

Abstract
The introduction of Haemophilus influenzae serotype b (Hib) conjugate vaccines has changed the epidemiology of invasive H. influenzae disease, with a shift in the predominant serotype from Hib to nonencapsulated H. influenzae (ncHi). The objective of this study was to identify the genotypes/clones associated with invasive H. influenzae disease in Italy. Eighty-seven H. influenzae strains isolated in the years 2009 to 2011 within the National Surveillance of Invasive Bacterial Disease program were analyzed. Strains were characterized by serotyping, antimicrobial susceptibility testing, and multilocus sequence typing (MLST). Genetic polymorphisms in the blaTEM gene promoter region as well as the occurrence of both adhesin genes (hmwA and hia) and the IgA1 protease-encoding gene (igaB) were also investigated. Of 87 strains, 67 were ncHi and 20 were encapsulated. Eleven strains were β-lactamase positive, harboring the blaTEM gene. Most blaTEM genes (10/11) were associated with a Pdel promoter region exhibiting a 135-bp deletion; the remaining strain possessed the Pa/Pb overlapping promoter. MLST analysis showed that encapsulated isolates were clonal, with each serotype sharing a few related sequence types (STs). Forty-six different STs were identified among the 67 ncHi strains. Despite this heterogeneity, a group of closely related STs (ST103, ST139, and ST145) encompassed almost 25% of all ncHi strains and 45.5% of the β-lactamase producers carrying the Pdel promoter. These major ST clones were found to be associated with the hmwA gene but not with the igaB gene. To conclude, although the heterogeneity of the ncHi population was confirmed, diffusion of major successful ST clones was documented.
INTRODUCTION
Haemophilus influenzae, a small Gram-negative bacillus causing a variety of localized respiratory tract infections as well as invasive disease, is characterized by the presence or absence of a polysaccharide capsule. Encapsulated strains can express six antigenically and structurally distinct capsular polysaccharides, designated serotypes a to f (1). Nonencapsulated strains fail to agglutinate with specific typing antisera and are commonly defined as nontypeable H. influenzae (NTHi). More recently, the use of capsular genotyping methods able to specifically detect the presence or absence of capsular genes has allowed us to define nonencapsulated H. influenzae (ncHi) strains as strains actually lacking capsular biosynthetic genes (2).
In Italy, data on cases of invasive H. influenzae disease are detected through the National Surveillance of Invasive Bacterial Disease program, a passive reporting system (3). Hospital laboratories are requested to send invasive H. influenzae isolates to the National Reference Laboratory (NRL) at the Istituto Superiore di Sanità, where capsular genotyping is performed and they are further characterized. As reported in a previous study of ours, in Italy, the introduction of conjugate vaccines against H. influenzae type b (Hib) has resulted in a drastic reduction of invasive Hib disease. This success has been accompanied by some changes in the epidemiological characteristics of invasive H. influenzae disease, with the predominant serotype changing from Hib to ncHi (3). The same has been observed in other European countries with established Hib immunization programs, where most invasive disease cases are now caused by ncHi (4–6).
The monitoring of the circulating non-vaccine-preventable strains (ncHi and encapsulated isolates other than serotype b isolates) with the objective to promptly identify the emergence of potentially successful clones is a “hot issue” for current surveillance of invasive H. influenzae disease. Multilocus sequence typing (MLST) is an appropriate tool for this aim, since it allows comparisons of the major genotypes associated with invasive disease, even in different settings and countries.
Another task of careful surveillance is the monitoring of the genes associated with the major antibiotic resistance traits of the microorganism under study. In H. influenzae, resistance to β-lactam antibiotics due to β-lactamase production by either the blaTEM or blaROB gene is the main resistance mechanism observed (3, 7, 8). Polymorphism in the blaTEM gene promoter sequence has been detected previously, but there is no knowledge regarding the distribution of the different promoter types among H. influenzae isolates from Italy (9).
Finally, investigations on bacterial factors that can affect the virulence of ncHi isolates causing invasive disease are increasingly important, since no vaccine against this capsular type is currently available. Therefore, in this view, it may be of interest to find correlations between specific virulence traits, such as the presence of both adherence factors and molecules that resist host immune defenses (IgA1 proteases), and specific clones circulating in Italy.
The main objective of this study was to identify the major genotypes associated with invasive H. influenzae disease in Italy, with particular regard to non-vaccine-preventable strains. Moreover, variability in the blaTEM gene promoter region as well as the presence of some potential virulence traits were investigated with the aim to discover whether or not there is an association between such specific characteristics and specific clones.
MATERIALS AND METHODS
Bacterial strains.
During the period of 1 January 2009 through 31 December 2011, a total of 87 H. influenzae strains isolated from patients with invasive disease detected through the National Surveillance of Invasive Bacterial Disease program were sent to the National Reference Laboratory at the Istituto Superiore di Sanità, Rome, Italy, and were included in this study. Thirty-one strains had been isolated from cerebrospinal fluid (CSF), and 56 were isolates from blood. For each strain, the capsular serotype was determined by PCR capsular genotyping, a method able to differentiate between ncHi strains actually lacking capsular biosynthetic genes and spontaneous capsule-deficient mutants of encapsulated strains (2).
Antimicrobial susceptibility testing.
The MICs of ampicillin, azithromycin, cefotaxime, chloramphenicol, and ciprofloxacin were determined by Etest (AB Biodisk, Solna, Sweden), according to the manufacturer's recommendations; the interpretative breakpoints were based on European Committee on Antimicrobial Susceptibility Testing (EUCAST) criteria (http://www.eucast.org/clinical breakpoints/). β-Lactamase activity was revealed by using nitrocefin sticks (Oxoid Ltd., Basingstoke, Hampshire, England).
Detection of blaTEM and blaROB genes by PCR and sequencing of the blaTEM gene promoter region.
In the ampicillin-resistant isolates, the presence of the blaTEM and blaROB genes was investigated by PCR, as previously described (10). The blaTEM gene PCR products were subjected to DNA sequencing to determine promoter region types. Comparative analysis of the blaTEM nucleotide sequences was performed by the advanced BLAST search program 2.2 at the National Center for Biotechnology Information website (http://www.ncbi.nlm.nih.gov/blast/).
MLST.
MLST analysis was carried out according to the H. influenzae MLST website scheme (http://haemophilus.mlst.net/). Briefly, approximately 450-bp internal fragments of 7 housekeeping genes (adk, atpG, frdB, fucK, mdh, pgi, and recA) were amplified by PCR using primers and methods previously described (11). Sequences were submitted to the MLST website for allele and sequence type (ST) assignments.
Phylogenetic analysis.
The nucleotide sequences of the seven gene fragments from each unique ST were concatenated in the order adk, atpG, frdB, fucK, mdh, pgi, and recA, using the concatenation tool at the MLST website. The 3,057-bp sequences obtained from all different STs of our collection of strains were aligned to infer their phylogenetic relatedness. The neighbor-joining method was used to generate a phylogenetic tree with MEGA5 software (12). The statistical robustness and reliability of the branching order within each phylogenetic tree were confirmed by bootstrap analysis. The bootstrap consensus tree was inferred from 1,000 replicates. The evolutionary distances were computed by using the maximum composite likelihood method and are in units of the number of base substitutions per site.
PFGE.
Genetic relatedness among ncHi strains belonging to ST103, ST139, and ST145 was further investigated by pulsed-field gel electrophoresis (PFGE) after digestion of DNA with SmaI, according to procedures previously described (13). Similarity analysis was performed with Dice's coefficient, and clustering was carried out by means of the unweighted-pair group mean association (UPGMA) with GelCompar II v.6.0 (Applied Maths, Sint-Martens-Latem, Belgium). Isolates showing a coefficient of similarity of ≥80% were considered to belong to the same cluster.
Detection of hmwA and hia adhesin genes by PCR.
PCR amplification of the hmwA and hia adhesin genes was carried out by using primers and conditions previously reported (14).
Detection of the igaB gene by PCR.
The presence of an additional IgA1 protease-encoding gene, igaB, was investigated by a PCR-based screening method using primers and procedures previously described (15).
RESULTS
By PCR capsular genotyping, 67 H. influenzae isolates (67/87; 77%) were identified as ncHi lacking capsular biosynthetic genes, 6 (6/87; 6.9%) were identified as type b, 5 (5/87; 5.8%) were identified as type e, and 9 (9/87; 10.3%) were identified as type f, while no capsule-deficient mutant of encapsulated strains was detected. Of the 87 cases, 32 were diagnosed as meningitis (36.8%), 42 as bacteremia (48.3%), 12 as bacteremic pneumonia (13.8%), and 1 as epiglottitis (1.1%) (clinical presentation by serotype is shown in Table S1 in the supplemental material).
The antimicrobial susceptibility results of the 87 H. influenzae strains are shown in Table S2 in the supplemental material. Briefly, ampicillin resistance and azithromycin resistance were the most frequently detected (14.9% and 11.5%, respectively), followed by chloramphenicol resistance (4.6%). Only one strain was found to be resistant to cefotaxime (1.1%), and no strain was resistant to ciprofloxacin. Among ampicillin-resistant strains, 11 (all ncHi) were β-lactamase producers and harbored a blaTEM gene (MIC range, 4 μg/ml to 256 μg/ml), while 2 ncHi strains showed a β-lactamase-negative ampicillin-resistant (BLNAR) phenotype (MIC of 1.5 μg/ml and MIC of 3 μg/ml, respectively). No strains possessed the blaROB gene. A genetic polymorphism in the blaTEM gene promoter region was detected. Most blaTEM genes were associated with a Pdel promoter region (10/11; 90.9%) exhibiting a 135-bp deletion (G23 to C157 included) compared with the conventional P3 promoter and a nucleotide mutation in the −10 region, as previously described (9) (Table 1). The Pa/Pb promoter (exhibiting a single-base-pair difference, C32T, compared to the P3 promoter but producing an overlapping promoter) was detected in only one strain (9). No strains had the P3 promoter. Strains with the Pdel promoter exhibited a wide range of ampicillin MICs (4 μg/ml to ≥256 μg/ml) (Table 1). The single strain with the Pa/Pb promoter showed an ampicillin MIC of ≥256 μg/ml (Table 1).
Table 1.
Main characteristics of β-lactamase-positive ncHi strains isolated from patients with invasive disease in Italy, 2009 to 2011
| Strain | Yr | Promoter type | Ampicillin MIC (μg/ml) | Sequence type |
|---|---|---|---|---|
| Hi319 | 2009 | Pdel | ≥256 | 3 |
| Hi378 | 2010 | Pdel | ≥256 | 103 |
| Hi390 | 2011 | Pdel | 64 | 103 |
| Hi396 | 2011 | Pdel | 48 | 103 |
| Hi402 | 2011 | Pdel | 32 | 103 |
| Hi406 | 2011 | Pdel | ≥256 | 103 |
| Hi374 | 2010 | Pdel | ≥256 | 156 |
| Hi337 | 2010 | Pdel | 48 | 165 |
| Hi373 | 2010 | Pa/Pb | ≥256 | 1132 |
| Hi391 | 2011 | Pdel | 48 | 1133 |
| Hi397 | 2011 | Pdel | 4 | 1134 |
Two strains were found to be not typeable by MLST, since no sequences were obtained for the fucK gene, in agreement with previously reported observations that occasional H. influenzae isolates lack the entire fucose operon (16). The remaining 85 strains were distributed into 54 different STs (Table 2). Forty-two STs included a single strain, and 12 STs were comprised of two or more strains. Ten STs were novel, being new combinations of known alleles. New STs were named ST1132 through ST1134 and ST1208 through ST1214. Regarding encapsulated strains, each capsular type was distributed into a few STs (Table 2). Most Hib strains (5/6) belonged to ST6, while the remaining strain was of ST1210, which differed from ST6 at 1 (single-locus variant) of the 7 MLST loci. Among Hie strains, 2 strains belonged to ST69, and the remaining 3 strains were classified as ST18, ST621, and ST1208, which were single- or double-locus variants of ST69. The majority of the Hif strains (8/9) belonged to ST124, and 1 strain was of ST598, a single-locus variant of the former.
Table 2.
Distribution of STs and adhesin (hmwA and hia) and igaB genes among H. influenzae strains isolated from patients with invasive disease in Italy, 2009 to 2011
| Serotype | ST (no. of isolates) | Housekeeping gene allele no. |
No. of isolates positive for: |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| adk | atpG | frdB | fucK | mdh | pgi | recA | hmw | hia | igaB | ||
| b | 6 (5) | 10 | 14 | 4 | 5 | 4 | 7 | 8 | |||
| 1210 (1) | 10 | 14 | 4 | 18 | 4 | 7 | 8 | ||||
| e | 18 (1) | 18 | 6 | 3 | 7 | 10 | 28 | 12 | |||
| 69 (2) | 18 | 6 | 3 | 22 | 10 | 28 | 12 | ||||
| 621 (1) | 18 | 6 | 3 | 15 | 10 | 28 | 12 | ||||
| 1208 (1) | 18 | 14 | 3 | 11 | 10 | 28 | 12 | ||||
| f | 124 (8) | 22 | 19 | 11 | 11 | 22 | 19 | 15 | 7 | ||
| 598 (1) | 22 | 4 | 11 | 11 | 22 | 19 | 15 | ||||
| ncHi | 3 (1) | 1 | 1 | 1 | 1 | 1 | 1 | 5 | 1 | ||
| 34 (1) | 11 | 2 | 15 | 8 | 28 | 26 | 3 | 1 | |||
| 46 (2) | 1 | 25 | 1 | 14 | 15 | 1 | 5 | 2 | |||
| 57 (1) | 14 | 7 | 13 | 7 | 17 | 13 | 17 | 1 | 1 | ||
| 103 (6) | 1 | 1 | 1 | 14 | 9 | 14 | 13 | 6 | |||
| 105 (2) | 3 | 9 | 8 | 2 | 14 | 8 | 4 | ||||
| 134 (1) | 1 | 1 | 1 | 14 | 53 | 14 | 13 | 1 | |||
| 137 (1) | 1 | 1 | 1 | 1 | 73 | 1 | 5 | 1 | |||
| 139 (5) | 1 | 1 | 1 | 14 | 45 | 14 | 21 | 5 | |||
| 142 (2) | 1 | 1 | 1 | 35 | 15 | 53 | 38 | 2 | |||
| 145 (5) | 1 | 8 | 1 | 14 | 22 | 14 | 13 | 5 | |||
| 147 (1) | 1 | 24 | 18 | 14 | 55 | 1 | 5 | 1 | |||
| 149 (1) | 3 | 9 | 8 | 13 | 14 | 8 | 4 | 1 | |||
| 156 (1) | 26 | 2 | 15 | 7 | 22 | 56 | 3 | 1 | |||
| 159 (1) | 33 | 8 | 16 | 16 | 17 | 2 | 29 | 1 | 1 | ||
| 160 (1) | 40 | 1 | 1 | 14 | 1 | 59 | 3 | ||||
| 161 (1) | 41 | 8 | 29 | 9 | 68 | 57 | 8 | 1 | |||
| 165 (1) | 44 | 2 | 16 | 37 | 17 | 2 | 3 | 1 | |||
| 183 (1) | 14 | 44 | 1 | 1 | 22 | 1 | 5 | 1 | |||
| 201 (1) | 33 | 8 | 8 | 16 | 1 | 2 | 3 | 1 | |||
| 203 (1) | 52 | 1 | 1 | 14 | 89 | 62 | 37 | 1 | |||
| 259 (1) | 1 | 1 | 1 | 14 | 89 | 62 | 37 | 1 | |||
| 367 (2) | 1 | 1 | 1 | 1 | 67 | 1 | 5 | 2 | |||
| 368 (1) | 3 | 4 | 17 | 11 | 54 | 3 | 34 | 1 | |||
| 388 (1) | 60 | 51 | 16 | 48 | 15 | 1 | 31 | 1 | |||
| 425 (2) | 39 | 8 | 35 | 11 | 65 | 48 | 19 | 2 | |||
| 427 (1) | 47 | 60 | 19 | 1 | 26 | 90 | 62 | ||||
| 448 (1) | 14 | 7 | 1 | 7 | 17 | 13 | 17 | 1 | |||
| 474 (2) | 3 | 54 | 65 | 1 | 64 | 78 | 48 | ||||
| 513 (1) | 93 | 41 | 41 | 8 | 69 | 60 | 29 | 1 | |||
| 531 (1) | 93 | 46 | 72 | 11 | 95 | 70 | 45 | 1 | |||
| 582 (1) | 1 | 80 | 1 | 1 | 1 | 13 | 5 | 1 | |||
| 691 (1) | 17 | 4 | 17 | 70 | 7 | 12 | 11 | 1 | |||
| 695 (1) | 131 | 2 | 15 | 8 | 26 | 61 | 3 | 1 | |||
| 832 (1) | 51 | 36 | 1 | 81 | 158 | 38 | 18 | 1 | |||
| 958 (1) | 1 | 1 | 137 | 13 | 13 | 25 | 16 | 1 | |||
| 1043 (1) | 50 | 12 | 32 | 50 | 147 | 193 | 125 | 1 | |||
| 1145 (1) | 37 | 35 | 57 | 14 | 202 | 183 | 13 | ||||
| 1132 (1) | 1 | 43 | 38 | 7 | 77 | 165 | 37 | 1 | |||
| 1133 (1) | 6 | 43 | 7 | 7 | 77 | 55 | 43 | ||||
| 1134 (1) | 5 | 33 | 7 | 10 | 202 | 10 | 29 | 1 | |||
| 1209 (1) | 40 | 18 | 53 | 15 | 86 | 14 | 23 | 1 | |||
| 1211 (1) | 1 | 1 | 1 | 14 | 89 | 1 | 37 | 1 | |||
| 1212 (1) | 1 | 3 | 1 | 1 | 31 | 14 | 5 | 1 | |||
| 1213 (1) | 1 | 3 | 1 | 1 | 15 | 78 | 5 | 1 | |||
| 1214 (1) | 25 | 1 | 1 | 1 | 190 | 58 | 5 | 1 | |||
Overall, the 67 ncHi strains were distributed into 46 different STs (Table 2). The most frequent STs were ST103 (5 isolates from blood and 1 from CSF), ST139, and ST145 (each including 5 isolates, 3 from CSF and 2 from blood), with the latter 2 STs being double-locus variants of ST103. Of note, 5 of the 10 β-lactamase-positive strains carrying the Pdel promoter belonged to ST103, while the other β-lactamase producers were dispersed among different minor STs (ST3, ST156, ST165, ST1133, and ST1134) (Table 1). The single strain carrying the Pa/Pb promoter belonged to ST1132. The 2 BLNAR strains belonged to different STs (ST367 and ST368).
The phylogenetic tree constructed based on the concatenated MLST sequences showed clustering of STs derived from encapsulated strains into separate branches according to capsular types (Fig. 1). Hif and Hie strains showed close phylogenetic relationships since they belonged to a single major clade (clade A) also including some ncHi strains. Overall, small clusters of phylogenetically related ncHi STs were identified, but they included only 2 or 3 strains (Fig. 1). Notably, the exception was the clustering of ST103, ST139, and ST145 in a separate branch (cluster 1) also including ST134 and ST1212 (with ST134 being a single-locus variant of ST103 and ST1212 sharing 3 alleles in common with ST103). Cluster 1 encompassed 18 of 67 (26.9%) ncHi strains. When the ncHi strains belonging to the major ncHi STs (ST103, ST139, and ST145) were analyzed by PFGE, cluster analysis of the profiles showed the presence of variants within each ST, although minor clusters were identified (clusters A, B, C, and D) (Fig. 2). ST139 appeared more genetically homogeneous since it segregated in a separate branch, while ST103 and ST145 appeared intermixed between each other.
Fig 1.
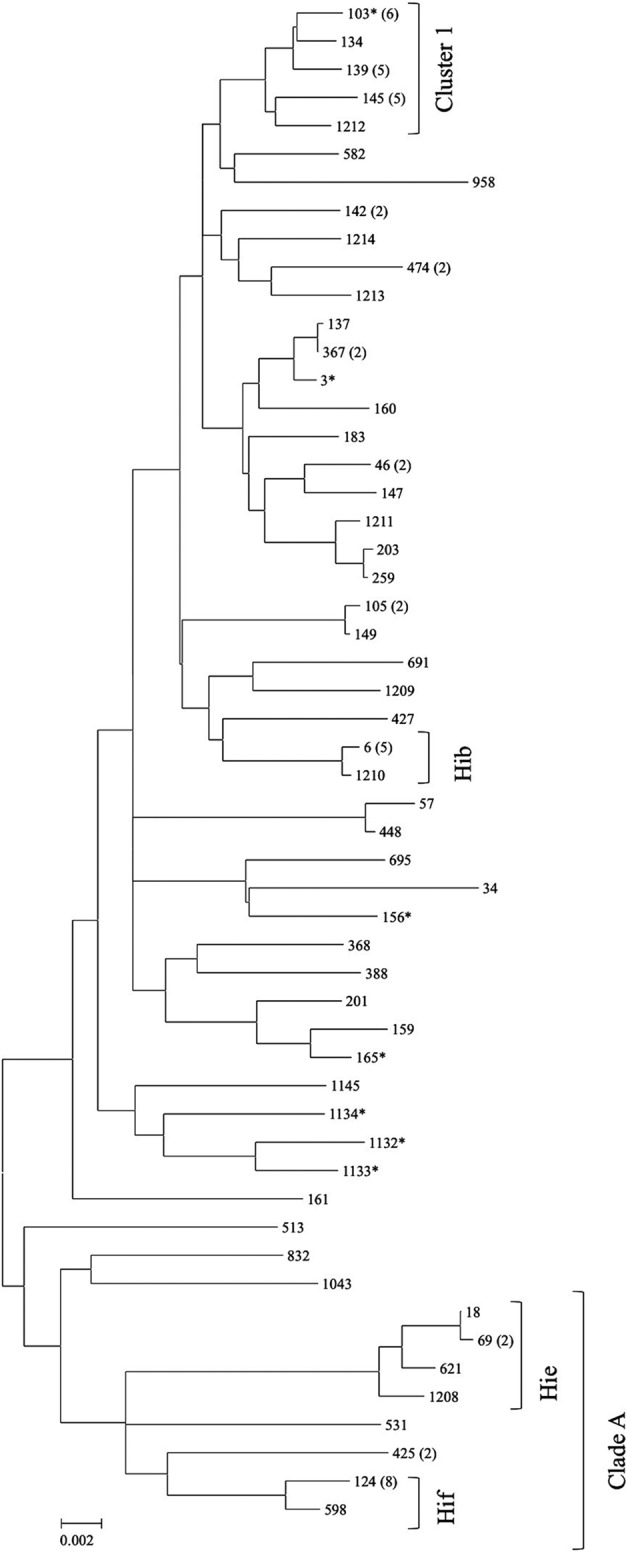
Neighbor-joining phylogenetic tree of 54 concatenated MLST sequences obtained from 85 H. influenzae strains isolated from patients with invasive disease in Italy. The evolutionary distances were computed by using the maximum composite likelihood method and were drawn to scale, with the bar at the bottom indicating 0.02 nucleotide substitutions per site. On the right, the sequence type (ST) is reported. The number of strains for each ST including more than one isolate is shown in parentheses. The main clade (clade A), the main cluster (cluster 1), and the serotype of the encapsulated strains are indicated. STs associated with ampicillin-resistant strains are marked with an asterisk.
Fig 2.
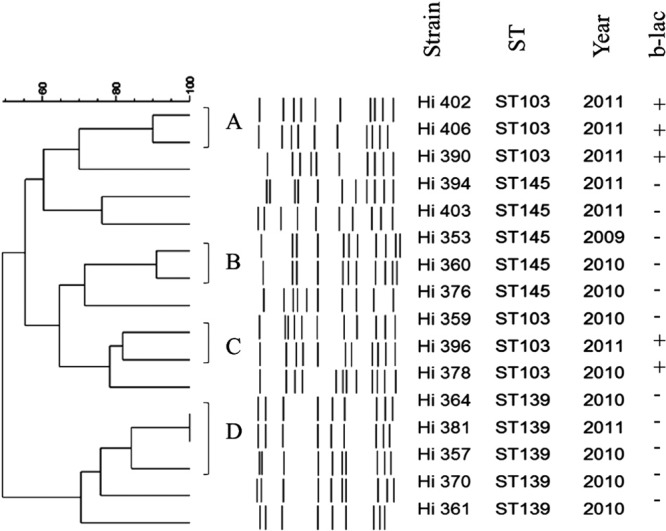
Genetic relatedness among the 16 ncHi strains belonging to ST103, ST145, and ST139 by cluster analysis of the PFGE patterns. Similarity analysis was performed by using Dice's coefficient, and clustering was carried out by means of the unweighted-pair group mean association (UPGMA) method. Strains with a coefficient of similarity value of ≥80% were considered to belong to the same cluster (clusters A, B, C, and D). On the right, the strain, the sequence type (ST), the year of strain isolation, and the β-lactamase result (+ or −) are reported.
No association was found between specific STs and geographical area or sites of isolation of strains (CSF/blood) when ST103, ST139, and ST145 were also considered a group. Strains included in the same clusters were isolated from invasive H. influenzae cases that were epidemiologically unrelated. Looking at the presence of the genes encoding the HMW and/or Hia adhesin (which are the major adherence factors of ncHi), overall, most ncHi strains possessed hmwA-related sequences (47/67 ncHi strains; 70.1%), 10 strains (10/67 ncHi; 14.9%) contained the hia gene, and 10 (10/67 ncHi; 14.9%) lacked both hmwA and hia. The hmwA-positive strains were distributed among 30 different ncHi STs, but they gathered mainly into cluster 1, in which all strains (18 strains) possessed hmwA sequences (P = 0.001) (Table 2). On the contrary, no association was found between specific ST clones and the hia gene. Among encapsulated strains, as expected, no strain possessed hmwA sequences, but homologs of hia were present in 7 strains (7/20; 35%), all belonging to serotype f and comprising ST124 (Table 2).
Finally, PCR screening for the igaB gene revealed the presence of only 3 positive ncHi strains belonging to three different STs (ST57, ST159, and ST388), which were not genetically related to each other (Table 2).
DISCUSSION
In a previous study of ours, we demonstrated that in Italy nowadays, most invasive H. influenzae infections are caused by ncHi strains, as also observed throughout Europe (3–6). However, although monitoring of the emerging non-vaccine-preventable strains has been considered an essential aim of present-day surveillance, little information is available on the major MLST clones associated with invasive ncHi strains circulating in Italy as well as in other European countries (7).
In this investigation, overall, ncHi strains were distributed among a large number of STs, which were often unrelated in the phylogenetic tree. This result is not surprising and confirms previous observations demonstrating the genetic diversity of ncHi strains (17, 18). However, and notably, a group of closely related STs (ST103, ST139, and ST145) was identified, together including almost 25% of all ncHi strains analyzed and about half (5/11; 45.5%) of the β-lactamase-producing strains. In the phylogenetic tree, this group segregated with 2 other, minor STs (ST134 and ST1212) into cluster 1, suggesting that a common origin may exist. Although not so high, the percentage of strains associated with the above-mentioned linked 5 STs may be considered relevant, taking into account the general diversity of the ncHi population. In fact, consistent with such a high level of diversity, almost 70% (690/1,003) of all STs associated with known capsular types and currently included in the Haemophilus MLST database (accessed 8 May 2013) belonged to ncHi. Regarding the clustering of closely related STs, our data are in agreement with a previous study by Erwin et al., who investigated the phylogeny of all STs submitted to the Haemophilus MLST website at that time (September 2006, a total of 372 different STs) by using the E-burst program (18). In the study by Erwin et al., the major STs identified here (ST103, ST139, ST145, and ST134) were members of the same E-burst group, group 2 (18). Reviewing the literature, ST103 was previously found among strains from either carriage or middle ear fluid in children and infants in the United States or invasive disease in Canada (19–22). ST139 and ST145 have also been reported among strains from cases of invasive disease in Canada (21). Whether these ST clones have or have not emerged as significant successful pathogens in invasive infections due to ncHi requires further studies and surveillance. In any case, as observed for some major ST clones identified in other bacterial species, ST103, ST139, and ST145 can be subdivided into variants by PFGE, indicating that independent selection from a common ancestor rather than the simple spread of a single “strain” occurred.
As far as encapsulated strains are concerned, overall, our findings are in agreement with previous, more comprehensive investigations of H. influenzae phylogeny, showing clustering according to the capsular type due to the clonal structure of the encapsulated strains (23). Looking at the single STs associated with the different capsular types, the main STs that we observed were previously reported to be associated with the same serotypes in other studies. In particular, ST6 has been found to be dominant among Hib strains (21, 24), both ST18 and ST69 are dominant among Hie strains (22, 25), and ST124 is dominant among Hif strains (21, 25).
In our phylogenetic tree, Hie and Hif strains were more genetically related (they belonged to the same clade, clade A) than Hib strains, confirming previous results obtained by Erwin et al. demonstrating clustering of both Hie and Hif isolates in the same clade by MLST (18). These data are not in agreement with a previous report using multilocus enzyme electrophoresis, in which encapsulated strains were divided into two major phylogenetic divisions, with Hie and most Hib strains belonging to the same division, division I, and Hif belonging to division II (26). On the other hand, Hie strains were previously found to share some features of Hif division II strains, such as the chromosomal location of the capsulation (cap) locus and the lack of association of the cap sequences with the IS1016 insertion element (27). Since some ncHi strains were also in clade A, we searched for their STs in the MLST database, but they were found to be associated only with ncHi strains and not with Hie or Hif strains, suggesting that if diversification from a common ancestor occurred, it was not a recent event. Overall, none of the 67 ncHi strains belonged to STs that were shared by encapsulated strains, and each encapsulated strain was included in their own unique STs according to capsular type. This finding confirms that, so far, there is no evidence of capsular switching in H. influenzae (21).
Results of the present study confirmed the assumption that the main antimicrobial resistance mechanism among invasive H. influenzae strains was β-lactamase production. A side objective of the present study was to discover whether or not there is an association between specific ST clones and ampicillin resistance/blaTEM gene/promoter types. Our results demonstrated that all β-lactamase producers analyzed here contained a blaTEM gene. Although the blaTEM gene is generally carried by a plasmid, which is a mobile element by definition, we found it to be frequently associated with a specific successful ST clone, ST103, in almost half of the β-lactamase-producing strains. When we looked at polymorphisms in the blaTEM gene promoter region, we found that the Pdel promoter type, which was originally identified in β-lactamase-producing H. influenzae isolates from Australia, has also become strongly prevalent in Italy (9).
Considering that H. influenzae infection can be mediated by multiple potential virulence factors, we investigated the possible association between the presence of some virulence traits and specific clones, with particular regard to ST clones included in cluster 1.
As far as adhesins are concerned, most ncHi strains contained hmwA-related sequences. Although the overall percentage that we found (70.1%) is similar to that previously reported in other studies, a strong, significant association between the presence of the hmwA genes and the major invasive ncHi clones circulating in Italy (ST103, ST139, ST145, ST134, and ST1212) was demonstrated (28, 29). Whether the occurrence of HMW adhesins may play a contributing role in making an invasive ncHi clone “successful” requires further studies to be elucidated.
Bacterial IgA1 proteases cleave human IgA1 and are supposed to play a role in invasion and pathogenesis of H. influenzae infection (30). Nearly all H. influenzae strains contain the iga gene, which encodes an IgA1 protease, but recently, a second protease gene, igaB, was identified, encoding an additional IgA1 protease among isolates from both sputum samples of patients with chronic obstructive pulmonary disease (COPD) (especially strains belonging to ST159) and middle ear fluid (15, 31). In the present study, a very small number of ncHi strains (3.4%) was found to possess the igaB gene, and no presence of this gene was detected in the major invasive clones included in cluster 1, although the previously described association between igaB and ST159 was confirmed (the single ST159 strain that we detected actually contained the igaB gene) (31).
Finally, as a preliminary approach, we also looked at the polymorphonuclear leukocyte (PMN)-mediated killing of a subgroup of H. influenzae strains (3 ncHi strains belonging to the major ST clones comprising cluster 1, 3 ncHi strains belonging to minor ST clones, and 2 Hib strains). Although the total number of strains that we tested was too small for us to draw firm conclusions, our results suggested that the PMN-mediated killing of the ncHi strains was strain dependent and overall poor compared to that of Hib strains (using heat-inactivated normal human serum), in agreement with a previous investigation (32). In particular, the ncHi strains that we analyzed differently decreased (or even did not decrease) in viable counts during phagocytosis according to the single strain. However, the mean percentage of killing for ncHi was significantly lower than that for Hib strains (mean ± standard deviation [SD] = 15.58% ± 23.34% versus 75.13% ± 9.12%, respectively; P = 0.0016), possibly in relation to the presence of Hib antibodies in the pool of adult sera used (data not shown). Of note, no significant difference was observed in the mean percentage of killed bacteria in the group of ncHi strains belonging to the major ST clones in comparison to that of the remaining ncHi strains, considered as a group (mean ± SD = 7.22% ± 8.40% versus 25.62% ± 34.0%, respectively; P = 0.2295) (data not shown). Therefore, we could not find any association between specific successful clones and phagocytosis resistance, but further investigation is necessary to address this issue.
In conclusion, this study allows us to identify, for the first time, the major ST clones associated with invasive H. influenzae disease in Italy. Although a diversity of ST clones was observed for ncHi, a group of genetically linked STs was discovered, which accounted for a significant portion of strains, including β-lactamase producers. Determination of whether dissemination of these or other clones will occur requires continuous surveillance, including close monitoring of circulating ncHi strains.
Supplementary Material
ACKNOWLEDGMENTS
This work was partially supported by the Ministry of Health-Centro Controllo Malattie project Surveillance of Invasive Bacterial Diseases Caused by Neisseria meningitidis, Streptococcus pneumoniae and Haemophilus influenzae (project no. 1M12).
We thank Tonino Sofia for editorial assistance. We are very grateful to Roberto Nisini (Istituto Superiore di Sanità) for helpful discussion and comments.
We declare that we have no conflicts of interest.
Footnotes
Published ahead of print 12 June 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/CVI.00028-13.
REFERENCES
- 1.Pittman M. 1931. Variation and type specificity in the bacterial species Haemophilus influenzae. J. Exp. Med. 53:471–492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Falla TJ, Crook DWM, Brophy LN, Maskell D, Kroll JS, Moxon ER. 1994. PCR for capsular typing of Haemophilus influenzae. J. Clin. Microbiol. 32:2382–2386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Giufrè M, Cardines R, Caporali MG, Accogli M, D'Ancona F, Cerquetti M. 2011. Ten years of Hib vaccination in Italy: prevalence of non-encapsulated Haemophilus influenzae among invasive isolates and the possible impact on antibiotic resistance. Vaccine 29:3857–3862 [DOI] [PubMed] [Google Scholar]
- 4.Ladhani S, Slack MP, Heath PT, von Gottberg A, Chandra M, Ramsay ME, European Union Invasive Bacterial Infection Surveillance Participants 2010. Invasive Haemophilus influenzae disease, Europe, 1996-2006. Emerg. Infect. Dis. 16:455–463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dworkin MS, Park L, Borchardt SM. 2007. The changing epidemiology of invasive Haemophilus influenzae disease, especially in persons ≥65 years old. Clin. Infect. Dis. 44:810–816 [DOI] [PubMed] [Google Scholar]
- 6.Kalies H, Siedler A, Grondahl B, Grote V, Milde-Busch A, von Kries R. 2009. Invasive Haemophilus influenzae infections in Germany: impact of non-type b serotypes in the post-vaccine era. BMC Infect. Dis. 9:45. 10.1186/1471-2334-9-45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Resman F, Ristovski M, Forsgren A, Kaijser B, Kronvall G, Medstrand P, Melander E, Odenholt I, Riesbeck K. 2012. Increase of β-lactam-resistant invasive Haemophilus influenzae in Sweden, 1997 to 2010. Antimicrob. Agents Chemother. 56:4408–4415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tristram S, Jacobs MR, Appelbaum PC. 2007. Antimicrobial resistance in Haemophilus influenzae. Clin. Microbiol. Rev. 20:368–389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tristram SG, Hawes R, Souprounov J. 2005. Variation in selected regions of blaTEM genes and promoters in Haemophilus influenzae. J. Antimicrob. Chemother. 56:481–484 [DOI] [PubMed] [Google Scholar]
- 10.Cerquetti M, Cardines R, Giufrè M, Mastrantonio P, HI Study Group 2004. Antimicrobial susceptibility of Haemophilus influenzae strains isolated from invasive disease in Italy. J. Antimicrob. Chemother. 54:1139–1143 [DOI] [PubMed] [Google Scholar]
- 11.Meats E, Feil EJ, Stringer S, Cody AJ, Goldstein R, Kroll JS, Popovic T, Spratt BG. 2003. Characterization of encapsulated and noncapsulated Haemophilus influenzae and determination of phylogenetic relationships by multilocus sequence typing. J. Clin. Microbiol. 41:1623–1636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. 2011. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28:2731–2739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cerquetti M, Ciofi degli Atti ML, Renna G, Tozzi AE, Garlaschi ML, Mastrantonio P, HI Study Group 2000. Characterization of non-type B Haemophilus influenzae strains isolated from patients with invasive disease. J. Clin. Microbiol. 38:4649–4652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cardines R, Giufrè M, Mastrantonio P, Ciofi degli Atti ML, Cerquetti M. 2007. Nontypeable Haemophilus influenzae meningitis in children: phenotypic and genotypic characterization of isolates. Pediatr. Infect. Dis. J. 26:577–582 [DOI] [PubMed] [Google Scholar]
- 15.Fernaays MM, Lesse AJ, Cai X, Murphy TF. 2006. Characterization of igaB, a second immunoglobulin A1 protease gene in nontypeable Haemophilus influenzae. Infect. Immun. 74:5860–5870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shuel ML, Karlowsky KE, Law DK, Tsang RS. 2011. Nonencapsulated or nontypeable Haemophilus influenzae are more likely than their encapsulated or serotypeable counterparts to have mutations in their fucose operon. Can. J. Microbiol. 57:982–986 [DOI] [PubMed] [Google Scholar]
- 17.Smith-Vaughan HC, Sriprakash KS, Leach AJ, Mathews JD, Kemp DJ. 1998. Low genetic diversity of Haemophilus influenzae type b compared to nonencapsulated H. influenzae in a population in which H. influenzae is highly endemic. Infect. Immun. 66:3403–3409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Erwin AL, Sandstedt SA, Bonthuis PJ, Geelhood JL, Nelson KL, Unrath WC, Diggle MA, Theodore MJ, Pleatman CR, Mothershed EA, Sacchi CT, Mayer LW, Gilsdorf JR, Smith AL. 2008. Analysis of genetic relatedness of Haemophilus influenzae isolates by multilocus sequence typing. J. Bacteriol. 190:1473–1483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schumacher SK, Marchant CD, Loughlin AM, Bouchet V, Stevenson A, Pelton SI. 2012. Prevalence and genetic diversity of nontypeable Haemophilus influenzae in the respiratory tract of infants and primary caregivers. Pediatr. Infect. Dis. J. 31:145–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kaur R, Chang A, Xu Q, Casey JR, Pichichero ME. 2011. Phylogenetic relatedness and diversity of non-typable Haemophilus influenzae in the nasopharynx and middle ear fluid of children with acute otitis media. J. Med. Microbiol. 60:1841–1848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shuel M, Law D, Skinner S, Wylie J, Karlowsky J, Tsang RS. 2010. Characterization of nontypeable Haemophilus influenzae collected from respiratory infections and invasive disease cases in Manitoba, Canada. FEMS Immunol. Med. Microbiol. 58:277–284 [DOI] [PubMed] [Google Scholar]
- 22.Sill ML, Law DK, Zhou J, Skinner S, Wylie J, Tsang RS. 2007. Population genetics and antibiotic susceptibility of invasive Haemophilus influenzae in Manitoba, Canada, from 2000 to 2006. FEMS Immunol. Med. Microbiol. 51:270–276 [DOI] [PubMed] [Google Scholar]
- 23.Musser JM, Kroll JS, Moxon ER, Selander RK. 1988. Clonal population structure of encapsulated Haemophilus influenzae. Infect. Immun. 56:1837–1845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schouls LM, van der Ende A, van de Pol I, Schot C, Spanjaard L, Vauterin P, Wilderbeek D, Witteveen SJ. 2005. Increase in genetic diversity of Haemophilus influenzae serotype b (Hib) strains after introduction of Hib vaccination in The Netherlands. J. Clin. Microbiol. 43:2741–2749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ladhani SN, Collins S, Vickers A, Litt DJ, Crawford C, Ramsay ME, Slack MP. 2012. Invasive Haemophilus influenzae serotype e and f disease, England and Wales. Emerg. Infect. Dis. 18:725–732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Musser JM, Kroll JS, Moxon ER, Selander RK. 1988. Evolutionary genetics of the encapsulated strains of Haemophilus influenzae. Proc. Natl. Acad. Sci. U. S. A. 85:7758–7762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Giufrè M, Cardines R, Mastrantonio P, Cerquetti M. 2010. Genetic characterization of the capsulation locus of Haemophilus influenzae serotype e. J. Clin. Microbiol. 48:1404–1407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.St Geme JW, III, Kumar VV, Cutter D, Barenkamp SJ. 1998. Prevalence and distribution of the hmw and hia genes and the HMW and Hia adhesins among genetically diverse strains of nontypeable Haemophilus influenzae. Infect. Immun. 66:364–368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ecevit IZ, McCrea KW, Pettigrew MM, Sen A, Marrs CF, Gilsdorf JR. 2004. Prevalence of the hifBC, hmw1A, hmw2A, hmwC, and hia genes in Haemophilus influenzae isolates. J. Clin. Microbiol. 42:3065–3072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kilian M, Mestecky J, Schrohenloher JE. 1979. Pathogenic species of the genus Haemophilus and Streptococcus pneumoniae produce immunoglobulin A1 protease. Infect. Immun. 26:143–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Murphy TF, Lesse AJ, Kirkham C, Zhong H, Sethi S, Munson RS., Jr 2011. A clonal group of nontypeable Haemophilus influenzae with two IgA proteases is adapted to infection in chronic obstructive pulmonary disease. PLoS One 6:e25923. 10.1371/journal.pone.0025923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vogel L, van Alphen L, Geluk F, Troelstra A, Martin E, Bredius R, Eijk P, Jansen H, Dankert J. 1994. Quantitative flow cytometric analysis of opsonophagocytosis and killing of nonencapsulated Haemophilus influenzae by human polymorphonuclear leukocytes. Clin. Diagn. Lab. Immunol. 1:394–400 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.