

Abstract
Influenza A viruses cause annual epidemics and occasionally pandemics. Antibodies directed to the conserved viral nucleoprotein (NP) may play a role in immunity against various influenza A virus subtypes. Here, we assessed the immunological significance of a human monoclonal antibody directed to NP in vitro. This antibody bound to virus-infected cells but did not display virus-neutralizing activity, complement-dependent cell cytotoxicity, or opsonization of viral antigen for improved antigen presentation to CD8+ T cells by dendritic cells.
TEXT
Influenza A viruses cause epidemics, infecting millions of people worldwide and causing 250,000 to 500,000 deaths annually (1). In addition to annual epidemics, influenza A viruses cause pandemics occasionally. Recently, a pandemic was caused by the influenza virus A(H1N1)pdm09, which originated from swine. This recent pandemic and the ongoing circulation of other potentially pandemic influenza A viruses of the H5N1, H7N7, H7N9, H3N2v, and H9N2 subtypes have highlighted the importance of the development of vaccines that induce immunity against various influenza A virus subtypes, so-called heterosubtypic immunity. The presence of heterosubtypic immunity has been demonstrated in humans and various animal models, but our knowledge of the arms of the immune system and their viral targets that contribute to heterosubtypic immunity is still incomplete (2–6). Studies performed with animal models have demonstrated that virus-specific T cells contribute to heterosubtypic immunity since they are able to recognize conserved epitopes present in the internal proteins of influenza A virus (6–9). In addition, antibodies against the extracellular domain of the matrix 2 protein (M2e) and the stalk region of the hemagglutinin contribute to heterosubtypic immunity (10–12).
The role of antibodies to the influenza A virus nucleoprotein (NP) in heterosubtypic immunity is largely unknown, although studies in mice suggest that they can afford some degree of protection (13, 14). More than 2 decades ago, it was demonstrated that NP-specific monoclonal antibodies (MoAbs) were able to bind to virus-infected cells, suggesting that viral NP was present on the surface of the cell (15). Furthermore, the protection against infection observed in mice associated with NP-specific IgG antibodies correlated with accelerated viral clearance and involved Fc receptors and CD8+ cells (13, 14). Although human subjects with a history of influenza virus infection all develop influenza A virus NP-specific antibodies and the generation of a human monoclonal antibody against the influenza A virus NP has been described decades ago (16, 17), it is currently unknown if these antibodies have immunological significance and afford any cross-protective immunity in humans.
In the present paper, we assessed the biological activity of an influenza A virus NP-specific human monoclonal antibody in vitro. Methods used to obtain and select the human NP-specific monoclonal antibody (D1-11; MoAb NP) have been described previously (18). This study was preapproved by the institutional review boards of Emory University School of Medicine and the Oklahoma Medical Research Foundation (18). In brief, blood was collected from a healthy individual 7 days after vaccination with a seasonal influenza vaccine and single IgG+ antibody-secreting cells were sorted. Subsequently, a reverse transcription-PCR (RT-PCR) was performed on transcripts of VHDJH and VκJκ genes of these single-sorted cells, and after amplification by specific PCR, VHDJH and VκJκ genes were cloned into expression vectors containing constant human IgG (Cγ1) or Igκ (Cκ) regions. Plasmids of the heavy and light genes were cotransfected in 293T cells using the calcium phosphate precipitation method, and antibodies secreted in the supernatant were purified essentially as described previously (18, 19). The purity of the MoAb NP was tested by SDS-PAGE (data not shown), and the protein concentration was determined using a bicinchoninic acid (BCA) protein assay kit (Thermo Fisher Scientific Pierce Protein Biology Products, Rockford, IL). Subsequently, the specificity of the antibody for the influenza A virus NP was confirmed using an influenza A virus NP competitive enzyme-linked immunosorbent assay (ELISA) (Idexx, Hoofddorp, the Netherlands) according to the instructions of the manufacturer. In brief, purified MoAb was diluted 10 times in assay buffer and transferred to wells of a 96-well plate precoated with influenza A virus NP. After incubation for 1 h, various washing steps, and incubation with anti-influenza virus NP, horseradish peroxidase, and 3,3′,5,5′-tetramethylbenzidine, plates were read with a Tecan Infinite 200 reader (Tecan, Giessen, the Netherlands). In addition to the controls provided by the manufacturer, negative-control ferret serum and positive-control human serum were used as controls. A proportional difference in optical density (OD) value between sample and negative control of <0.6 was interpreted as positive as indicated by the manufacturer.
To test whether the monoclonal antibody was able to bind to NP present on the surface of influenza A virus-infected cells, A549 adenocarcinoma human alveolar basal epithelial cells, Epstein-Barr virus (EBV)-transformed human B cells, and Madin-Darby canine kidney (MDCK) cells were inoculated with influenza A(H3N2) virus RESVIR-9 with a multiplicity of infection (MOI) of 3. Influenza A(H3N2) virus RESVIR-9 is an influenza vaccine strain obtained after reassortment between influenza virus A/Nanchang/933/95 (HA, NA, NP) and influenza virus A/PR/8/34 (the remaining five gene segments). After incubation for 16 to 18 h at 37°C with 5% CO2, cells were trypsinized and subsequently incubated for 30 min with 10, 20, and 40 μg/ml of MoAb NP or isotype control (BD, Alphen a/d Rijn, the Netherlands) at 4°C. After washing, cells were stained with fluorescein isothiocyanate (FITC)-labeled MoAb directed against human IgG (Dako, Glostrup, Denmark). Subsequently, cells were fixed with Cytofix (BD, Alphen a/d Rijn, the Netherlands) and then assessed by flow cytometry with a FACSCanto II cytometer. The data were analyzed with FACSDiva software (BD, Alphen a/d Rijn, the Netherlands). All experiments were performed in duplicate, and uninfected cells were used as negative controls. As shown in Fig. 1, MoAb NP bound to virus-infected cells of all three cell lines, while no binding was observed to uninfected control cells. In addition, virtually no binding of the isotype control antibodies was observed (Fig. 1A). In addition, time course experiments indicated that NP could already be detected on the surface of infected cells within 1 h after inoculation (data not shown), suggesting the binding of free NP present in the inoculum. These findings confirm those originally obtained by Yewdell et al., who demonstrated the presence of NP on the surface of infected P815 cells by using a MoAb NP of murine origin (15).
Fig 1.
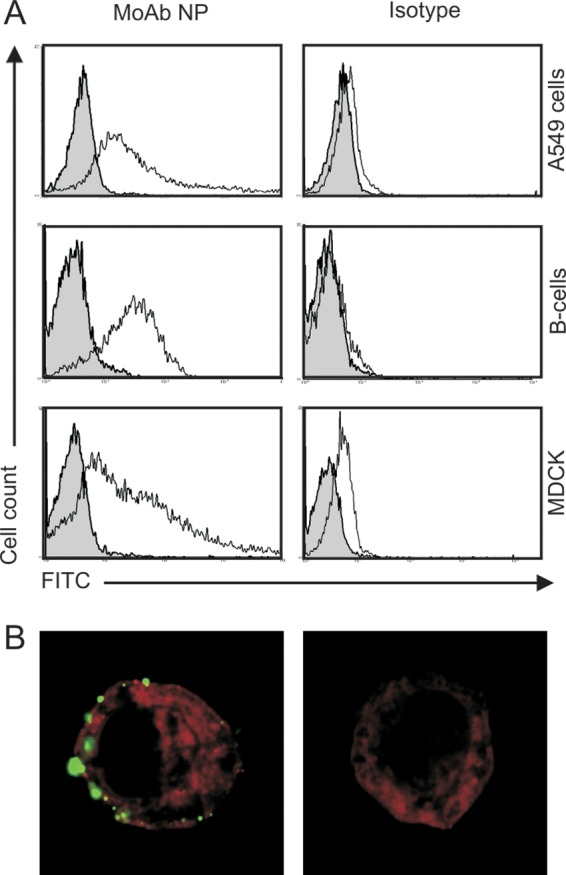
Detection of influenza A virus NP on the surface of influenza virus-infected A549 cells, B cells, and MDCK cells by MoAb NP. Cells were infected with influenza virus RESVIR-9 (H3N2) at an MOI of 3 (white) or not infected (gray) and after 16 to 18 h of incubation subsequently incubated with MoAb NP or isotype control antibody (20 μg/ml) as indicated, and binding of these antibodies was assessed after subsequent incubation with an FITC-labeled MoAb directed against human IgG by flow cytometry (A) or confocal microscopy (B).
In addition, the cellular localization of NP in virus-infected A549 cells was analyzed by confocal microscopy using a Zeiss LSM700 microscope in combination with Zeiss LSM Zen 2010 software (Carl Zeiss, Sliedrecht, the Netherlands). The same procedure as that described for flow cytometry was used, except that cells were also incubated with PKH-26 (Sigma-Aldrich, St. Louis, MO) to visualize cellular membranes. Also using this method, binding of MoAb NP to extracellular NP was observed on virus-infected cells, while virtually no binding was observed with the isotype control (Fig. 1B). Of interest, binding of MoAb NP to NP was not evenly distributed over the whole cell surface but displayed a patchy pattern, which is in accordance with the pattern of NP staining of infected mouse cells with a mouse monoclonal antibody specific for NP (15). Permeabilization of the infected cells prior to incubation with the NP-specific antibody resulted in the typical nuclear and cytoplasmic staining of NP, and also on these cells, binding of MoAb NP on the surface of cells was observed (data not shown).
To assess the broadness of reactivity of MoAb NP, A549 cells were inoculated with influenza A viruses A/Puerto Rico/8/1934 (H1N1), A/Swine/Netherlands/25/1980 (H1N1), A/Netherlands/246/2008 (H1N1), A/Netherlands/602/2009 (H1N1pdm09), A/HongKong/2/1968 (H3N2), and A/Netherlands/348/2007 (H3N2) and influenza B viruses B/Netherlands/151/2001 (Victoria lineage) and B/Netherlands/080/2002 (Yamagata lineage) at an MOI of 3 and subsequently tested by flow cytometry as described above. FITC-labeled MoAbs directed against the NP of either influenza A or influenza B virus (Imagen immunofluorescence test; Thermo Fisher Scientific, Waltham, MA) were used as positive controls.
Binding of the MoAb NP was observed to cells infected with any of the influenza A viruses but not to those infected with influenza B viruses. This confirms the broad reactivity of this antibody for influenza A viruses. However, differences were observed in the extent of binding, which might be due to differences in the biology of infection or due to differences in the affinity of the antibody for the respective NPs (Fig. 2) (20, 21).
Fig 2.
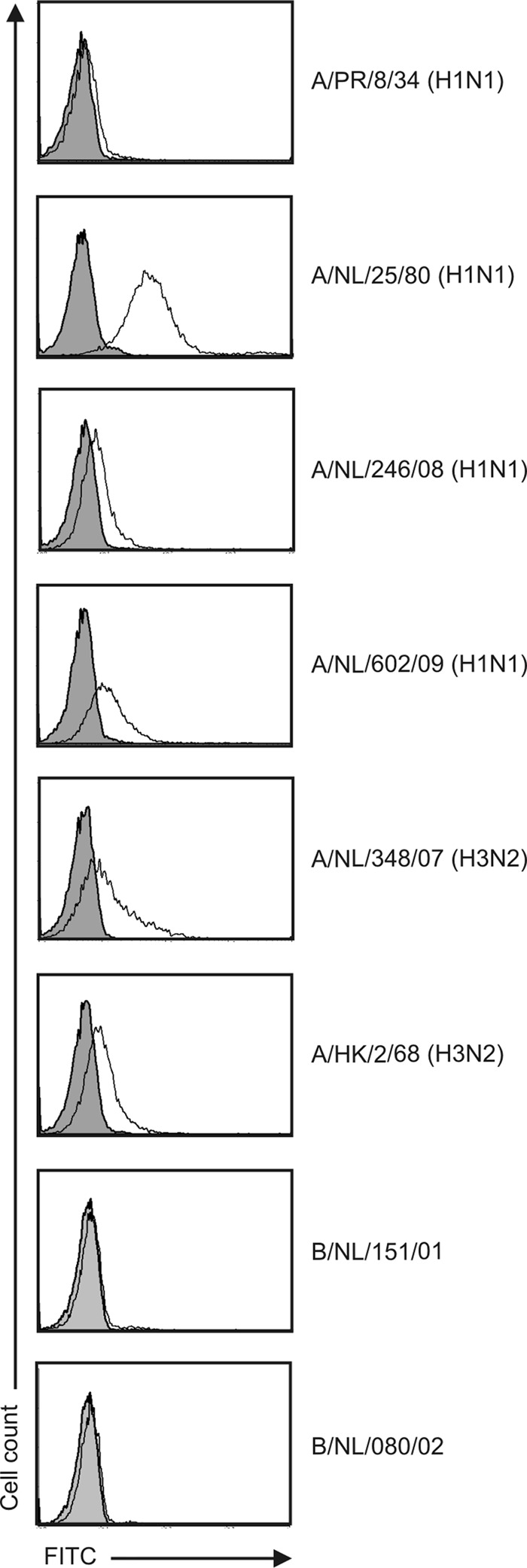
Detection of influenza virus NP on the surface of A549 cells infected with various influenza A and B viruses. Cells were infected with various influenza viruses at an MOI of 3 and incubated for 16 to 18 h. Subsequently, cells were incubated with MoAb NP (white) or isotype control antibody (gray; 20 μg/ml), and binding of these antibodies was assessed after subsequent incubation with an FITC-labeled MoAb directed against human IgG by flow cytometry.
Although the MoAb NP is highly cross-reactive and can bind to virus-infected cells, the role of this NP-specific MoAb in immunity against influenza remains elusive. Therefore, we performed in vitro experiments to assess possible roles of the MoAb NP in immunity against influenza A virus.
First, the virus-neutralizing capacity of the MoAb NP was tested using the infection reduction assay with influenza A viruses RESVIR-9 (H3N2) and A/PR/8/34 (H1N1) as described previously (22). Using this assay, which is more sensitive than the conventional virus neutralization assay (22), no reduction of infection was observed using 2.5, 5.0, 10.0, and 20.0 μg/ml of MoAb NP in contrast to postinfection ferret sera raised against the homologous strains that were used as positive controls. These findings indicate that this NP-specific MoAb is not capable of preventing infection of cells, which confirms previous results obtained with murine monoclonal antibodies specific for NP (Fig. 3A) (15).
Fig 3.
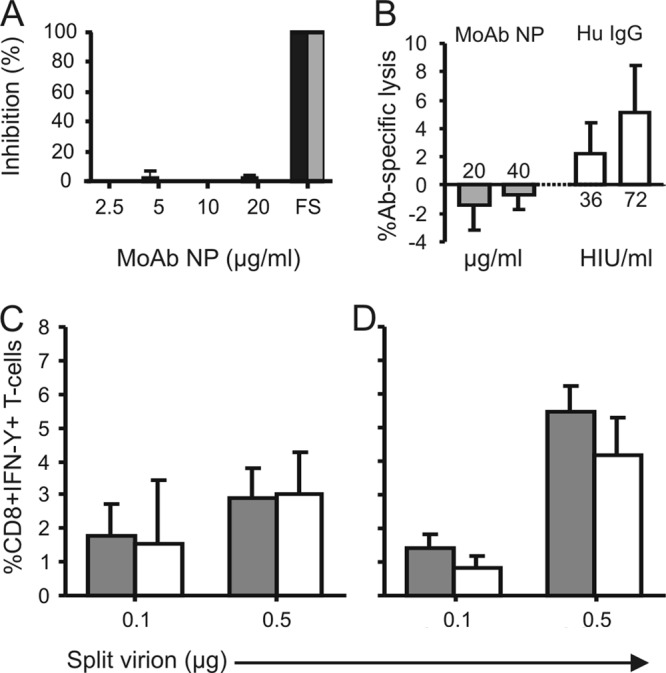
Immunologic activity of human NP-specific MoAb in vitro. (A) The virus-neutralizing activity of MoAb NP against influenza virus A/PR/8/34 (H1N1, black bars) or RESVIR-9 (H3N2, gray bars) was tested using an infectivity reduction assay as described previously (22) at various concentrations as indicated. No virus-neutralizing activity was observed, in contrast to the use of homologous positive-control postinfection ferret sera (FS). (B) MoAb NP did not mediate complement-dependent cytotoxicity of A549 cells (gray bars), in contrast to an IgG antibody preparation obtained from human serum with HI activity against the virus used (RESVIR-9 [H3N2]) (white bars). Since equivalent volumes were used for the human (Hu) IgG and the MoAb NP, the concentration of MoAb NP (μg/ml) is indicated or the multiplicity of the volume of human IgG that was able to prevent hemagglutination of 4 hemagglutinin units as measured in the HI assay (HI units [U]/ml) is indicated on the x axis. (C and D) Activation of virus-specific CD8+ T cells (CD8+ T cell clone specific for the HLA-B*3501-restricted epitope NP418–426 [LPFEKSTVM]) by DC incubated with various concentrations of split virion antigen preparation as measured by intracellular IFN-γ in the absence (gray bars) or presence (white bars) of MoAb NP with DC obtained from two different blood donors. Indicated are mean values ± standard deviations. The presence of MoAb NP did not have an effect on the activation of the NP418–426-specific CD8+ T cells.
Second, we tested whether the MoAb against NP could mediate complement-dependent cellular cytotoxicity (CDCC) as described previously for antibodies against the matrix 2 protein and the conserved region of the stalk protein of the hemagglutinin (10, 23). To this end, A549 cells were infected with influenza A virus RESVIR-9 (H3N2) at an MOI of 3 and 30,000 cells were incubated per well of a V-bottom 96-well plate for 3 h at 37°C in a humid atmosphere of 5% CO2. An interval of 3 h was used since it was demonstrated in pilot experiments that the recognition of NP on the surface of cells reached a plateau within 3 h after inoculation and this relatively short duration of infection resulted in the highest number of live cells with NP on the surface. Subsequently, cells were incubated with either 0, 20, or 40 μg/ml of MoAb NP for 30 min in phenol red-free RPMI medium containing 5% fetal calf serum, 100 IU/ml penicillin, 100 μg/ml streptomycin, 2 mM glutamine, and 20 μM β-mercaptoethanol. As a positive control, we used purified IgG of a human donor in which the presence of antibodies against the hemagglutinin of the RESVIR-9 virus was demonstrated using the hemagglutination inhibition (HI) assay (HI titer of 40). Then, 1:20-diluted, Low-Tox guinea pig complement (Cedarlane, Burlington, Ontario, Canada) was added, and after incubation for 2 h, complement-dependent lysis of cells was determined using the Cytotox 96 nonradioactive cytotoxicity assay (Promega, Leiden, the Netherlands) according to the manufacturer's recommendations. All experiments were performed in quintuplicate, and uninfected cells were used as negative controls. Antibody-specific lysis was calculated by subtracting the percent specific lysis measured for uninfected cells from the percent specific lysis of infected cells. Percent specific lysis was calculated using the following formula: (% lysis by antibody and complement − % lysis by complement only)/(% maximum lysis − % lysis by complement). Also in this assay, no activity of the MoAb NP was observed despite the use of high antibody concentrations of MoAb NP, in contrast to equivalent volumes of the positive-control IgG (Fig. 3B). These findings suggest that this antibody to the NP does not mediate CDCC, in contrast to antibodies against the hemagglutinin or the matrix 2 protein (10, 23). The differences are most likely explained by the fact that the hemagglutinin and matrix 2 protein are anchored in the cell membrane, while this has not been demonstrated and most likely is not the case for the nucleoprotein. Of interest, Yewdell and colleagues observed low levels of CDCC using a murine MoAb directed against the NP, which is in contrast to our observations (15). This might be related to differences between the assays used or the MoAb NP used.
It has been suggested that the protective effect of NP-specific antibodies observed in mice can be attributed to the formation of immune complexes that are processed by antigen-presenting cells, which subsequently results in accelerated and enhanced virus-specific CD8+ T cell responses. Indeed no protection was observed in the absence of memory T cells (14, 24). Therefore, we wished to investigate the effect of MoAb NP on opsonization of viral antigen, possibly resulting in improved uptake, processing, and antigen presentation to virus-specific CD8+ T cells by professional antigen-presenting cells. To test this possible mode of action, a split virion preparation of influenza A virus RESVIR-9 was produced by treating concentrated and purified virus stocks with decanoyl-N-methylglucamide (Mega-10; Sigma-Aldrich, Zwijndrecht, the Netherlands) for 30 min. After dialysis against phosphate-buffered saline (PBS) for 24 h, the protein concentration was determined using the BCA protein assay kit (Thermo Scientific Pierce Protein Biology Products, Rockford, IL), and full inactivation of the virus was confirmed as described previously (25). An 0-, 0.5-, or 1-μg amount of the split virion preparation was incubated with or without 1 μg MoAb NP for 1 h at 37°C and subsequently added to 100,000 human monocyte-derived dendritic cells (DC) from HLA-B*3501+ donors, which were prepared as described previously with monocytes derived from two different healthy blood donors (26). After incubation of DC with antigen and with or without MoAb NP for 16 to 18 h at 37°C, 30,000 cells of a CD8+ T cell clone specific for the HLA-B*3501-restricted epitope NP418-426 (LPFEKSTVM) (27) were added. Following incubation for 6 h in the presence of Golgistop (BD, Alphen a/d Rijn, the Netherlands), the production of gamma interferon (IFN-γ) by cells of the CD8+ T cell clone was assessed by flow cytometry using a FACSCanto II cytometer. The data were analyzed by FACSDiva software essentially as described previously, and specific production of IFN-γ was calculated by subtracting the percentage of IFN-γ+ T cells of medium controls from the DC incubated with the split virion preparation (28). Stimulation of the NP-specific CD8+ T cells with dendritic cells incubated with the split virion preparation induced IFN-γ production. However, preincubation of the split virion antigen preparation with MoAb NP did not increase the percentage of IFN-γ-producing T cells (Fig. 3C and D). These findings suggest that this MoAb NP does not improve uptake, processing, and antigen presentation to virus-specific CD8+ T cells by DC in vitro. In addition, no increased upregulation of costimulatory molecules CD80, CD83, and CD86 was observed after incubation of DC with split virion and MoAb NP compared to that with DC incubated with the split virion preparation only (data not shown). However, MoAb NP might improve endocytosis and presentation via the major histocompatibility complex (MHC) class II molecule to CD4+ T cells. Collectively, we have demonstrated that MoAb NP bound to viral nucleoprotein on the surface of virus-infected cells, but we were not able to demonstrate biological activity of immunological significance for this human antibody directed to the NP in various in vitro test systems.
The exact source and trafficking route of NP resulting in the patchy appearance on the surface of virus-infected cells are still unknown. In pilot studies using an NP-specific mouse monoclonal antibody, we observed that NP was already detectable on the surface of infected cells within 1 h after inoculation. Furthermore, we were not able to prevent this appearance by using the protein transport inhibitor brefeldin A, suggesting that it is not an active process, which was already suggested previously (15). Possibly, NP detected on the cell surface is acquired after release by other virus-infected cells or accumulated from the viral inoculum.
Of course, the in vitro test systems used here do not reflect the situation in vivo. In addition, the biological effect of NP-specific antibodies may be dependent on the epitopes that are recognized, and more antibodies against the NP should be tested to address this issue. Furthermore, the heavy chain constant region of the MoAb of the present study was of the IgG1 subclass, which may not be the same as that of the original antibody. However, IgG1, which is the most abundant subclass, is able to activate complement and to bind to Fc receptors on various cells of the immune system (29, 30). It cannot be excluded, however, that other subclasses may display different biological activity, and therefore. more subclasses should be tested. Thus, antibodies against the influenza A virus nucleoprotein might still play a role in heterosubtypic immunity, for example by cooperating with T cells to eliminate virus-infected cells in the lung (14). More research is necessary to address the potential of NP-specific antibodies to afford heterosubtypic immunity in humans, to elucidate their relative contribution to this type of immunity, and to elucidate the mechanisms of protection.
Footnotes
Published ahead of print 12 June 2013
REFERENCES
- 1.World Health Organization 2009. Influenza (seasonal) fact sheet no. 211. World Health Organization, Geneva, Switzerland: http://www.who.int/mediacentre/factsheets/fs211/en/index.html [Google Scholar]
- 2.Epstein SL. 2006. Prior H1N1 influenza infection and susceptibility of Cleveland Family Study participants during the H2N2 pandemic of 1957: an experiment of nature. J. Infect. Dis. 193:49–53 [DOI] [PubMed] [Google Scholar]
- 3.Cowling BJ, Ng S, Ma ES, Cheng CK, Wai W, Fang VJ, Chan KH, Ip DK, Chiu SS, Peiris JS, Leung GM. 2010. Protective efficacy of seasonal influenza vaccination against seasonal and pandemic influenza virus infection during 2009 in Hong Kong. Clin. Infect. Dis. 51:1370–1379 [DOI] [PubMed] [Google Scholar]
- 4.Bodewes R, Osterhaus AD, Rimmelzwaan GF. 2010. Targets for the induction of protective immunity against influenza A viruses. Viruses 2:166–188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kreijtz JH, Fouchier RA, Rimmelzwaan GF. 2011. Immune responses to influenza virus infection. Virus Res. 162:19–30 [DOI] [PubMed] [Google Scholar]
- 6.Grebe KM, Yewdell JW, Bennink JR. 2008. Heterosubtypic immunity to influenza A virus: where do we stand? Microbes Infect. 10:1024–1029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wilkinson TM, Li CK, Chui CS, Huang AK, Perkins M, Liebner JC, Lambkin-Williams R, Gilbert A, Oxford J, Nicholas B, Staples KJ, Dong T, Douek DC, McMichael AJ, Xu XN. 2012. Preexisting influenza-specific CD4+ T cells correlate with disease protection against influenza challenge in humans. Nat. Med. 18:274–280 [DOI] [PubMed] [Google Scholar]
- 8.Kreijtz JH, de Mutsert G, van Baalen CA, Fouchier RA, Osterhaus AD, Rimmelzwaan GF. 2008. Cross-recognition of avian H5N1 influenza virus by human cytotoxic T-lymphocyte populations directed to human influenza A virus. J. Virol. 82:5161–5166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hillaire ML, Vogelzang-van Trierum SE, Kreijtz JH, de Mutsert G, Fouchier RA, Osterhaus AD, Rimmelzwaan GF. 2013. Human T cells directed to seasonal influenza A virus cross-react with A(H1N1)pdm09 and swine origin triple reassortant H3N2 influenza viruses. J. Gen. Virol. 94:583–592 [DOI] [PubMed] [Google Scholar]
- 10.Grandea AG, III, Olsen OA, Cox TC, Renshaw M, Hammond PW, Chan-Hui PY, Mitcham JL, Cieplak W, Stewart SM, Grantham ML, Pekosz A, Kiso M, Shinya K, Hatta M, Kawaoka Y, Moyle M. 2010. Human antibodies reveal a protective epitope that is highly conserved among human and nonhuman influenza A viruses. Proc. Natl. Acad. Sci. U. S. A. 107:12658–12663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Steel J, Lowen AC, Wang TT, Yondola M, Gao Q, Haye K, Garcia-Sastre A, Palese P. 2010. Influenza virus vaccine based on the conserved hemagglutinin stalk domain. mBio 1(1):pii:e00018-10. 10.1128/mBio.00018-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kaminski DA, Lee FE. 2011. Antibodies against conserved antigens provide opportunities for reform in influenza vaccine design. Front. Immunol. 2:76. 10.3389/fimmu.2011.00076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Carragher DM, Kaminski DA, Moquin A, Hartson L, Randall TD. 2008. A novel role for non-neutralizing antibodies against nucleoprotein in facilitating resistance to influenza virus. J. Immunol. 181:4168–4176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.LaMere MW, Lam HT, Moquin A, Haynes L, Lund FE, Randall TD, Kaminski DA. 2011. Contributions of antinucleoprotein IgG to heterosubtypic immunity against influenza virus. J. Immunol. 186:4331–4339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yewdell JW, Frank E, Gerhard W. 1981. Expression of influenza A virus internal antigens on the surface of infected P815 cells. J. Immunol. 126:1814–1819 [PubMed] [Google Scholar]
- 16.Voeten JT, Groen J, van Alphen D, Claas EC, de Groot R, Osterhaus AD, Rimmelzwaan GF. 1998. Use of recombinant nucleoproteins in enzyme-linked immunosorbent assays for detection of virus-specific immunoglobulin A (IgA) and IgG antibodies in influenza virus A- or B-infected patients. J. Clin. Microbiol. 36:3527–3531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Crawford DH, Callard RE, Muggeridge MI, Mitchell DM, Zanders ED, Beverley PC. 1983. Production of human monoclonal antibody to X31 influenza virus nucleoprotein. J. Gen. Virol. 64:697–700 [DOI] [PubMed] [Google Scholar]
- 18.Wrammert J, Smith K, Miller J, Langley WA, Kokko K, Larsen C, Zheng NY, Mays I, Garman L, Helms C, James J, Air GM, Capra JD, Ahmed R, Wilson PC. 2008. Rapid cloning of high-affinity human monoclonal antibodies against influenza virus. Nature 453:667–671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Smith K, Garman L, Wrammert J, Zheng NY, Capra JD, Ahmed R, Wilson PC. 2009. Rapid generation of fully human monoclonal antibodies specific to a vaccinating antigen. Nat. Protoc. 4:372–384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.van Wyke KL, Hinshaw VS, Bean WJ, Jr, Webster RG. 1980. Antigenic variation of influenza A virus nucleoprotein detected with monoclonal antibodies. J. Virol. 35:24–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Miyoshi-Akiyama T, Yamashiro T, Mai le Q, NaraharaK, Miyamoto A, Shinagawa S, Mori S, Kitajima H, Kirikae T. 2012. Discrimination of influenza A subtype by antibodies recognizing host-specific amino acids in the viral nucleoprotein. Influenza Other Respi. Viruses 6:434–441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hillaire ML, van Eijk M, van Trierum SE, van Riel D, Saelens X, Romijn RA, Hemrika W, Fouchier RA, Kuiken T, Osterhaus AD, Haagsman HP, Rimmelzwaan GF. 2011. Assessment of the antiviral properties of recombinant porcine SP-D against various influenza A viruses in vitro. PLoS One 6:e25005. 10.1371/journal.pone.0025005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Terajima M, Cruz J, Co MD, Lee JH, Kaur K, Wrammert J, Wilson PC, Ennis FA. 2011. Complement-dependent lysis of influenza a virus-infected cells by broadly cross-reactive human monoclonal antibodies. J. Virol. 85:13463–13467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zheng B, Zhang Y, He H, Marinova E, Switzer K, Wansley D, Mbawuike I, Han S. 2007. Rectification of age-associated deficiency in cytotoxic T cell response to influenza A virus by immunization with immune complexes. J. Immunol. 179:6153–6159 [DOI] [PubMed] [Google Scholar]
- 25.Bodewes R, Kreijtz JH, Hillaire ML, Geelhoed-Mieras MM, Fouchier RA, Osterhaus AD, Rimmelzwaan GF. 2010. Vaccination with whole inactivated virus vaccine affects the induction of heterosubtypic immunity against influenza virus A/H5N1 and immunodominance of virus-specific CD8+ T-cell responses in mice. J. Gen. Virol. 91:1743–1753 [DOI] [PubMed] [Google Scholar]
- 26.Bodewes R, Geelhoed-Mieras MM, Heldens JG, Glover J, Lambrecht BN, Fouchier RA, Osterhaus AD, Rimmelzwaan GF. 2009. The novel adjuvant CoVaccineHT increases the immunogenicity of cell-culture derived influenza A/H5N1 vaccine and induces the maturation of murine and human dendritic cells in vitro. Vaccine 27:6833–6839 [DOI] [PubMed] [Google Scholar]
- 27.Boon AC, de Mutsert G, Graus YM, Fouchier RA, Sintnicolaas K, Osterhaus AD, Rimmelzwaan GF. 2002. Sequence variation in a newly identified HLA-B35-restricted epitope in the influenza A virus nucleoprotein associated with escape from cytotoxic T lymphocytes. J. Virol. 76:2567–2572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Berkhoff EG, Boon AC, Nieuwkoop NJ, Fouchier RA, Sintnicolaas K, Osterhaus AD, Rimmelzwaan GF. 2004. A mutation in the HLA-B*2705-restricted NP383-391 epitope affects the human influenza A virus-specific cytotoxic T-lymphocyte response in vitro. J. Virol. 78:5216–5222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jefferis R, Lund J. 2002. Interaction sites on human IgG-Fc for FcgammaR: current models. Immunol. Lett. 82:57–65 [DOI] [PubMed] [Google Scholar]
- 30.Schroeder HW, Jr, Cavacini L. 2010. Structure and function of immunoglobulins. J. Allergy Clin. Immunol. 125:S41–S52 [DOI] [PMC free article] [PubMed] [Google Scholar]