856. doi: 10.1038/nbt1117. [DOI] [PubMed] [Google Scholar]
- 25.Vunjak-Novakovic G, Lui KO, Tandon N, et al. Bioengineering heart muscle: A paradigm for regenerative medicine. Annu Rev Biomed Eng. 2011;13:245–267. doi: 10.1146/annurev-bioeng-071910-124701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tiburcy M, Didie M, Boy O, et al. Terminal differentiation, advanced organotypic maturation, and modeling of hypertrophic growth in engineered heart tissue. Circ Res. 2011;109:1105–1114. doi: 10.1161/CIRCRESAHA.111.251843. [DOI] [PubMed] [Google Scholar]
- 27.Morritt AN, Bortolotto SK, Dilley RJ, et al. Cardiac tissue engineering in an in vivo vascularized chamber. Circulation. 2007;115:353–360. doi: 10.1161/CIRCULATIONAHA.106.657379. [DOI] [PubMed] [Google Scholar]
- 28.Choi YS, Matsuda K, Dusting GJ, et al. Engineering cardiac tissue in vivo from human adipose-derived stem cells. Biomaterials. 2010;31:2236–2242. doi: 10.1016/j.biomaterials.2009.11.097. [DOI] [PubMed] [Google Scholar]
- 29.Blazeski A, Zhu R, Hunter DW, et al. Cardiomyocytes derived from human induced pluripotent stem cells as models for normal and diseased cardiac electrophysiology and contractility. Prog Biophys Mol Biol. 2012;110:166–177. doi: 10.1016/j.pbiomolbio.2012.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mehta A, Chung YY, Ng A, et al. Pharmacological response of human cardiomyocytes derived from virus-free induced pluripotent stem cells. Cardiovasc Res. 2011;91:577–586. doi: 10.1093/cvr/cvr132. [DOI] [PubMed] [Google Scholar]
- 31.Radisic M, Malda J, Epping E, et al. Oxygen gradients correlate with cell density and cell viability in engineered cardiac tissue. Biotechnol Bioeng. 2006;93:332–343. doi: 10.1002/bit.20722. [DOI] [PubMed] [Google Scholar]
- 32.Tee R, Morrison WA, Dusting GJ, et al. Transplantation of engineered cardiac muscle flaps in syngeneic rats. Tissue Eng Part A. 2012;18:1992–1999. doi: 10.1089/ten.tea.2012.0151. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.