

Abstract
Brucellosis is an important zoonotic infectious disease of humans and livestock with worldwide distribution and is caused by bacteria of the genus Brucella. The diagnosis of brucellosis always requires laboratory confirmation by either isolation of pathogens or detection of specific antibodies. The conventional serological tests available for the diagnosis of brucellosis are less specific and show cross-reactivity with other closely related organisms. These tests also necessitate the handling of Brucella species for antigen preparation. Therefore, there is a need to develop reliable, rapid, and user-friendly systems for disease diagnosis and alternatives to vaccine approaches. Keeping in mind the importance of brucellosis as an emerging infection and the prevalence in India, we carried out the present study to compare the recombinant antigens with the native antigens (cell envelope and sonicated antigen) of Brucella for diagnosis of human brucellosis by an indirect plate enzyme-linked immunosorbent assay (ELISA). Recombinant outer membrane protein 28 (rOmp28) and rOmp31 antigens were cloned, expressed, and purified in the bacterial expression system, and the purified proteins were used as antigens. Indirect plate ELISAs were then performed and standardized for comparison of the reactivities of recombinant and native antigens against the 433 clinical samples submitted for brucellosis testing, 15 culture-positive samples, and 20 healthy donor samples. The samples were separated into four groups based on their positivity to rose bengal plate agglutination tests (RBPTs), standard tube agglutination tests (STATs), and 2-mercaptoethanol (2ME) tests. The sensitivities and specificities of all the antigens were calculated, and the rOmp28 antigen was found to be more suitable for the clinical diagnosis of brucellosis than the rOmp31 antigen and native antigens. The rOmp28-based ELISA showed a very high degree of agreement with the conventional agglutination tests and promising results for further use in clinical screening and serodiagnosis of human brucellosis.
INTRODUCTION
Brucellosis is one of the world's major zoonoses, caused by bacteria of the genus Brucella. It is a serious public health problem in India and in other developing countries. The transmission of Brucella infection and its prevalence in a region depend upon several factors, like food habits, methods of processing milk and milk products, social customs, husbandry practices, climatic conditions, socioeconomic status, and environment hygiene (1). The disease has been recognized as one of the most common laboratory-acquired infections; it has been reported to occur in clinical research and production laboratories. Human brucellosis is a multisystem disease that may present with a broad spectrum of clinical manifestations, and its diagnosis requires microbiological confirmations by means of isolation from blood cultures or demonstration of the presence of specific antibodies by serological tests (2). Blood cultures provide definite proof of brucellosis but may not provide positive results for all patients, even under ideal conditions (3). Brucella is a slow-growing organism, and cultures are rarely positive (4) and should be kept at least 45 days before the culture can be considered conclusively negative. Serological tests are used in the diagnosis of brucellosis; the most commonly used are the standard tube agglutination test (STAT), the Coombs anti-Brucella test, the rose bengal plate agglutination test (RBPT), the complement fixation test (CFT), and the 2-mercaptoethanol (2ME) test. The STAT measures the total quantity of agglutinating antibodies (IgM and IgG), and the quantity of specific IgG is determined by the 2ME test (5).
Outer membrane proteins (Omps) are structural constituents of the cell and not likely to function as virulence factors; they also act as immunodominant antigens for vaccine potential (6). Indirect enzyme-linked immunosorbent assays (ELISAs) typically use these outer membrane and cytoplasmic proteins as antigens and measure IgG, IgM, and IgA, which allows for a better interpretation of the clinical situation than the STAT and other conventional tests (7). The serological tests based on whole-cell extracts or lipopolysaccharides (LPSs) are not completely specific and cannot always distinguish reactions due to Brucella melitensis or due to cross-reactions to other bacteria, particularly Yersinia enterocolitica O:9 (8). To overcome these problems and to increase the sensitivity and specificity of the test system, this study was designed to use recombinant proteins as antigens in ELISAs. The cloning and expression of the recombinant Omp28 (rOmp28) protein of B. melitensis and screening for its diagnostic potential were reported in our previous study (9). In the present study, we have compared the efficacies of rOmp28 and rOmp31 proteins with those of the cell envelope and whole-cell sonicated antigen by indirect plate ELISAs and also with conventional agglutination tests for the serodiagnosis of human brucellosis.
MATERIALS AND METHODS
Bacterial strains.
The B. melitensis 16M strain was used in this study for cloning of the omp28 and omp31 genes and also for the preparation of native antigens. Brucella strains were routinely cultured in Brucella broth (Difco Laboratories) and maintained at −20°C in 30% glycerol. The pQE30UA vector and host cell Escherichia coli strain M15 were purchased from Qiagen. The host cells and recombinant clones were grown routinely in Luria broth (Difco), and when an antibiotic was needed, kanamycin (Sigma) at 25 μg/ml or ampicillin (Sigma) at 100 μg/ml was also added to the medium.
Samples.
The clinical serum samples were collected from individual patients reporting to medical college hospitals from different regions in India where brucellosis is endemic and also from field laboratories. A total of 433 serum samples were collected and initially tested by RBPTs, STATs, and 2ME tests. Based on these conventional standard serological tests, the samples were separated into four groups: group I (n = 409), consisting of clinical samples and healthy donor serum samples that were negative by all three standard conventional agglutination tests (RBPTs, STATs, and 2ME tests); group II (n = 9), consisting of samples positive by RBPTs and negative by STATs and 2ME tests; group III (n = 17), consisting of samples positive by RBPTs and STATs and negative by 2ME tests; and group IV (n = 33), consisting of clinical samples positive by all the three tests and serum samples from culture-positive cases which were also positive by all three tests (Table 1).
Table 1.
Total numbers of serum samples grouped and evaluated based on agglutination assays
| Group | Serological characteristics of samples | Total no. of samples |
|---|---|---|
| I | RBPT negative, STAT negative, 2ME test negative, and healthy donors (n = 20) | 409 |
| II | RBPT positive, STAT negative, and 2ME test negative | 9 |
| III | RBPT positive, STAT positive, and 2ME test negative | 17 |
| IV | RBPT positive, STAT positive, and 2ME test positive and culture-positive cases (n = 15) | 33 |
| Total | 468 |
Preparation of sonicated antigen.
The B. melitensis 16M strain was subcultured by inoculation into 5 ml brain heart infusion (BHI) broth and incubated in a shaker incubator (Labcon) overnight at 37°C and 180 rpm. This overnight-grown culture was used for bulk growth by inoculation into 100 ml BHI broth and again incubated in a shaker incubator overnight at 37°C and 180 rpm. Then, 1 ml formaldehyde was added to the overnight-grown culture and incubated for 1 h to inactivate the bacteria. After inactivation, the culture was again streaked onto fresh BHI agar plates to confirm the complete inactivation. The inactivated culture was pelleted by centrifugation in a Sorvall RC5C centrifuge at 10,000 rpm for 20 min at room temperature. The supernatant was discarded, and the pellet was washed three times with 1× phosphate-buffered saline (PBS) and resuspended in 10 ml 1× PBS. The bacterial suspension was sonicated by using a Vibrocell (Sonics) sonicator for 5 cycles of 5 min each with a pulse after every 8 s and amplitude of 40 W. The sonicated suspension was again centrifuged at 10,000 rpm and 4°C for 30 min. The supernatant containing the soluble proteins was preserved, and the pellet containing the insoluble proteins was discarded. The protein in the supernatant was further purified by ammonium sulfate (80%) precipitation. The supernatant was stirred in a magnetic stirrer, and ammonium sulfate was added slowly so that the final concentration of the solution reached 80%. The supernatant was subjected to centrifugation at 10,000 rpm and 4°C for 30 min, and the precipitate was washed twice with an 80% ammonium sulfate solution, dissolved in sterile PBS, and stored at −20°C until further use.
Preparation of cell envelope antigen.
Five milliliters of an overnight-grown culture of B. melitensis was inoculated into 100 ml of BHI broth and incubated overnight in a shaker incubator at 37°C with constant shaking (180 rpm). One milliliter formaldehyde (10 μl formaldehyde/ml of culture) was added to the overnight-grown culture and incubated for 1 h to kill the bacteria. The inactivated bacterial culture was pelleted at 10,000 rpm for 15 min at room temperature. The bacterial pellet was washed twice with 1× sterile PBS and resuspended in 100 ml of buffer 1 (15 mM Tris-HCl, 0.45 mM sucrose, 8 mM EDTA, 0.4 mg/ml lysozyme). The bacterial suspension was incubated in a water bath at 37°C for 15 min and then centrifuged at 8,000 × g (10,000 rpm). The pellet was resuspended in 10 to 15 ml buffer 2 (50 mM Tris-HCl, 5 mM MgCl2, 2 mM phenylmethylsulfonyl fluoride [PMSF]), chilled on ice, and sonicated as described previously. The sonicated suspension was centrifuged at 3,000 × g (6,000 rpm) for 30 min at 4°C, and the supernatant was subjected to ultracentrifugation at 43,900 rpm and 4°C for 90 min (Sorvall Ultra Pro ultracentrifuge). Finally, the pellet was resuspended in 1.5 ml of buffer 3 (50 mM Tris-HCl, 2 mM PMSF). The suspension was centrifuged again at 10,000 rpm for 15 min, and the supernatant was stored at −20°C until further use. SDS-PAGE was performed using a discontinuous buffer system per a previously described procedure (10) with minor modifications to resolve the different banding patterns of proteins in the prepared antigens.
Expression and purification of recombinant Omp28 and Omp31 antigens.
The omp28 gene of B. melitensis was cloned in pQE30UA in our previous study, and purified rOmp28 protein was prepared as described earlier (9, 11). The clone was inoculated into 10 ml LB medium containing kanamycin (25 μg/ml) to prepare a starter culture for the expression and purification of the rOmp28 antigen. This 10-ml overnight-grown culture was used as an inoculum for 250 ml medium containing an antibiotic and incubated in a shake flask at 37°C until the optical density (OD) of the culture reached 0.6 for induction. Thereafter, the culture was induced with 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG) and incubated for 5 h; the cells were then harvested, and rOmp28 was purified under native conditions using nickel-nitrilotriacetic acid (Ni-NTA) affinity chromatography. The purified protein was dialyzed against 1× PBS (pH 7.2), and the final concentration of rOmp28 was estimated by a method described previously (12).
For preparation of the rOmp31 protein, the 723-bp omp31 gene (GenBank accession no. AF076290) encoding an ∼27-kDa protein was amplified by PCR from bacterial genomic DNA from the B. melitensis 16M strain using the forward primer 5′-AAA TCC GTA ATT TTG GCG-3′ and the reverse primer 5′-TTA GAA CTT GTA GTT CAG A-3′ . The amplified product was cloned in the pQE30UA vector (Qiagen, Germany) per the manufacturer's protocol, and the protein was expressed in E. coli strain M15 host cells by using IPTG induction at 1.5 mM for 6 h. The induced cells were then harvested, and the protein was purified under denaturing conditions by Ni-NTA affinity chromatography. The purified protein was dialyzed against 1× PBS and analyzed on SDS-PAGE gels. The final concentration of protein was also estimated, and the protein was stored at −20°C until further use.
Western blotting with positive and negative samples.
The recombinant purified proteins were then subjected to Western blotting per a procedure described previously (11). The electrophoretic transfer of the protein from the polyacrylamide gel to the nitrocellulose membrane was carried out by using Tris-glycine buffer (pH 8.3) with 20% methanol (13). Then, the protein-free sites of the nitrocellulose membrane were blocked by 3% bovine serum albumin (BSA) in PBS by overnight incubation at 4°C. Thereafter, the membrane was washed three times with 1× phosphate buffer with 0.05% Tween 20 (PBS-T) for 15 min each. The membrane was then incubated with anti-His conjugate, the pooled culture was confirmed to be human positive, and negative sera and rabbit polyclonal antibodies were generated against the sonicated whole-cell antigen of B. melitensis for 1 h at 37°C. The membranes were again thoroughly washed three times with PBS-T for 15 min each and then developed with 3′,3′-diaminobenzidine (DAB)-H2O2.
Agglutination tests for antibody detection.
For the RBPT, 20 μl of RBPT antigen was added to a drop of serum sample on a glass slide, mixed with a sterile toothpick, and observed for an agglutination reaction. The STAT was also performed with and without 2-mercaptoethanol (2ME) per the standard protocol (14).
Indirect microplate ELISA for IgG antibody detection.
An indirect plate ELISA was standardized for screening of serum samples using purified rOmp28, rOmp31, cell envelope (CE), and whole-cell sonicated antigen (SA). The proteins were diluted to final concentrations of 25 μg/ml in 0.05 M carbonate bicarbonate buffer, and 100 μl/well was added to ELISA plates (Nunc) in three replicates and incubated at 37°C for 1 h. The plates were washed three times with 1× PBS-T and then blocked with 200 μl of 1% BSA at 37°C for 1 h. Thereafter, the plates were washed three times with PBS-T, and 2 μl of serum samples was added to 200 μl sterile 1× PBS buffer in individual wells and incubated for 1 h at 37°C (a pooled culture confirmed that positive- and negative-control serum samples were also included on each plate to ensure the accuracy of the test and allow for comparative analysis). Again, the plates were washed three times with PBS-T and incubated with 100 μl of polyclonal anti-human IgG-horseradish peroxidase (HRP) conjugate (Dako) at a 1:1,000 dilution in 1× PBS for 1 h at 37°C. After a final wash with PBS-T, the reaction was developed by the addition of 100 μl of developing solution consisting of o-phenylenediamine and H2O2, and the plates were incubated for 5 to 10 min in the dark for color development. The reaction was stopped by adding 10 μl of 1 N H2SO4 per well, and absorbance was measured at 495 nm. All the tests were run in three replicates and three times to confirm the reproducibility of the assay, and the mean values were used for calculations.
RESULTS
Preparation and characterization of native antigens.
The whole-cell sonicated component and cell envelope component of the B. melitensis 16M strain were extracted and prepared per the protocol described and used as protein antigens in ELISAs. The estimated protein concentrations were 200 mg/liter and 20 mg/liter for sonicated antigen and cell envelope antigen, respectively. These antigens were also analyzed by SDS-PAGE, giving resolved protein bands of different sizes in both sonicated and cell envelope antigens (Fig. 1). The prepared antigens were then stored in small aliquots at −20°C until further use.
Fig 1.
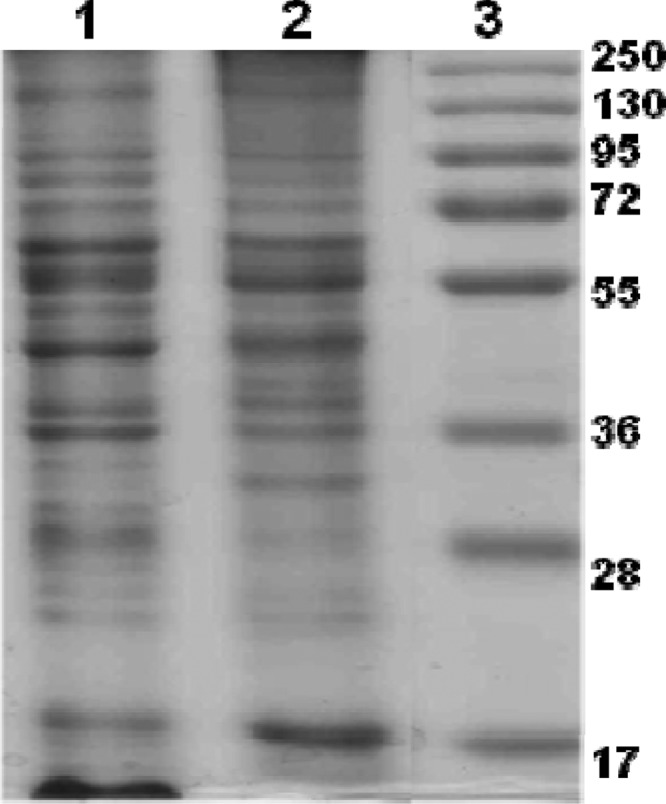
SDS-PAGE analysis of native antigens (CE antigen and SA). Lane 1, CE of B. melitensis; lane 2, SA of B. melitensis; lane 3, prestained protein marker (Fermentas no. SM1811). Molecular mass markers are shown on the right.
Purification of rOmp28 and rOmp31 proteins under denaturing conditions.
The rOmp28 and rOmp31 proteins were purified under denaturing conditions by Ni-NTA affinity chromatography, and the eluted proteins along with different fractions were examined by SDS-PAGE. Single bands of 30 kDa of Omp28 (data shown in previous studies) (9, 11) and of 29 kDa of Omp31 were observed in gels because of the coexpression of an additional 26 amino acids from the pQE30UA vector, including a 6×His tag (Fig. 2). The purified rOmp31 and rOmp28 proteins were characterized by Western blot analysis as shown in Fig. 3 and in our previous studies of rOmp28 (9, 11).
Fig 2.
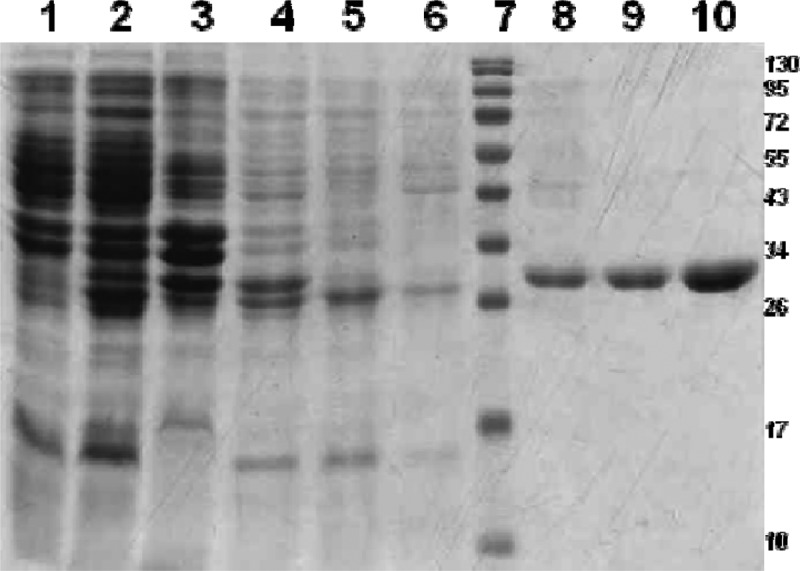
Expression and purification of rOmp31 antigen in the pQE30UA vector. Lane 1, uninduced; lane 2, induced; lane 3, pellet; lane 4, lysate; lane 5, flowthrough; lane 6, wash; lane 7, prestained protein marker (Fermentas no. SM0671); lane 8, eluate 1; lane 9, eluate 2; lane 10, eluate 3. Molecular mass markers are shown on the right.
Fig 3.
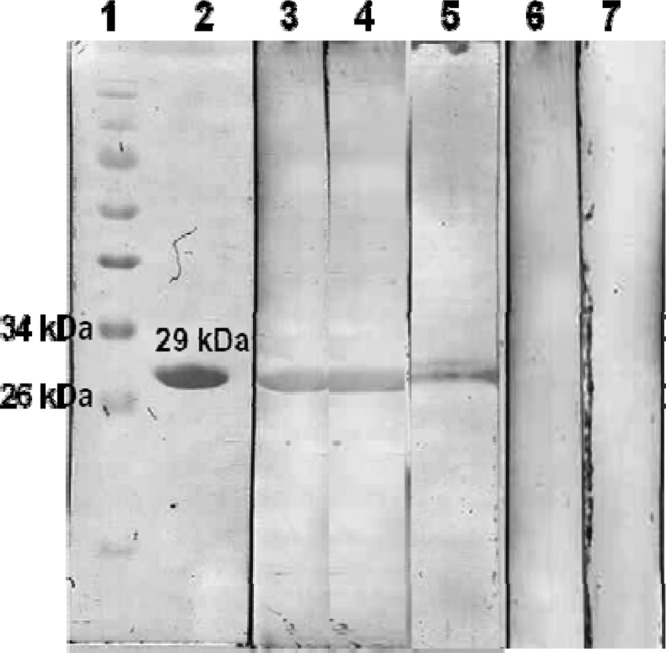
Characterization of rOmp31 protein by Western blotting. Lane 1, prestained protein marker (Fermentas no. SM0671); lane 2, reactivity with anti-His-HRP antibody conjugate; lanes 3 and 4, positive human sera; lane 5, rabbit polyclonal antibody sera generated against the sonicated whole-cell antigen of B. melitensis; lanes 6 and 7, negative human sera. Specific molecular mass markers are shown.
Agglutination tests for detection of clinical human samples.
The classical rose bengal plate agglutination test (RBPT) and standard tube agglutination test (STAT) are often used as rapid screening tests for the diagnosis of brucellosis and are based on the agglutination of serum antibodies with a stained whole-cell preparation of heat-killed bacteria. The tests were performed on 433 clinical samples submitted for brucellosis testing, out of which 44 samples were found positive by RBPT, 35 samples were positive by STAT, and 18 samples were positive by the 2ME test (Table 1).
Indirect plate ELISAs of native and recombinant antigens.
All the serum samples collected from individuals with suspected cases of brucellosis and from healthy individuals and those with culture-confirmed positive cases were analyzed by indirect plate ELISAs using native (CE and SA) and recombinant (rOmp28 and rOmp31) antigens. Cutoff values for the screening of suspected samples were calculated by estimating the mean optical densities (ODs) and standard deviations (SDs) for healthy individuals and were found to be 0.26 ± 0.14, 0.26 ± 0.10, 0.14 ± 0.07, and 0.16 ± 0.06 for CE, SA, rOmp28, and rOmp31, respectively. A sample was considered positive if the OD value of the sample was greater than the mean OD value plus 3 SDs for the healthy controls. Thus, the cutoff values for the four antigens were calculated to be 0.68, 0.56, 0.35, and 0.34 for CE, SA, rOmp28, and rOmp31, respectively. Out of the 409 samples in group I (RBPT, STAT, and 2ME test negative and healthy donors), 36, 20, 10, and 23 were found positive by CE, SA, rOmp28, and rOmp31, respectively (Fig. 4A). Out of the 9 samples in group II (RBPT positive but STAT and 2ME test negative), 3, 1, 1, and 1 were found positive by CE, SA, rOmp28, and rOmp31, respectively (Fig. 4B). Out of the 17 samples in group III (RBPT and STAT positive but 2ME test negative), 3, 4, 4, and 1 were found positive by CE, SA, rOmp28, and rOmp31, respectively (Fig. 4C). In group IV (positive by the RBPT, STAT, and 2ME test and culture positive), which consisted of 33 samples, 18, 20, 30, and 13 were found positive by CE, SA, rOmp28, and rOmp31, respectively (Fig. 4D) (Table 2).
Fig 4.

Total numbers of positive (P) and negative (N) samples tested in indirect plate ELISAs with the different antigens (CE, SA, rOmp28, and rOmp31). (A) Group I; (B) group II; (C) group III; (D) group IV.
Table 2.
Performance of ELISAs using native and recombinant antigens
| Group | Results (no. [%]) of ELISAs using: |
|||||||
|---|---|---|---|---|---|---|---|---|
| CE |
SA |
rOmp28 |
rOmp31 |
|||||
| Positive | Negative | Positive | Negative | Positive | Negative | Positive | Negative | |
| I | 36 (8.9) | 373 (91.1) | 20 (4.9) | 389 (95.1) | 10 (2.4) | 399 (97.6) | 23 (5.7) | 386 (94.3) |
| II | 3 (33.3) | 6 (66.7) | 1 (11.1) | 8 (88.9) | 1 (11.1) | 8 (88.9) | 1 (11.1) | 8 (88.9) |
| III | 3 (16.7) | 15 (83.3) | 4 (22.2) | 14 (77.8) | 4 (22.2) | 14 (77.8) | 1 (5.6) | 17 (94.4) |
| IV | 18 (54.6) | 15 (45.4) | 20 (60.7) | 13 (39.3) | 30 (90.9) | 3 (9.1) | 13 (39.3) | 20 (60.7) |
Sensitivity and specificity.
The sensitivities and specificities of indirect plate ELISAs using native and recombinant antigens were calculated; true-positive samples were defined as samples confirmed by culture to be positive or found positive by all three tests (RBPT, STAT, and 2ME test), and true-negative samples (clinical or healthy donor) were negative by all three agglutination tests. False-negative samples were defined as those that were positive by culture but negative by ELISAs, and false-positive samples were those that were negative by all three agglutination tests but positive by ELISAs. The sensitivities of the indirect ELISAs were 68.7%, 71.7%, 91.6%, and 62.2% for CE, SA, rOmp28, and rOmp31, respectively, whereas the specificities were 91.9%, 95.3%, 97.6%, and 94.7% for CE, SA, rOmp28, and rOmp31, respectively. Correlations of ELISAs with RBPTs, STATs, and 2ME tests using all four antigens were also calculated and are shown in Table 3.
Table 3.
Comparison of sensitivities and specificities of indirect ELISAs using native and recombinant antigens
| Calculation | Antigen |
|||
|---|---|---|---|---|
| CE | SA | rOmp28 | rOmp31 | |
| Sensitivity (%)a | 68.7 | 71.7 | 91.6 | 62.2 |
| Specificity (%)b | 91.9 | 95.3 | 97.6 | 94.7 |
| Correlation (%) with RBPTc | 84.8 | 88.5 | 92.7 | 85.7 |
| Correlation (%) with STATc | 85.5 | 89.7 | 94.2 | 87.2 |
| Correlation (%) with 2ME testc | 87.8 | 91.9 | 96.2 | 90.4 |
| Likelihood ratio for positive resultd | 8.5 | 15.3 | 38.2 | 11.7 |
| Likelihood ratio for negative resulte | 2.9 | 3.4 | 11.6 | 2.5 |
Sensitivity = [true positives/(true positives + false negatives)] × 100.
Specificity = [true negatives/(true negatives + false positives)] × 100.
Correlation = [(number of samples positive by both tests + number of samples negative by both tests)/total number of samples] × 100.
Likelihood ratio for positive result = sensitivity/1 − specificity.
Likelihood ratio for negative result = specificity/1 − sensitivity.
DISCUSSION
In studies conducted earlier, several proteins were identified as potential markers for the diagnosis of brucellosis, as antibodies against these markers have been demonstrated in culture-confirmed positive human serum samples (15, 16). Antigen detection for the diagnosis of brucellosis has not been in practical use, and estimation of antibodies remains the only viable alternative. The antibodies generated against the brucellosis antigens can be detected after a few days of infection and up to 3 months. In the earlier studies, native antigens were used in the standard serological tests for brucellosis, and most of these antigens are in the forms of whole cells, cell sonic extracts, lipopolysaccharides, or proteins extracted from the whole-cell sonicated extracts. The rose bengal plate agglutination test (RBPT) is often used for the rapid screening of samples for brucellosis and is based on the agglutination of serum antibodies with a stained whole-cell preparation of heat-killed acidified bacteria (17). The sensitivity of the RBPT is high, but the specificity is low, as all these antigens show cross-reactivity with many clinically important bacteria, like E. coli O:159, Y. enterocolitica O:9, Vibrio cholerae, and Salmonella spp. (18). To overcome this problem, efforts have been made to improve the serodiagnosis of brucellosis by replacing the native antigens with highly purified specific recombinant antigens (9, 19). The major outer membrane proteins are the potential targets as they react to all the Brucella spp. including Brucella canis and Brucella ovis (20, 21). The cell envelope proteome of Brucella abortus has been analyzed and found to consist of 164 proteins, and based on the reactivity with the positive bovine and human serum samples, reactive proteins like OMP31, Omp2b porin, and GroEL were identified (16, 22). In this study, the efficacies of four antigens (two recombinant and two native) were evaluated in indirect ELISAs; rOmp28 showed promising results, as the sensitivity (91.6%) and specificity (97.6%) of ELISAs using this antigen were found to be higher than those for native and recombinant Omp31 antigens. rOmp28 ELISAs also showed maximal agreement with the conventional agglutination tests (RBPT, STAT, and 2ME test), as 379 out of 389 samples from group I were found negative by rOmp28; i.e., 10 (2.5%) samples were found false positive by rOmp28, whereas 23 (5.9%), 36 (9.2%), and 39 (10.02%) samples in group I were found false positive by rOmp31, CE, and SA, respectively. Similarly, the highest number of positive samples in group IV was found by rOmp28 (16/18), followed by SA (9/18), CE (6/18), and rOmp31 (4/18). However, from groups II and III, most of the positive samples (8/9 from group II and 13/17 from group III) were found negative by rOmp28. In addition, the lesser sensitivities observed in groups II and III compared with that in group IV needs further investigation, and the rOmp28-based ELISA needs to be evaluated on large sets of clinical samples from these groups representing different geographical regions. No promising results were observed in the cases of native and recombinant Omp31 antigens in ELISAs with samples of suspected brucellosis from the four groups (data shown in Fig. 3). Similarly, correlations of ELISAs with RBPTs, STATs, and 2ME tests using the rOmp28 antigen were 92.7%, 94.2%, and 96.2%, respectively, higher than those for rOmp31 and native antigens (Table 2). The poor correlation of ELISAs with RBPTs and STATs prior to 2ME treatment observed in this study may be attributed to the fact that RBPTs and STATs measure total amounts of agglutinating antibodies (IgM and IgG), whereas 2ME tests and ELISAs measure IgG antibodies only. The rOmp28 ELISA needs to be modified further for the detection of both IgM and IgG simultaneously or individually for differentiation of acute and chronic cases of brucellosis. However, these results suggest that rOmp28 has an immense diagnostic potential for the screening of clinical samples of suspected brucellosis. Therefore, in comparison with rOmp31 and native antigens, rOmp28 was found to be the ideal candidate for development of a serodiagnostic test system for human brucellosis. Additionally, the diagnostic test developed with this recombinant antigen will not only provide high sensitivity and specificity but also take care of the safety aspects associated with handling Brucella species and the preparation of native antigens in the laboratory. The commercial kits available for the diagnosis of brucellosis are not very popular for routine use in the developing world due to their high costs. The high sensitivity and specificity of the indirect plate ELISA, together with its speed, capability for processing large numbers of samples simultaneously, risk reduction for laboratory personnel, and cost-effectiveness, make it a very useful tool for the serodiagnosis of brucellosis.
In conclusion, keeping in mind the importance of brucellosis as an emerging infection and the prevalence in India, we carried out the present study to compare recombinant antigens (rOmp28 and rOmp31) with native antigens (CE and SA) of Brucella for the serodiagnosis of human brucellosis by an indirect plate ELISA. The development of sensitive and specific ELISA techniques with the rOmp28 antigen will help in the early detection and management of this disease worldwide. However, large numbers of clinical samples from different geographical areas still need to be evaluated in the indirect plate ELISA system to determine the usefulness of these proteins in the serodiagnosis of human brucellosis.
ACKNOWLEDGMENTS
We thank M. P. Kaushik and G. P. Rai, Microbiology division, Defence R&D Establishment, Gwalior, India, for their valuable support for this study. Sapana Tiwari thanks the Indian Council of Medical Research, New Delhi, India, for providing her Senior Research Fellowship.
Footnotes
Published ahead of print 12 June 2013
REFERENCES
- 1. Corbel M. 2006. Brucellosis in humans and animals. World Health Organization, Geneva, Switzerland [Google Scholar]
- 2. Corbel MJ. 1997. Brucellosis: an overview. Emerg. Infect. Dis. 3: 213– 221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Yagupsky P. 1999. Detection of Brucellae in blood cultures. J. Clin. Microbiol. 37: 3437– 3442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yagupsky P. 1994. Detection of Brucella melitensis by Bactec NR660 blood culture system. J. Clin. Microbiol. 32: 1899– 1901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Buchanan TM, Faber LC. 1980. 2-Mercaptoethanol Brucella agglutination test: usefulness for predicting recovery from brucellosis. J. Clin. Microbiol. 11: 691– 693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Vizcaíno N, Cloeckaert A, Zygmunt MS, Dubray G. 1996. Cloning, nucleotide sequence, and expression of the Brucella melitensis omp31 gene coding for an immunogenic major outer membrane protein. Infect. Immun. 64: 3744– 3751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gad El-Rab MG, Kambal AM. 1998. Evaluation of a Brucella enzyme immunoassay test (ELISA) in comparison with bacteriological culture and agglutination. J. Infect. 36: 197– 201 [DOI] [PubMed] [Google Scholar]
- 8. Baldi PC, Giambartolomei GH, Goldbaum FA, Abdon LF, Velikovsky CA, Kittelberger R, Fossati CA. 1996. Humoral immune response against lipopolysaccharide and cytoplasmic proteins of Brucella abortus in cattle vaccinated with B. abortus S19 or experimentally infected with Yersinia enterocolitica serotype O:9. Clin. Diagn. Lab. Immunol. 3: 472– 476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Thavaselvam D, Kumar A, Tiwari S, Mishra M, Prakash A. 2010. Cloning and expression of immunoreactive Brucella melitensis 28 kDa outer membrane protein (Omp28) gene and evaluation of its potential for clinical diagnosis of brucellosis. J. Med. Microbiol. 59: 421– 428 [DOI] [PubMed] [Google Scholar]
- 10. Laemmli UK. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227: 680– 685 [DOI] [PubMed] [Google Scholar]
- 11. Kumar A, Tiwari S, Thavaselvam D, Sathyaseelan K, Prakash A, Barua A, Arora S, Rao MK. 2012. Optimization and efficient purification of recombinant Omp28 protein of Brucella melitensis using Triton X-100 and β-mercaptoethanol. Protein Expr. Purif. 83: 226– 232 [DOI] [PubMed] [Google Scholar]
- 12. Lowry DH, Rosebrough NJ, Farr AI, Randall EJ. 1951. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 193: 265– 275 [PubMed] [Google Scholar]
- 13. Towbin H, Staehelin H, Gordon J. 1979. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc. Natl. Acad. Sci. U.S.A. 76: 4350– 4354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Alton GG, Jones LM, Pietz DE. 1975. Laboratory techniques in brucellosis, 2nd ed. World Health Organization, Geneva, Switzerland: [PubMed] [Google Scholar]
- 15. Wagner MA, Eschenbrenner M, Horn TA, Kraycer JA, Mujer CV, Hagius S, Elzer P, DelVecchio VG. 2002. Global analysis of the Brucella melitensis proteome: identification of proteins expressed in laboratory grown culture. Proteomics 2: 1047– 1055 [DOI] [PubMed] [Google Scholar]
- 16. Connolly JP, Comerci D, Alefantis TG, Walz A, Quan M, Chafin R, Grewal P, Mujer CV, Ugald RA, Delvecchio VG. 2006. Proteomic analysis of Brucella abortus cell envelope and identification of immunogenic candidate protein for vaccine development. Proteomics 6: 3767– 3780 [DOI] [PubMed] [Google Scholar]
- 17. Ruiz-Mesa JD, Sanchez-Gonzalez J, Reguera JM, Martin L, Lopez-Palmero S, Colmenero JD. 2005. Rose Bengal test: diagnostic yield and use for the rapid diagnosis of human brucellosis in emergency departments in endemic areas. Clin. Microbiol. Infect. 11: 221– 225 [DOI] [PubMed] [Google Scholar]
- 18. Muñoz PM, Martin CM, Morreal D, Gonzales D, Garin-Bastuji B, Diaz R. 2005. Efficacy of several serological tests and antigens for diagnosis of bovine brucellosis in the presence of false positive serological tests due to Yersinia enterocolitica O:9. Clin. Diagn. Lab. Immunol. 12: 141– 145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Araj GF, Lulu AR, Mustafa MY, Khateeb MI. 1986. Evaluation of ELISA in the diagnosis of acute and chronic brucellosis in human beings. J. Hyg. (Lond.) 97: 457– 469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hunter SB, Bibb WF, Shih CN, Kaufmann AF, Mitchell JR, Mckinney RM. 1986. Enzyme-linked immunosorbent assay with major outer membrane proteins of Brucella melitensis to measure immune response to Brucella species. J. Clin. Microbiol. 24: 566– 568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cloeckaert A, Vizcaino N, Paquet JY, Bowden Elzer RA HR. 2002. Major outer membrane proteins of Brucella spp: past, present and future. Vet. Microbiol. 90: 229– 247 [DOI] [PubMed] [Google Scholar]
- 22. Al Dahouk S, Nöckler K, Scholz CH, Tomaso H, Bogumil R, Neubauer 2006. Immunoproteomic characterization of Brucella abortus 1119-3 preparations used for the serodiagnosis of Brucella infections. J. Immunol. Methods 309: 34– 37 [DOI] [PubMed] [Google Scholar]