

Abstract
A live attenuated vaccine candidate strain (M2) of human metapneumovirus (hMPV) was generated by removing the N-linked carbohydrate at amino acid 172 in the fusion (F) protein. Previously, replication of M2 in mouse lungs could be detected by molecular assays but not by viral titration. In the present study, the protective effects of M2 against infection by homologous or heterologous viruses were evaluated in BALB/c mice. Immunization with M2 produced a high titer of serum virus-neutralizing antibodies in BALB/c mice at 4 and 8 weeks postimmunization, with the titers against the homologous virus being higher than those against the heterologous virus. Challenges at 4 and 8 weeks postinoculation with M2 or wild-type virus led to no replication when mice were challenged with a homologous virus and extremely reduced replication when mice were challenged with a heterologous virus, as determined by the detection of viral genomic RNA copies in the lungs, as well as significantly milder pulmonary pathology. Thus, M2, with only one N-linked carbohydrate removed in the F protein, provides complete protection from homologous virus infection and substantial cross-protection from heterologous virus infection for at least 56 days after inoculation. This vaccine strain may therefore be a candidate for further preclinical study. Furthermore, this attenuating strategy (changing the glycosylation of a major viral protein) may be useful in the development of other viral vaccines.
INTRODUCTION
Human metapneumovirus (hMPV) was first isolated from the nasopharyngeal aspirates of young children suffering from acute respiratory tract diseases in the Netherlands in 2001 (1). It has been characterized as the only human respiratory pathogen in the Metapneumovirus genus of the Paramyxoviridae family. Sequence analysis of hMPV isolates from various parts of the world has revealed two major genetic lineages (lineages A and B), each of which can be further divided into two sublineages (sublineages A1 and A2 and sublineages B1 and B2). The two main lineages, with prototype viruses NL/1/00 and NL/1/99 for lineages A and B, respectively, have been found to differ in antigenicity, which may lead to periodic reinfection and circulation around the world (1–6).
The clinical severity of hMPV warrants the development of vaccines, particularly for the pediatric population, immunocompromised individuals, and the elderly. Since the discovery of hMPV, a variety of studies on vaccines for hMPV have been conducted in rodents and nonhuman primates (7). These have included live attenuated vaccines (8–11), subunit vaccines (4, 5, 12), a T-cell epitope vaccine (13), heat-inactivated vaccines (14), and formalin-inactivated (FI) vaccines (15, 16). Some studies on FI vaccines have indicated that classical inactivated vaccines for hMPV might predispose the host to enhanced pulmonary disease, as is the case with the vaccine for a close relative of hMPV, the FI respiratory syncytial virus (RSV) vaccine (17, 18). Subunit vaccines usually induce conservative protective antibodies, which provide complete or nearly complete protection of the host from hMPV infection over time (4, 12). However, no licensed vaccine has thus far been developed for clinical use against this human pathogen.
Live attenuated viruses have the advantage of mimicking a natural infection and thus can provide better protection against subsequent infections in immunologically naive individuals (8–11). Therefore, live attenuated vaccines may be more useful for priming or boosting hMPV-specific immune responses in young children. We previously generated a live attenuated recombinant vaccine candidate strain of hMPV, designated M2, by removing the N-linked carbohydrate at amino acid 172 in the fusion (F) protein (19). M2 led to a profoundly impaired growth capacity compared with that of wild-type recombinant virus NL/1/00 (the prototype strain of lineage A) both in Vero E6 cells and in mouse lungs. At the same time, pulmonary pathology following M2 infection was markedly milder than that following infection by the parent virus of M2, wild-type (WT) recombinant hMPV strain NL/1/00 expressing green fluorescent protein (GFP), referred to as NL/1/00-GFP. Thus, M2 has been considered to be substantially attenuated and therefore a candidate vaccine strain for hMPV. In the present study, we evaluated the protective effect of immunization with M2 against infection with hMPV of both lineages in BALB/c mice.
MATERIALS AND METHODS
Cells and viruses.
Vero E6 (African green monkey kidney) cells were purchased from the American Type Culture Collection (ATCC) and were grown in Dulbecco's minimum essential medium (DMEM; Gibco) containing 5% fetal bovine serum (FBS; Gibco), 2 mM l-glutamine, 100 μg/ml streptomycin, and 100 IU/ml penicillin. Recombinant NL/1/00-GFP (WT) and recombinant NL/1/99 (without GFP) viruses were recovered from cloned cDNA, as described previously (19). The former was used for the initial inoculation and the subsequent postinoculation challenge, and the latter was used only for the postinoculation challenge. Recombinant live attenuated hMPV (M2) was prepared as previously described (19). Briefly, the asparagine at position 172 in the F gene of WT was mutated into glutamine using a QuikChange mutagenesis kit (Stratagene), which led to the removal of an N-linked carbohydrate and extensive attenuation of the recovered virus. The GFP was inserted between the P and M protein genes of the viral genome to prepare purified viral stocks. Viruses were cultured in the infection medium, which consisted of DMEM supplemented with 3% FBS, 100 IU/ml penicillin, 100 μg/ml streptomycin, 2 mM glutamine, and 0.25 mg/ml trypsin (Sigma), until the cytopathic effect reached 80 to 90%. After one freeze-thaw cycle, cell-free supernatants were purified and concentrated using a 35% (wt/wt) sucrose gradient. Viral titers were determined by plaque assay as described below and expressed as the numbers of PFU per milliliter. More rounds of propagation were required to make a stock of M2 with a titer of over 107 PFU/ml. M2 was also propagated in Vero E6 cells 30 times. The full-length F-gene sequences of the input virus and the output virus at the endpoint were identical, and no additional mutations were found in over 30 passages, indicating that the genetic alteration in the F gene of M2 was stable.
Full-length sequence analysis of WT and M2 genomic RNA.
The WT and M2 strains were propagated 30 times in vitro, and then the viruses were purified using a 35% (wt/wt) sucrose gradient. RNA was isolated from highly purified WT and M2 virus strains using a QIAamp viral RNA minikit (Qiagen, CA), according to the manufacturer's instructions. RNA was extracted from a 140-μl sample and resuspended in 40 μl. Then, total RNA was reversibly transcribed into cDNA using a PrimeScript reverse transcription (RT) kit (TaKaRa, Shiga, Japan). Subsequently, the whole genomic sequence was amplified by serial overlapping PCR and sequenced by the Sanger method, and the 13.5-kb genes were amplified with 14 pairs of primers (data not shown). The PCR amplification conditions were as follows: 98°C for 30 s; 28 cycles of 98°C for 10 s, 58°C for 20 s, and 72°C for 30 s; and 72°C for 5 min. The sequencing was done with an ABI 3730 XL instrument (Applied Biosystems). For all of the PCR fragments, both strands were sequenced at least twice. The complete protein from the N terminus to the C terminus was constructed from the overlapping PCR fragments using the DNAStar program (version 8.0). Nucleic acid and amino acid sequences were analyzed by BLAST analysis of the sequences in GenBank and DNAMAN (version 6.0). In the case of sequence pairs with different bases, the fragments were amplified more than three times to make sure that the final information was accurate.
Study design.
Specific-pathogen-free, 6- to 7-week-old female BALB/c mice were purchased and housed under pathogen-free conditions in the animal research facility of Chongqing Medical University. One hundred ninety-two mice were assigned to 24 groups (n = 8 per group), and each group was housed separately. While they were under anesthesia, mice were intranasally inoculated with 107 PFU of WT or M2 virus in 100 μl of medium. As a control, mice were inoculated with 100 μl of DMEM. The mice in groups 1 to 12 were sacrificed at days 5, 14, 28, and 56 postinoculation (p.i.) to investigate viral growth and the production of neutralizing antibodies. At day 28 or 56 p.i., the mice in the other groups were challenged intranasally with 100 μl recombinant NL/1/00 or recombinant NL/1/99 (107 PFU). Five days later, the animals were sacrificed. The left lungs were collected for histopathology and RNA extraction, and the right ones were weighed for viral isolation and titration (Fig. 1). The study protocols were approved by The Ethics Committee of Chongqing Medical University.
Fig 1.
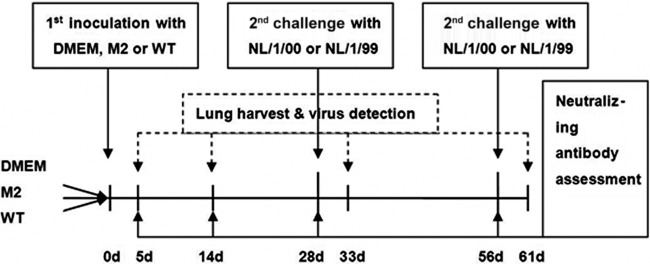
Study design. Schematic overview of the study. The time scale represents the number of days (d) after the initial inoculation with DMEM, M2, or WT. Eight mice in each group were sacrificed at 5, 14, 28, and 56 days p.i. At 28 and 56 days p.i., mice in six groups were challenged intranasally with 100 μl of 107 PFU of recombinant NL/1/00 or NL/1/99. Five days later, the animals were sacrificed.
Viral titration of harvested lungs.
A fluorescent plaque assay was used to titrate the viruses in the mouse lungs. Briefly, Vero E6 cells were plated into 6-well plates in DMEM containing 5% FBS (Gibco) and 2 mM l-glutamine. On the next day, the confluence of Vero E6 cells reached 60 to 70%. Clarified lung homogenates were serially diluted, added into the wells containing the Vero E6 cells, and then incubated for 2 h. The overlay, containing equal volumes of DMEM with 0.25% SeaPlaque GTG agarose (Lonza) and 2% FBS, was added to each well, and the plates were incubated for 4 days at 37°C. Then, green fluorescent plaques were counted under a fluorescence microscope. The resulting titers for each sample were divided by the lung weight and are reported as the numbers of PFU per gram of lung.
RT-PCR assay for viral genomic RNA in mouse lungs.
Total RNA was extracted from the mouse lungs with an EZgeno total RNA extraction kit (Genemega) according to the manufacturer's instructions. Total RNA was reversibly transcribed into cDNA using a PrimeScript RT kit (TaKaRa Bio Inc., Shiga, Japan). The F gene was amplified with the following primers: 5′-GTC TTG GAA AGT GGT GAT CAT-3′ (forward) and 5′-ACT GTG TCT GCG TCT TGG TT-3′ (reverse) (GenBank accession number AF371337) (20). At the same time, β-actin, an internal reference, was also amplified using primers 5′-GTG ACG AGG CCC AGA GCA AGA G-3′ (forward) and 5′-AGG GGC CGG ACT CAT CGT ACT C-3′ (reverse) under the following cycling conditions: 95°C for 4 min; 35 cycles of 94°C for 1 min, 55°C for 30 s, and 72°C for 1 min; and 72°C for 10 min. The expected sizes of the F and β-actin genes were 450 bp and 940 bp, respectively. The RT-PCR products were subjected to 1.5% agarose gel electrophoresis at 130 V for 40 min and photographed on a UV transilluminator.
Assessment of viral genomic RNA of recombinant hMPV NL/1/99 in mouse lungs by real-time RT-PCR.
In previous studies, recombinant hMPV NL/1/99 was detected by real-time RT-PCR assay or determination of the 50% tissue culture infective dose (4, 5, 21, 22). The recombinant NL/1/99 virus used in our lab, unfortunately, was not a GFP-expressing virus. To verify the presence of hMPV recombinant NL/1/99 in the lungs and the protective effect of M2 virus, a real-time RT-PCR assay, instead of a viral titration one, was used to quantitatively detect the replication of recombinant NL/1/99 in the mouse lungs. The primers were (GenBank accession number DQ336144) 5′-CGT TTC TTA CAT GCC GAC ATC TG-3′ (forward) and 5′-GCT CCC GTA GAC CCC TAT CAG-3′ (reverse), and the probe was 5′-(6-carboxyfluorescein) CCC TTT CTT CGC ACC ATC GCA CGG-3′ (Eclipse) (23). Each lung sample was tested three times by real-time PCR, and the mean copy number was obtained by using a standard curve generated by RNA runoff transcripts of a PCR product.
Microneutralization assay.
Serum virus-neutralizing (VN) antibody titers were determined by a microneutralization assay as described elsewhere, with slight modifications (24). Briefly, nine serial 2-fold dilutions from 1:20 to 1:5,120 were prepared in 50 μl of DMEM in 96-well plates. Fifty PFU of NL/1/00 or NL/1/99 virus in 50 μl of medium was added to the serum dilutions. All serum samples were tested in the presence of 10% guinea pig complement (Sigma). The serum-virus mixtures were incubated at 37°C in an atmosphere with 5% CO2 for 1 h. Positive-control wells without serum and negative-control wells without virus or serum were included in triplicate in each plate. Fifty microliters of the serum-virus mixture was added to Vero E6 cell monolayers in 96-well plates, and the plates were incubated for 1 h. The supernatants were then removed. Plates were blotted and washed with 100 μl per well DMEM, which was then replaced by fresh viral growth medium, followed by incubation at 37°C in an atmosphere with 5% CO2 for 4 days. Then, cells were fixed in 80% acetone in phosphate-buffered saline (PBS) for 15 min at 4°C, air dried, and blocked for 1 h in PBS containing 0.5% Tween 20 and 2% FBS. A monoclonal antibody against hMPV F protein (FAb 80120; Millipore) was diluted to 1:1,000 in PBS containing 1% FBS and 0.5% Tween 20 and added to the cells, followed by incubation at 37°C for 2 h. Cells were washed and treated with horseradish peroxidase-conjugated goat anti-mouse IgG (1:5,000) in PBS with 1% FBS–0.5% Tween 20 (50 μl/well) for 2 h at room temperature. After washing, 3′,3′,5′,5′-tetramethylbenzidine was added at 100 μl/well, followed by incubation at 37°C for 30 min. Then, H2SO4 was added at 35 μl/well to stop the reaction, and the neutralization titer was defined as the titer of serum that reduced color development by 50% normalized by that of the positive-control wells.
IFA to measure antibodies in sera from infected mice.
Briefly, Vero E6 cells were infected by recombinant NL/1/00 or NL/1/99 viruses and used as antigens for indirect fluorescence assay (IFA), as described by Ebihara et al (25). The Vero E6 cell suspension was prepared and washed. First, cell smears were created by spotting one drop of the cell suspension onto a glass slide, which was then air dried and fixed in acetone for 10 min. Smears were incubated with serum from all groups of mice for 30 min at 37°C, and those treated with monoclonal antibody against the hMPV F protein (FAb 80120; Millipore) served as positive controls. These smears were then incubated with fluorescein-conjugated Affinipure goat anti-mouse IgG (1:50) for 30 min at 37°C. After washing four times in PBS (5 min each), smears were air dried, stained with 0.001% Evans blue for 5 min, and then mounted with 35% glycerol in PBS. Under a fluorescence microscope, the presence of positive staining in at least one cell was defined as a positive result.
Pulmonary histopathology.
Left lungs were collected and fixed in 4% paraformaldehyde. Lungs were embedded in paraffin, cut into 4-μm sections, and stained with hematoxylin-eosin. The histopathologic score (HPS) was determined by a pathologist in a blind manner using the method described by Cimolai et al. (26).
Immunohistochemistry (IHC).
The lung sections were placed on slides and baked at 70°C for 48 h to remove the remaining paraffin. Then, the slides were heated in 2 mM citric acid (pH 7.4) in a microwave oven for 10 min for antigen retrieval. After cooling, they were rinsed four times with 0.01 M PBS containing 0.05% Tween 20. Nonspecific protein binding was blocked by goat serum. The slides were incubated at 4°C for 12 h with monoclonal antibody (diluted 1:1,000) against the hMPV F protein (FAb 80120; Millipore). Slides were successively incubated with biotinylated rabbit anti-mouse IgG (ZSGB-BIO, China) at 37°C for 1 h with horseradish peroxidase streptavidin at 37°C for 30 min and at room temperature with 3,3′-diaminobenzidine (DAB) for 3 min, rinsed with 0.01 M PBS four times, and stained with hematoxylin. Sections of positive samples without primary antibody during incubation or sections without challenge virus served as negative controls.
Statistical analysis.
Data are presented as the mean ± standard deviation (SD). If the viral titers were 0, the minimum detectable titer was assigned for statistical analysis. Statistical significance was determined using Student's t test and one-way analysis of variance (ANOVA), where a P value of <0.05 was considered statistically significant. All statistical analyses were performed with the SPSS program, version 2.
RESULTS
Sequence analysis.
Whole-gene sequences were constructed by assembling overlapping PCR fragments until the entire protein-encoding sequence from the N terminus to the C terminus was obtained. The presence of a GFP gene (750-bp) insertion between genes P and M was confirmed in WT and M2 strains. No additional gene mutation was found in the F gene, except for one artificial mutation in WT and three artificial mutations in M2 (Table 1). At position 3352, both the WT and M2 sequences were mutated from T to C, causing the substitution S101P. The virus with the S101P mutation did not require trypsin for growth in tissue culture (27, 28). Comparison of the WT and M2 sequences from nucleotides (nt) 3365 to 3367 showed that AAT mutated into CAG, resulting in the mutation N172Q in the F protein. This resulted in a lower replication efficiency in M2. The F protein was highly conserved, and its sequence was inconsistent with that of the F protein in pneumovirus, which has a functionally and structurally restricted amino acid mutation (29). There were four mutations (three missense mutations and one synonymous mutation) in the large polymerase (L) gene of virus M2 and two mutations (one missense mutation and one synonymous mutation) in the L protein gene of the WT virus (see Table 3). There were no mutations in domain III (amino acids 625 to 847 in the hMPV L protein open reading frame [ORF]) in the L protein, which was thought to be essential for a functional hMPV L ORF since it contained four core polymerase motifs (30, 31). One insertion was found in the noncoding region between the F and M2 genes. No deletions were detected.
Table 1.
Sequence analysisa
| Gene | Position | Nucleotide sequence or amino acid |
||
|---|---|---|---|---|
| NL/1/00 | WT | M2 | ||
| F | 3352 | TCT | CCT | CCT |
| 3565 | AAT | CAG | ||
| 3567 | AAT | CAG | ||
| L | 8335 | TGG | TTG | TTG |
| 10040 | AAT | AAA | ||
| 10045 | GCG | GAG | ||
| 12203 | TGC | TGT | TGT | |
| F/M2 | 4690 | Ins C | ||
| 4693 | Ins T | |||
| 4695 | Ins A | |||
| 4696 | Ins A | |||
| 4697 | Ins A | Ins A | ||
| 4699 | Ins A | |||
| 4700 | Ins A | |||
| 4701 | Ins A | |||
| 4703 | Ins T | |||
| CDS | 101 | S | P | P |
| 172 | N | Q | ||
| 172 | N | Q | ||
| 386 | W | L | L | |
| 958 | N | K | ||
| 973 | A | E | ||
| 1679 | C | C | C | |
For the F and L genes and the noncoding region between the F and M2 (F/M2), the nucleotide position is numbered from the 3′ end of the negative-sense RNA (GenBank accession no. AF371337). For CDS, the position is the protein position from the N terminus. Underlining indicates the nucleotide changes resulting in a mutated amino acid. F, fusion protein; L, large polymerase protein; M2, putative protein with a molecular mass of 22 kDa; NL/1/00, wild-type strain NL/1/00; Ins, insertion.
Table 3.
Viral titers in mouse lungs 5 days after challenge with recombinant NL/1/00a
| Time of challenge (day p.i.) | Inoculum | Mean viral titer (log10 PFU/g ± SD) |
|---|---|---|
| 28 | DMEM | 4.11 ± 0.28 |
| 28 | M2 | 0 |
| 28 | WT | 0 |
| 56 | DMEM | 4.56 ± 0.21 |
| 56 | M2 | 0 |
| 56 | WT | 0 |
All of the mice inoculated with the WT or M2 virus or mock inoculated were challenged with recombinant hMPV strain NL/1/00 at 28 or 56 days after the initial inoculation. Five days later, the mouse lungs were harvested for viral titration by fluorescent plaque assay. If the viral titers were 0, the minimum detectable titer was assigned for statistical analysis. At 28 and 56 days p.i., P < 0.001 for the M2 and WT groups versus the DMEM group; at 28 and 56 days p.i., P = 1 for the WT group versus the M2 group. All P values were determined by one-way ANOVA. The limit of detection was 100 PFU/g.
Replication of WT and M2 viruses in mouse lungs.
Our previous studies showed that the titer of the WT virus in BALB/c mouse lungs peaked on the 5th day postinfection, whereas at the same multiplicity of infection, M2 virus was undetectable in the mouse lungs 5 days after infection by plaque assay and viral isolation. However, molecular methods such as RT-PCR or real-time PCR were able to identify the viral RNA in the lungs. On the other hand, in severe combined immunodeficient (SCID) mice or cyclophosphamide-treated mice in our previous study, M2 was able to replicate at a low level and was detectable by plaque assay or culture. In the present study, the kinetics of replication of WT and M2 were assessed at 5, 14, 28, and 56 days p.i. The WT virus replicated to a mean titer of 4.94 log10 PFU/g on the 5th day p.i. On day 14 p.i., the WT virus was undetectable by plaque assay (Table 2) and viral culture but displayed a weak band following electrophoresis after amplification by PCR. M2 was detected at a very low level by the same PCR assay on the 5th day p.i. but was undetectable on the 14th day p.i. The findings presented above were consistent with those of our previous studies, indicating that the replication capacity of M2 was profoundly impaired compared with that of the WT in the mouse lungs. We also demonstrated that the replication level of the WT was close to that of recombinant NL/1/00 (without GFP; data not shown), indicating that the insertion of the GFP gene between the P and M genes did not have any significant influence on replication capacity.
Table 2.
Viral titers of the mouse lungs at 5 and 14 days p.i. with WT or M2a
| Day p.i. | Inoculum | Mean viral titer (log10 PFU/g ± SD) |
|---|---|---|
| 5 | DMEM | 0 |
| 5 | M2 | 0 |
| 5 | WT | 4.94 ± 0.56 |
| 14 | DMEM | 0 |
| 14 | M2 | 0 |
| 14 | WT | 0 |
Eight mice in each group were inoculated with DMEM (negative control) or the WT or M2 virus. The viral titer was measured by florescent plaque assay at 5 and 14 days p.i. If the viral titer was 0, the minimum detectable titer was assigned for the statistical analysis. P < 0.001 for the M2 and DMEM group versus the WT group; P = 1 for the DMEM group versus the M2 group. All P values were determined by one-way ANOVA. The limit of detection was 100 PFU/g.
Pulmonary histopathology at inoculation.
WT-infected mice had a higher HPS than M2-infected mice at 5 days p.i. (16.25 ± 1.04 versus 10.75 ± 1.04; P < 0.05) (Fig. 2). The abnormalities of the lungs of WT-infected mice were histopathologically characterized by abundant perivascular lymphocytic infiltrate and moderate peribronchiolar and bronchiolar lymphocytic infiltrates. These inflammatory responses were alleviated at 24 days p.i. The lungs of M2-infected mice had mild peribronchiolitis and perivasculitis at 5 days p.i. which had largely resolved at 14 days p.i. In the control group (inoculated with DMEM only), mice presented clear alveoli without an inflammatory response around the bronchioles or vessels and a low HPS of 2.63 ± 0.52. The HPS was consistent with the results of immunohistochemistry (IHC) and indirect fluorescence assay (IFA). More antigens were detected in lung sections from mice inoculated with the WT or recombinant NL/1/00 (without GFP) than in those from mice inoculated with M2 (Fig. 3), although this result was more qualitative than quantitative. Both recombinant NL/1/00 and WT replicated well in BALB/c mouse lungs. More positive fluorescence was detected in the membrane and cytoplasm of the epithelial cells of lungs infected with WT or recombinant NL/1/00 than in those of lungs infected with M2 (detected by confocal microscopy; data not shown).
Fig 2.
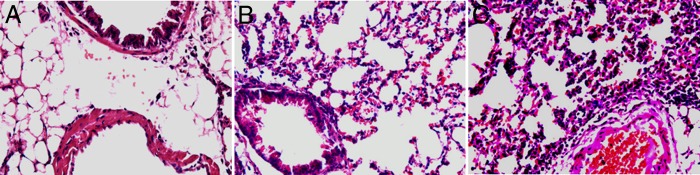
Histopathology. (A) BALB/c mouse lung inoculated with DMEM. At 5 days p.i., the lung was free of inflammatory cell infiltrates with normal pulmonary alveoli. The HPS was 2.63 ± 0.52. (B and C) BALB/c mouse lung inoculated with M2 (B) or WT (C) and at 5 days p.i. showing infiltration of mononuclear cells. The HPSs were 10.75 ± 1.04 (M2) and 16.25 ± 1.0 (WT). P < 0.05 for M2 versus WT (Student's t test). Hematoxylin-eosin stain was used. Magnifications, ×200 (A) and ×100 (B and C).
Fig 3.
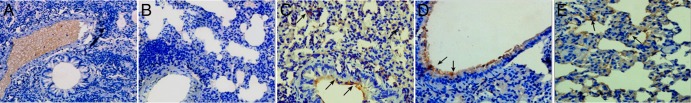
Immunohistochemistry. BALB/c mice were sacrificed at day 5 after the initial inoculation. (A) Lung from BALB/c mouse inoculated with DMEM only (no virus); (B) lung from BALB/c mouse inoculated with virus but not incubated with primary antibody; (C) NL/1/00-infected BALB/c mouse lung (arrows, plentiful antigens detected at the luminal surface and alveoli); (D) M2-infected BALB/c mouse lung (arrows, antigens detected at the luminal surface; fewer antigens were detected in alveoli); (E) WT-infected BALB/c mouse lung (arrows, antigens detected at the luminal surface). Magnifications, ×100 (A and B) and ×200 (C to E).
Neutralization antibody titers in serum of infected mice.
All serum samples collected from the infected mice at 5, 14, 28, and 56 days p.i. were subjected to measurement of titers of neutralizing antibodies against recombinant NL/1/00 and recombinant NL/1/99 (Fig. 4) by microneutralization assay. All the DMEM-inoculated mice were negative for neutralizing antibody to hMPV. Serum neutralizing antibodies against hMPV NL/1/00 were initially detectable at 5 days p.i. By 14 days p.i., the mean neutralizing titer continued to rise, reaching a plateau (means, 10.95 log2 units against NL/1/00 and 8.32 log2 units against NL/1/99) at 28 days p.i. that was maintained (approximately 10.57 log2 units against NL/1/00 and 7.90 log2 units against NL/1/99) throughout the 56 days p.i. Mice that received M2 inoculation had neutralizing antibody titers similar to those of mice infected with WT virus (P > 0.05). The titers of neutralizing antibodies against NL/1/00 were generally higher than those of antibodies against NL/1/99, with the highest homologous titers being in the WT-immunized animals. Mean homologous VN antibody titers induced by M2 or WT inoculation were 4- to 10-fold higher than heterologous antibody titers (Fig. 4), indicating serological differences between the two main lineages of hMPV, as described previously (6). The difference in protective effect was reflected in the difference between homologous and heterologous neutralizing antibody titers, as homologously challenged animals were completely protected from lower respiratory tract infection, but for the NL/1/99-challenged animals, virus RNA was detectable in their lungs. In addition, after inoculation with M2 or WT, there were enough virus antibodies to protect the lower respiratory tract from homologous reinfection completely and from heterologous reinfection partly, at least until 56 days p.i.
Fig 4.
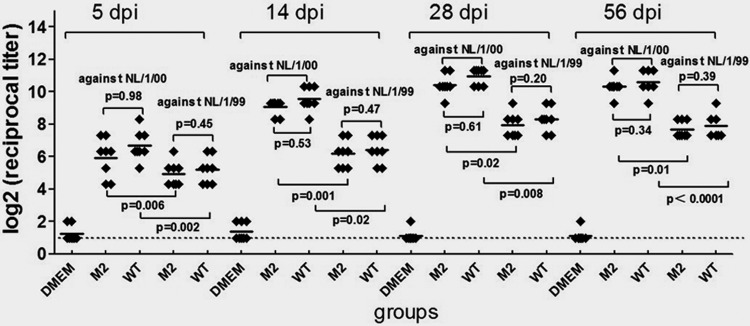
Microneutralization assay. The titers of antibodies against recombinant NL/1/00 and recombinant NL/1/99 after immunization with M2, WT, or DMEM are shown. Sera were collected at 5, 14, 28, and 56 days p.i. (dpi). All serum samples were tested in the microneutralization assay against NL/1/00 or against NL/1/99. Eight mice were in each group. P < 0.001 for the M2 and WT groups versus the DMEM group (one-way ANOVA, two-tailed). Dotted line, limit of detection; horizontal bars, means.
IFA to detect serum antibodies against recombinant NL/1/00 or recombinant NL/1/99 virus in infected mice.
IFA was used to visualize the patterns of the antibodies against both viruses in tissue. Sera from mock-infected mice did not recognize either recombinant NL/1/00- or recombinant NL/1/99-infected cells. Sera from WT- or M2-inoculated mice, however, invariably recognized both the recombinant NL/1/00 and recombinant NL/1/99 antigens, suggesting cross seroreactivity of the viruses from different subtypes. Since recombinant NL/1/99 replicated more slowly in Vero E6 cells in our lab, fewer antigens may have been present in the cells, which may have led to more sporadic fluorescence. The differences in the amounts of fluorescence from the sera collected at different time points were not significant, however, probably because this assay was not sensitive enough to detect such differences.
Viral replication of recombinant NL/1/00 in mouse lungs after challenge.
Viral replication in the mouse lungs was measured at 5 days after challenge with recombinant NL/1/00. As expected, for the recombinant NL/1/00-challenged animals, virus in the lungs of the WT groups and M2 groups was undetectable by plaque assay and viral isolation. In contrast, in the lungs of the DMEM-only group, the challenge virus replicated to mean titers of 4.11 log10 PFU/g and 4.56 log10 PFU/g at 28 and 56 days p.i., respectively (Table 3). Viral RNA was undetectable in the lungs of the WT groups and M2 groups at the same time points, but viral genomic RNA was readily detected in the lungs of the DMEM-only groups by PCR, indicating that M2 is able to provide complete protection against infection with homologous virus in BALB/c mice at levels similar to those conferred by WT vaccination.
Viral replication of recombinant NL/1/99 in mouse lungs after challenge.
The mice inoculated with the WT or M2 were challenged with the heterologous recombinant NL/1/99 virus at 28 and 56 days p.i. At 5 days after challenge, no recombinant NL/1/99 virus could be isolated from the lungs of mice inoculated with either WT or M2. In addition, no cytopathic effect (CPE) was seen and no fluorescence was detected by IFA. In contrast, only the control mice inoculated with DMEM showed viral replication by both CPE and IFA. Since the viral culture itself might not be sensitive enough to detect low-level viral replication, PCR and real-time PCR assays were employed to analyze viral genomic RNA from all groups challenged with recombinant NL/1/00 and recombinant NL/1/99 viruses. In accordance with the results of viral culture, the F fragment was not detected in the lungs of mice inoculated with the WT or M2 and then challenged with recombinant NL/1/00, but weak F-fragment expression was seen in mice inoculated with the WT or M2 and then challenged with the heterologous virus, recombinant NL/1/99. The expected bands of the F fragment that were amplified from the lungs of M2- and WT-immunized mice were significantly weaker than those amplified from the lungs of mice inoculated with DMEM only. Further, real-time RT-PCR revealed that the mean copy number in the lungs of the WT and M2 groups was reduced by at least 1,000-fold compared with that in the lungs of the DMEM-only group (Table 4), which indicated that M2 can provide partial protection against infection with hMPV of the heterologous genotype in BALB/c mice.
Table 4.
Number of copies of virus RNA in mouse lungs 5 days after challenge with recombinant NL/1/99a
| Time of challenge (day p.i.) | Inoculum | Mean log10 no. of copies/g ± SD) | Reduction of mean log10 no. of copies/g |
|---|---|---|---|
| 28 | DMEM | 8.18 ± 0.43 | |
| 28 | M2 | 4.66 ± 0.83 | 3.52 |
| 28 | WT | 4.31 ± 0.82 | 3.87 |
| 56 | DMEM | 8.23 ± 0.49 | |
| 56 | M2 | 4.70 ± 0.79 | 3.53 |
| 56 | WT | 4.79 ± 0.56 | 3.44 |
All of the mice inoculated with the WT or M2 virus or mock inoculated were challenged with recombinant hMPV strain NL/1/99 at 28 or 56 days after the initial inoculation. Five days later, mouse lungs were harvested for viral RNA measurement by real-time PCR. At 28 days p.i., P < 0.001 for the M2 and WT groups versus the DMEM group; at 28 days p.i., P = 0.34 for the WT group versus the M2 group. At 56 days p.i., P < 0.001 for the M2 and WT groups versus the DMEM group; at 56 days p.i., P = 0.77 for the WT group versus the M2 group. All P values were determined by one-way ANOVA.
Pulmonary pathology 5 days after challenge with recombinant NL/1/00 or NL/1/99.
To further assess the protective effects of M2 inoculation, pulmonary histopathology was examined at day 5 after challenge with recombinant NL/1/00. The lungs in the DMEM-only groups showed accentuated peribronchiolitis and perivasculitis, with high HPSs of 15.69 ± 0.80 (Fig. 5A) and 15.81 ± 0.75 (Fig. 5G), respectively. The lungs in the M2 groups showed significantly milder pulmonary pathological changes, with the HPSs being similar to those seen in the WT groups (6.19 ± 0.92 versus 5.5 ± 0.93 for challenge at day 28 [P = 0.14] and 6.38 ± 0.95 versus 6.06 ± 0.78 for challenge at day 56 [P = 0.46], respectively), indicating that both M2 and WT inoculation provided complete protection against recombinant NL/1/00. Viral titers were undetectable in the lungs of the WT groups and M2 groups, but a mild inflammatory response was noted, which may be attributed to immunopathogenesis or to transitory viral replication. Mice inoculated with M2 had a pathology similar to those inoculated with WT (P > 0.05). The degree of pathology in mice challenged with NL/1/99 at 28 or 56 days p.i. did not differ, but the HPSs these for mice were significantly higher than those for mice challenged with recombinant NL/1/00.
Fig 5.

Histopathology after challenge. (A, D, G, J) Control groups. Lung tissue from BALB/c mice initially inoculated with DMEM only and challenged with recombinant NL/1/00 (A and G) or recombinant NL/1/99 (D and J) at 28 days after the initial inoculation (days p.i.) (A and D) or 56 days p.i. (G and J); (B, E) lung tissue from BALB/c mice initially inoculated with M2 and challenged with recombinant NL/1/00 (B) or recombinant NL/1/99 (E) at 28 days p.i.; (H, K) lung tissue from BALB/c mice initially inoculated with M2 and challenged with recombinant NL/1/00 (H) or recombinant NL/1/99 (K) at 56 days p.i.; (C, F) lung tissue from BALB/c mice initially inoculated with WT and challenged with recombinant NL/1/00 (C) or recombinant NL/1/99 (F) at 28 days p.i.; (I, L) lung tissue from BALB/c mice initially inoculated with WT and challenged with recombinant NL/1/00 (I) or recombinant NL/1/99 (L) at 56 days p.i. For day 28 p.i., P = 0.14 for the M2 group versus the WT group after challenge with recombinant NL/1/00 and P = 0.1 for the M2 group versus the WT group after challenge with recombinant NL/1/99; for day 56 p.i., P = 0.46 for the M2 group versus the WT group after challenge with recombinant NL/1/00 and P = 0.69 for the M2 group versus the WT group after challenge with recombinant NL/1/99. Comparisons between groups were done by Student's t test. Magnifications, ×100.
DISCUSSION
Infants and young children are at increased risk for hMPV infection and related severe respiratory diseases, such as bronchiolitis and pneumonia (32–36). Since this population is most likely immunologically naive, traditional vaccines such as immunogenic protein-subunit and FI vaccines have been studied, but the latter were found to increase the likelihood of severe disease during subsequent infection after immunization (14–16). A study in cynomolgus macaques also found that the host-protective immunity conferred by these vaccines was transient (22). Therefore, an efficient candidate vaccine for hMPV should ideally be more immunogenic and protective than natural hMPV. Live attenuated vaccines contain most of the protective epitopes of WT virus, mimic the natural infection course, and are more likely to provide protection strong enough when used alone or in combination with other vaccination strategies.
hMPV has three viral transmembrane surface proteins: a small hydrophobic (SH) glycoprotein with unknown function, the G protein (a putative major attachment protein), and the fusion (F) protein, which mediates viral penetration and syncytium formation. F protein is synthesized as a precursor, and its activation requires cleavage by host cell protease (37, 38). Vector-expressed or mammalian cell-expressed F protein can induce high levels of neutralizing antibodies and produce moderate resistance to the replication of intranasally administered challenge virus, suggesting that F protein is one of the major antigens that induce protection against hMPV infection (4, 11, 12). In our previous study, we developed an attenuated hMPV NL/1/00 strain that has an N172Q substitution in the F protein that results in the removal of an N-linked carbohydrate. This virus, defined as M2, was genetically stable during multiple propagations and was able to express the F protein properly on the membrane of infected cells but showed a significantly impaired replication capacity both in vitro and in vivo. The immunoprotection conferred by M2 was similar to that conferred by WT virus. The present study demonstrated that M2 could induce high levels of neutralizing antibodies and produce complete resistance to challenge homologous virus, indicating that the removal of the carbohydrate at amino acid 172 altered the replication ability of the virus but did not alter immunoprotection. A good balance between attenuation and immunogenicity is usually difficult to obtain. However, the balance between the level of attenuation and induction of a specific immune response by M2 in BALB/c mice indicates that M2 may be an ideal vaccine candidate for hMPV.
Two major genetic subgroups of hMPV have been identified. They are antigenically related, on the basis of in vitro cross-neutralization, and some studies involving hamsters and primates have indicated that their similarity is as high as 48% or between 64% and 99%, respectively (39). The sequence of the F gene was highly conserved between the genetic subgroups compared with the level of conservation of the highly diverse membrane glycoproteins G and SH. F protein has therefore been thought to be a major antigen inducing cross-protection between the two genetic subgroups. Indeed, in several studies, vector-expressed F gene and purified F gene induced high levels of neutralizing antibodies and conferred protection against viral challenge to different degrees. Of note, the protection against homologous virus was higher than that against heterologous virus. It is still unclear whether a single-valent vaccine can provide satisfactory protection against both viral subgroups. Live attenuated viruses in which no whole-protein-encoding gene has been replaced provide a unique opportunity to assess the protective effect and the degree of cross-protection against all major genetic subgroups.
The growth of the recombinant hMPV NL/1/99 strain in mouse lungs was more troublesome. Lung samples did not yield clear plaques on Vero E6 cells by immunostaining. In addition, the recombinant NL/1/99 used in our lab was not a GFP-expressing virus, and thus, we used RT-PCR and real-time RT-PCR to evaluate the replication of this virus. The genome copy numbers in the groups immunized with M2 or WT were reduced by at least 1,000-fold compared with those in the group inoculated with DMEM only at 5, 28, and 56 days after challenge with recombinant NL/1/99. Meanwhile, the challenge recombinant NL/1/00 or NL/1/99 virus in the lungs of the immunized groups could not be detected by fluorescence plaque assay, whereas the DMEM-only group showed clear evidence of infection (for the DMEM groups, 4.11 log10 ± 0.28 PFU/g 5 days after challenge at day 28 and 4.56 log10 ± 0.21 PFU/g 5 days after challenge at day 56). In addition, several different detection methods indicated that immunization reduced the levels of both challenge viruses, recombinant hMPV NL/1/00 and recombinant NL/1/99. This indicates that M2 can hinder replication of hMPV lineages A and B upon subsequent infection, although the level of reduction was different.
M2 induced a high titer of neutralizing antibodies, as did WT virus. The titer peaked at 4 weeks p.i. and was maintained at >210 throughout 8 weeks p.i. The neutralizing antibody titer appeared to be higher than that induced by expressed F protein (40), probably because the whole virion more strongly stimulated the host and/or the BALB/c mice had a greater ability to produce antiviral antibodies. It is thought that there is a threshold of neutralizing antibody titers for protection against subsequent infection. Interestingly, M2 inoculation induced very high mean titers of neutralizing antibodies. The sera collected at 5 days, 2 weeks, 4 weeks, and 8 weeks p.i. could all recognize the virus, as demonstrated by IFA. The neutralizing antibody titer at 4 and 8 weeks p.i. was high enough to prevent infection by homologous virus, and thus, no virus was isolated from the lungs after 5 days postchallenge. However, the mean neutralizing antibody titer against recombinant NL/1/99 was significantly lower than that against recombinant NL/1/00. This was attributed to partial protection against challenge with recombinant NL/1/99. Due to the current lack of commercially available cotton rats in China, we had to choose mice as the animal model to assess the protective effect, and this may have led to a lower peak titer in the lungs at 5 days p.i. than that seen in cotton rats (41). Although the viral titer was approximately 4 log10 PFU/g, the absence of viruses in the lungs indicated that the protection provided by M2 immunization was complete. It remains unclear whether the high neutralizing antibody titer can be maintained and whether it will protect the host from challenge after 8 weeks p.i.
We neither assessed the viral replication in the upper respiratory tract after inoculation and challenge nor detected the virus-specific IgA level. Thus, it remains unknown whether M2 is capable of protecting rodents from hMPV infection of the upper respiratory tract. However, we can conclude that attenuated strain M2 provided complete protection against homologous virus infection and partial cross-protection against heterologous viral infection in the lower respiratory tract in BALB/c mice. It is essential to thoroughly study the immunogenicity, duration of immunity, and mechanism of immunity in a model closer to humans, such as nonhuman primates. Such research would help to evaluate whether M2 might indeed be a perfect attenuated vaccine for clinical use in the future.
ACKNOWLEDGMENTS
This work was supported by the National Natural Science Foundation of China (30730098, 30800972) and the Natural Science Foundation project of the Chongqing Scientific Committee (CSTC; 2010BB5372, 2012BB2145).
We appreciate Qiang-lin Duan from Tongji Hospital of Tongji University for critical reading of the manuscript. We thank Bu Zhigao and Ge Jinying (HarBin Veterinary Research Institute, Chinese Agricultural Academy of Science) for resuscitation of WT hMPV and mutant hMPV. We thank Ron A. M. Fouchier (Erasmus Medical Center Rotterdam, Rotterdam, Netherlands) for the rescue plasmids of NL/1/00.
Footnotes
Published ahead of print 12 June 2013
REFERENCES
- 1. Van den Hoogen BG, de Jong JC, Groen J, Kuiken T, de Groot R, Fouchier RA, Osterhaus AD. 2001. A newly discovered human pneumovirus isolated from young children with respiratory tract disease. Nat. Med. 7:719–724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Boivin G, Mackay I, Sloots TP, Madhi S, Freymuth F, Wolf D, Shemer-Avni Y, Ludewick H, Gray GC, LeBlanc E. 2004. Global genetic diversity of human metapneumovirus fusion gene. Emerg. Infect. Dis. 10:1154–1157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Galiano M, Videla C, Puch SS, Martínez A, Echavarría M, Carballal GE. 2004. Evidence of human metapneumovirus in children in Argentina. Med. Virol. 72:299–303 [DOI] [PubMed] [Google Scholar]
- 4. Herfst S, M de Graaf Schrauwen EJ, Ulbrandt ND, Barnes AS, Senthil K, Osterhaus AD, Fouchier RA, van den Hoogen BG. 2007. Immunization of Syrian golden hamsters with F subunit vaccine of human metapneumovirus induces protection against challenge with homologous or heterologous strains. J. Gen. Virol. 88:2702–2709 [DOI] [PubMed] [Google Scholar]
- 5. Herfst S, Schrauwen E, de Graaf M, van Amerongen G, van den Hoogen BG, de Swart RL, Osterhaus AD, Fouchier RA. 2008. Immunogenicity and efficacy of two candidate human metapneumovirus vaccines in cynomolgus macaques. Vaccine 26:4224–4230 [DOI] [PubMed] [Google Scholar]
- 6. van den Hoogen BG, Herfst S, Sprong L, Cane PA, Forleo E. 2004. Antigenic and genetic variability of human metapneumoviruses. Emerg. Infect. Dis. 10:658–666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Herfst S, Fouchier RA. 2008. Vaccination approaches to combat human metapneumovirus lower respiratory tract infections. J. Clin. Virol. 41:49–52 [DOI] [PubMed] [Google Scholar]
- 8. Biacchesi S, Pham QN, Skiadopoulos MH, Murphy BR, Collins PL, Buchholz UJ. 2005. Infection of nonhuman primates with recombinant human metapneumovirus lacking the SH, G, or M2-2 protein categorizes each as a nonessential accessory protein and identifies vaccine candidates. J. Virol. 79:12608–12613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Herfst S, de Graaf M, EJ Schrauwen, Sprong L, Hussain K, BG Van den Hoogen, Osterhaus AD, Fouchier RA. 2008. Generation of temperature-sensitive human metapneumovirus strains that provide protective immunity in hamsters. J. Gen. Virol. 89:1553–1562 [DOI] [PubMed] [Google Scholar]
- 10. Pham QN, Biacchesi S, Skiadopoulos MH, Murphy BR, Collins PL, Buchholz UJ. 2005. Chimeric recombinant human metapneumoviruses with the nucleoprotein or phosphoprotein open reading frame replaced by that of avian metapneumovirus exhibit improved growth in vitro and attenuation in vivo. J. Virol. 79:15114–15122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tang RS, Mahmood K, Macphail M, Guzzetta JM, Haller AA, Liu H, Kaur J, Stillman EA, Schickli JH, Fouchier RA, Osterhaus AD, Spaete RR. 2005. A host-range restricted parainfluenza virus type 3 (PIV3) expressing the human metapneumovirus (hMPV) fusion protein elicits protective immunity in African green monkeys. Vaccine 23:1657–1667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cseke G, Wright DW, Tollefson SJ, Johnson JE, Crowe JE, Williams JV. 2007. Human metapneumovirus fusion protein vaccines that are immunogenic and protective in cotton rats. J. Virol. 81:698–707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Herd KA, Mahalingam S, Mackay IM, Nissen M, Sloots TP, Tindle RW. 2006. Cytotoxic T-lymphocyte epitope vaccination protects against human metapneumovirus infection and disease in mice. J. Virol. 80:2034–2044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hamelin ME, Couture C, Sackett MK, Boivin G. 2007. Enhanced lung disease and Th2 response following human metapneumovirus infection in mice immunized with the inactivated virus. J. Gen. Virol. 88:3391–3400 [DOI] [PubMed] [Google Scholar]
- 15. De Swart RL, van den Hoogen BG, Kuiken T, Herfst S, van Amerongen G, Yuksel S. 2007. Immunization of macaques with formalin-inactivated human metapneumovirus induces hypersensitivity to hMPV infection. Vaccine 25:8518–8528 [DOI] [PubMed] [Google Scholar]
- 16. Yim KC, Cragin RP, Boukhvalova MS, Blanco JC, Hamlin ME, Boivin G, Porter DD, Prince GA. 2007. Human metapneumovirus: enhanced pulmonary disease in cotton rats immunized with formalin-inactivated virus vaccine and challenged. Vaccine 25:5034–5040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. De Swart RL, Kuiken T, Timmerman HH, van Amerongen G, Van Den Hoogen BG, Vos HW, Neijens HJ, Andeweg AC, Osterhaus AD. 2002. Immunization of macaques with formalin-inactivated respiratory syncytial virus (RSV) induces interleukin-13-associated hypersensitivity to subsequent RSV infection. J. Virol. 76:11561–11569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kim HW, Canchola JG, Brandt CD, Pyles G, Chanock RM, Jensen K, Parrott RH. 1969. Respiratory syncytial virus disease in infants despite prior administration of antigenic inactivated vaccine. Am. J. Epidemiol. 89:422–434 [DOI] [PubMed] [Google Scholar]
- 19. Zhang J, Dou Y, Wu J, She W, Luo L, Zhao Y, Liu P, Zhao X. 2011. Effects of N-linked glycosylation of the fusion protein on replication capacity of human metapneumovirus in vitro and in mouse lungs. J. Gen. Virol. 92:1666–1675 [DOI] [PubMed] [Google Scholar]
- 20. Peret TC, Boivin G, Li Y, Couillard M, Humphrey C, Osterhaus AD, Erdman DD, Anderson LJ. 2002. Characterization of human metapneumoviruses isolated from patients in North America. J. Infect. Dis. 185:1660–1663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Maertzdorf J, Wang CK, Brown JB, Quinto JD, Chu M, de Graaf M, van den Hoogen BG, Spaete R, Osterhaus AD, Fouchier RA. 2004. Real-time reverse transcriptase PCR assay for detection of human metapneumovirus from all known genetic lineages. J. Clin. Microbiol. 42:981–986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. van den Hoogen BG, Herfst S, de Graaf M Sprong L, van Lavieren R, van Amerongen G, Yüksel S, Fouchier RA, Osterhaus AD, de Swart RL. 2007. Experimental infection of macaques with human metapneumovirus induces transient protective immunity. J. Gen. Virol. 88:1251–1259 [DOI] [PubMed] [Google Scholar]
- 23. Chen X, Zhang Z, Zhao Y, Liu E, Zhao X. 2010. Acute lower respiratory tract infections by human metapneumovirus in children in Southwest China: a 2-year study. Pediatr. Pulmonol. 45:824–831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Falsey AR, Formica MA, Walsh EE. 2009. Microneutralization assay for the measurement of neutralizing antibodies to human metapneumovirus. J. Clin. Virol. 46:314–317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ebihara T, Endo R, Ma X, Ishiguro N, Kikuta H. 2005. Detection of human metapneumovirus antigens in nasopharyngeal secretions by an immunofluorescent antibody test. J. Clin. Microbiol. 43:1138–1141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cimolai N, Taylor G, Mah D, Morrison B. 1992. Definition and application of a histopathological scoring scheme for an animal model of acute Mycoplasma pneumoniae pulmonary infection. Microbiol. Immunol. 36:465–478 [DOI] [PubMed] [Google Scholar]
- 27. Schickli JH, Kaur J, Ulbrandt N, Spaete RR, Tang RS. 2005. An S101P substitution in the putative cleavage motif of the human metapneumovirus fusion protein is a major determinant for trypsin-independent growth in Vero cells and does not alter tissue tropism in hamsters. J. Virol. 79:10678–10689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Biacchesi S, Pham QN, Skiadopoulos MH, Murphy BR, Collins PL, Buchholz UJ. 2006. Modification of the trypsin-dependent cleavage activation site of the human metapneumovirus fusion protein to be trypsin independent does not increase replication or spread in rodents or nonhuman primates. J. Virol. 80:5798–5806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Spriggs MK, Olmsted RA, Venkatesan S, Coligan JE, Collins PL. 1986. Fusion glycoprotein of human parainfluenza virus type 3: nucleotide sequence of the gene, direct identification of the cleavage activation site, and comparison with other paramyxoviruses. Virology 152:241–251 [DOI] [PubMed] [Google Scholar]
- 30. Poch O, Blumberg BM, Bougueleret L, Tordo N. 1990. Sequence comparison of five polymerases (L proteins) of unsegmented negative-strand RNA viruses: theoretical assignment of functional domains. J. Gen. Virol. 71:1153–1162 [DOI] [PubMed] [Google Scholar]
- 31. Poch O, Sauvaget I, Delarue M, Tordo N. 1989. Identification of four conserved motifs among the RNA-dependent polymerase encoding elements. EMBO J. 8:3867–3874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Martinello RA, Esper F, Weibel C, Ferguson D, Landry ML, Kahn JS. 2006. Human metapneumovirus and exacerbations of chronic obstructive pulmonary disease. J. Infect. 53:248–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mullins JA, Erdman DD, Weinberg GA, Edwards K, Hall CB, Walker FJ, Iwane M, Anderson LJ. 2004. Human metapneumovirus infection among children hospitalized with acute respiratory illness. Emerg. Infect. Dis. 10:700–705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Vicente D, Montes M, Cilla G, Perez Trallero E. 2004. Human metapneumovirus and chronic obstructive pulmonary disease. Emerg. Infect. Dis. 10:1338–1339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Williams JV, Harris PA, Tollefson SJ, Halburnt-Rush LL, Pingsterhaus JM, Edwards KM, Wright PF, Crowe JE., Jr 2004. Human metapneumovirus and lower respiratory tract disease in otherwise healthy infants and children. N. Engl. J. Med. 350:443–450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Williams JV, Wang CK, Yang CF, Tollefson SJ, House FS, Eck JM, Chu M, Brown JB, Lintao LD, Quinto JD, Chu D, Spaete RR, Edwards KM, Wright PF, Crowe JE., Jr 2006. The role of human metapneumovirus in upper respiratory tract infections in children: a 20-year experience. J. Infect. Dis. 193:387–395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. De Graaf M, Herfst S, Schrauwen E, van Amerongen G, Osterhaus AD, Fouchier RA. 2009. Fusion protein is the main determinant of metapneumovirus host tropism. J. Gen. Virol. 90:1408–1416 [DOI] [PubMed] [Google Scholar]
- 38. Boivin G, Abed Y, Pelletier G, Ruel L, Moisan D, Côté S, Peret TC, Erdman DD, Anderson LJ. 2002. Virological features and clinical manifestations associated with human metapneumovirus: a new paramyxovirus responsible for acute respiratory-tract infections in all age groups. J. Infect. Dis. 186:1330–1334 [DOI] [PubMed] [Google Scholar]
- 39. Skiadopoulos MH, Biacchesi S, Buchholz UJ, Riggs JM, Surman SR, Amaro-Carambot E, McAuliffe JM, Elkins WR, St Claire M, Collins PL, Murphy BR. 2004. The two major human metapneumovirus genetic lineages are highly related antigenically, and the fusion (F) protein is a major contributor to this antigenic relatedness. J. Virol. 78:6927–6937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Williams JV, Chen Z, Cseke G, Wright DW, Keefer CJ, Hessell SJA, Podsiad A, Shepherd BE. 2007. A recombinant human monoclonal antibody to human metapneumovirus fusion protein that neutralizes virus in vitro and is effective therapeutically in vivo. J. Virol. 81:8315–8324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Williams JV, Tollefson SJ, Johnson JE, Crowe JE., Jr 2005. The cotton rat (Sigmodon hispidus) is a permissive small animal model of human metapneumovirus infection, pathogenesis, and protective immunity. J. Virol. 79:10944–10951 [DOI] [PMC free article] [PubMed] [Google Scholar]