

Abstract
Mycobacterium tuberculosis, the causative agent of tuberculosis, is one of the most important bacterial pathogens. Recent work has revealed that the natural bactericidal properties of copper are utilized by the host immune system to combat infections with bacteria, including M. tuberculosis. However, M. tuberculosis employs multiple mechanisms to reduce the internal copper amount by efflux and sequestration, which are required for virulence of M. tuberculosis. Here, we describe an alternative mechanism of copper resistance by M. tuberculosis. Deletion of the rv0846c gene increased the susceptibility of M. tuberculosis to copper at least 10-fold, establishing Rv0846c as a major component of copper resistance in M. tuberculosis. In vitro assays showed that Rv0846c oxidized organic substrates and Fe(II). Importantly, mutation of the predicted copper-coordinating cysteine 486 resulted in inactive Rv0846c protein which did not protect M. tuberculosis against copper stress. Hence, Rv0846c is a multicopper oxidase of M. tuberculosis and was renamed mycobacterial multicopper oxidase (MmcO). MmcO is membrane associated, probably by lipidation after export across the inner membrane by the twin-arginine translocation system. However, mutation of the lipidation site did not affect the oxidase activity or the copper protective function of MmcO. Our study revealed MmcO as an important copper resistance mechanism of M. tuberculosis, which possibly acts by oxidation of toxic Cu(I) in the periplasm.
INTRODUCTION
Mycobacterium tuberculosis, the causative agent of tuberculosis, kills over one million people each year, and the global occurrence of multidrug and extensively drug-resistant strains is increasing (1). Understanding the survival mechanisms of M. tuberculosis in the human host is key to controlling this devastating disease. In addition to the known mechanisms of the innate and adaptive immune response to infections (2, 3), it is increasingly recognized that copper is utilized by macrophages to kill pathogenic bacteria (4, 5). In most organisms, copper is used as a cofactor in a variety of enzymes, including cytochrome c oxidases, and is therefore an essential micronutrient (6). However, copper is also toxic by a variety of mechanisms: lipid peroxidation (7), metal ion replacement in proteins (8), formation of spurious disulfide bonds (9), and oxidation and degradation of iron-sulfur clusters in proteins (10). Hence, cells employ defense mechanisms against copper poisoning while maintaining sufficient intracellular copper levels (11, 12).
Copper also is utilized in host immune systems to prevent infection (reviewed in reference 13). Not only is copper required for proper development of the immune system (14), but also, new evidence shows that copper is employed at a cellular level to kill invading bacteria. Macrophages increase intracellular copper concentrations in response to multiple bacteria, including M. tuberculosis (5, 15, 16). Additionally, we have shown that copper accumulates in granulomas of guinea pigs infected with M. tuberculosis and that copper resistance is required for full virulence in M. tuberculosis (4).
The mechanisms of copper homeostasis in mycobacteria include copper efflux and sequestration of cytoplasmic copper by the metallothionein MymT (13, 17–19). Multicopper oxidases play a crucial role in copper detoxification in many bacteria, including Escherichia coli (20), Pseudomonas syringae (21), Salmonella enterica (22), and others (23, 24). Multicopper oxidases are also required for virulence in Salmonella (22) and Xanthomonas campestris (25). However, it is unknown whether the putative multicopper oxidase Rv0846c plays a role in copper resistance in M. tuberculosis. Multicopper oxidases catalyze the single-electron oxidation of four substrate equivalents coupled with the reduction of oxygen to water (26). Four copper ions coordinated in two centers comprise the multicopper oxidase active site (27). The type 1 copper center is the site of substrate oxidation, and the Cu coordination by cysteine gives multicopper oxidases their characteristic blue color (27). The coupled type 2 and type 3 copper centers are the site of oxygen reduction (27). Multicopper oxidases are conserved throughout all kingdoms of life, and prominent examples include fungal laccases and human ceruloplasmin (26, 27). Multicopper oxidases can oxidize a variety of substrates, including phenolic compounds such as siderophores and lignins (26).
Here we show that Rv0846c is a bona fide multicopper oxidase capable of oxidizing organic substrates and Fe(II). Thus, we have renamed Rv0846c MmcO, for mycobacterial multicopper oxidase. Importantly, the susceptibility of the ΔmmcO mutant to copper in vitro was increased more than 10-fold compared to that of wild-type M. tuberculosis, indicating that MmcO also oxidizes toxic Cu(I). We also showed that the conserved residue cysteine 486 in the active site is required for oxidase activity and copper resistance. MmcO is membrane associated, possibly through a lipidation site at cysteine 35. Together, these results demonstrate that MmcO plays an important role in copper resistance in M. tuberculosis.
MATERIALS AND METHODS
Chemicals, enzymes, and DNA.
Hygromycin B was purchased from Calbiochem. All other chemicals were purchased from Merck, Roche, or Sigma at the highest purity available. Enzymes for DNA restriction and modification were purchased from New England BioLabs. Isolation and modification of DNA was performed using standard protocols (28). Oligonucleotides were obtained from Integrated DNA Technologies (see Table 2).
Table 2.
Oligonucleotides used in this work
| Oligonucleotide | Sequence (5′→3′)a |
|---|---|
| CN110 | CGTTCTCGGCTCGATGATCC |
| CN145 | CGACCAGCACGGCATACATC |
| CN1695 | AAGCATGCGCAGAAAggaggttaatATGCCCGAGCTGGCCACGAG (SphI) |
| CN1698 | AAATGTCACAGAATGTAGTCCAG (SwaI half site) |
| CN1700 | AAATCTCGTCTGGATTTGGTCTCGCTC (SwaI half site) |
| CN1702 | ACGAGGTTAATTAACTACATTCTGTGACAGGCGGCTTG (PacI) |
| CN1703 | CATATGCATGGTGCTGGCCTGTACGCTAG (NsiI) |
| CN2398 | CATGGTTCGAAAAGCTTATTTAAATCTACTTCTCGAACTGCGGG (HindIII) |
| CN2484 | GGAGATATACATATGGCCTCGAAGCCCACGGCATCCGGCGCC (NdeI) |
| CN2863 | GCTTTGCGCTAGCCGCTGCTGCCTCGAAGC |
| CN2873 | GTGTGGGTGATGCACGCCCACAACAACTATCAC |
| CN3119 | ACTAGTCACCTTACCAGCGAGGGCTAG (SpeI) |
| CN3120 | TAGCTAAAGCTTATTTAAATCTACTTCTCGAACTGCGGGTGCGACCAAGCGGCCGCCAGAATGTAGTCCAGGCGGGTCGC (HindIII) |
Restriction sites are underlined. The sequence encoding the Strep-Tag II is in bold. Mutations are italicized and bold. The ribosome binding site is in lowercase.
Bacterial strains, media, and culture conditions.
Bacterial strains used in this work are described in Table 1. E. coli was grown routinely in LB medium at 37°C with shaking. M. tuberculosis H37Rv and its derivatives were grown in Middlebrook 7H9 liquid medium supplemented with 0.2% glycerol, OADC (8.5 g/liter NaCl, 20 g/liter dextrose, 50 g/liter bovine albumin [fraction V], 0.03 g/liter catalase, 0.6 ml/liter oleic acid), and 0.02% tyloxapol or on Middlebrook 7H10 agar supplemented with 0.5% glycerol using premixed powders (Difco). The avirulent M. tuberculosis strain mc26230 (kind gift from Bill Jacobs) and its derivatives were grown in the same media as H37Rv with the addition of 24 μg/ml pantothenate and 0.2% Casamino Acids (acid hydrolyzed). Copper was added when required in the form of CuSO4 at various concentrations. Antibiotics were used at the following concentrations when required: hygromycin (Hyg), 200 μg/ml for E. coli and 50 μg/ml for mycobacterial strains; kanamycin (Kan), 30 μg/ml.
Table 1.
Strains and plasmids used in this work
| Strain or plasmid | Parent strain and relevant genotype (strains) or parent vector, relevant genotype, and properties (plasmids)a | Source or reference |
|---|---|---|
| Strains | ||
| E. coli DH5α | recA1 endA1 gyrA96 thi relA1 hsdR17(rK− mK+) supE44 ϕ80ΔlacZΔM15 ΔlacZYA-argF(U169) | Sambrook et al. (28) |
| E. coli BL21(DE3) | fhuA2 [lon] ompT gal (λ DE3) [dcm] ΔhsdS λ DE3 | Novagen |
| M. tuberculosis H37Rv | Wild type | ATCC 25618 |
| M. tuberculosis mc26230 | H37Rv ΔRD1 ΔpanCD | Bill Jacobs (39) |
| M. tuberculosis ML1221 | mc26230 ΔmmcO::pML1649(loxP-gfpm2+-hyg-loxP) | This study |
| M. tuberculosis ML1222 | mc26230 ΔmmcO::loxP | This study |
| M. tuberculosis ML413 | mc26230 Δrv1698::loxP | This study |
| M. tuberculosis ML1223 | H37Rv ΔmmcO::pML1649(loxP-gfpm2+-hyg-loxP) | This study |
| M. tuberculosis ML1224 | H37Rv ΔmmcO::loxP | This study |
| Plasmids | ||
| pET-21(a+) | T7 promoter, transcription start, and terminator, His tag, lacI aph, pBR322 ORI; 5,443 bp | Novagen, WI |
| pCreSacB1 | pgroEL-cre oriE oriM sacR sacB aph; 7,891 bp | Adrie Steyn |
| pMS2 | ColE1 origin, hyg oriM; 5,229 bp | 74 |
| pMN016 | ColE1 origin, hyg oriM psmyc-mspA; 6,164 bp | 29 |
| pML523 | pUC origin, pAL5000ts origin; sacB xylE loxP-gfpm2+-hyg-loxP; 9,845 bp | 30 |
| pML1648 | pML523; mmcOup-loxP-gfpm2+-hyg-loxP; 10,875 bp | This study |
| pML1649 | pML1648; loxP-gfpm2+-hyg-loxP-mmcOdown; 11,857 bp | This study |
| pML1227 | pMN016; mmcO-Strep-Tag II; 7,055 bp | This study |
| pML1641 | pMN016; mmcO; 7,027 bp | This study |
| pML1252 | pMN016; mmcOC35A; 7,027 bp | This study |
| pML1262 | pMN016; mmcOC486A; 7,027 bp | This study |
| pML1231 | pET-21(a+); mmcOΔ2–35-Strep-Tag II; 6,836 bp | This study |
Gfpm2+ denotes a mutant Gfp with enhanced folding and fluorescence. The codon usage of the gfpm2+ gene was adapted for expression in mycobacteria as described by Steinhauer et al. (75). psmyc refers to the mycobacterial promoter smyc. The subscripts “up” and “down” refer to upstream and downstream homologous regions used for recombination and knockout generation.
Plasmid construction.
E. coli strain DH5α was routinely used for plasmid construction and propagation. Plasmids used in this work are described in Table 1; primers used are described in Table 2. Plasmid pML1641 was generated by PCR amplifying mmcO from the wild-type (wt) M. tuberculosis chromosome using primers CN1695 and CN1698, the product was digested with SphI, primer CN1698 was used to add a SwaI half site, and the product was ligated into pMN016 (29) which was digested with SphI and SwaI. Plasmid pML1648 was generated by amplifying the upstream homologous region of mmcO using primers CN1702 and CN1703, which added PacI and NsiI restriction sites, respectively; the PCR product and empty knockout vector pML523 (30) were digested with PacI and NsiI and ligated. The downstream homologous region of mmcO was amplified with primers CN3119 and CN1700, which added an SpeI and SwaI half site, respectively; the PCR product was digested with SpeI and the vector pML1648 was digested with SpeI and SwaI, and the products were ligated to generate pML1649, the final mmcO knockout vector. The mmcO knockout vector was designed such that 9 bp upstream of the mmcO start codon and the entire mmcO gene, except the last 13 bp, were deleted. Plasmid pML1227 was generated by amplifying mmcO from pML1641 using primers CN1695, which added an SphI site and ribosome binding site, and CN3120, which added Strep-Tag II and a HindIII restriction site; the PCR product and backbone vector pMN016 were digested with SphI and HindIII and ligated. Residue Cys35 was mutated to Ala by CCR (31) using primers CN145, CN110, and CN2863 (the mutagenesis primer); the product was generated by PCR amplification from pML1641, digested with SphI and HindIII, and ligated into pMN016 digested with the same enzymes, resulting in pML1252. Residue Cys486 was mutated to Ala by CCR (31) using primers CN145, CN110, and CN2873 (the mutagenesis primer); the sequence was amplified from pML1641. The product was digested with SphI and HindIII and ligated into pMN016 digested with the same enzymes, resulting in plasmid pML1262. Plasmid pML1231 was generated by amplifying mmcO without a TAT secretion signal (amino acid residues 2 to 35; designated mmcOΔ2-35) from pML1227 with primers CN2484 and CN2398, which added NdeI and HindIII, respectively; the PCR product and vector pET21(a+) were digested with NdeI and HindIII and ligated.
Construction of mutants of M. tuberculosis.
Mutants were generated through a plasmid-based homologous recombination scheme as described previously (32) with some modifications. M. tuberculosis mc26230 and M. tuberculosis H37Rv were transformed with knockout vector pML1649 and grown at 37°C on 7H9/OADC/Hyg medium (with additional Casamino Acids and pantothenate for mc26230 strains). Transformants were picked, grown in liquid cultures, and plated directly for double-crossover mutants (DCOs). DCO candidates were screened for the presence of xylE and gfp; DCO candidates were green fluorescent protein (GFP) positive and XylE negative. DCO candidates were then grown in liquid culture for approximately 5 days to prepare chromosomal DNA, at which point correct candidates were confirmed by Southern blotting. Marked mutants (ΔmmcO::loxP-gfpm2+-hyg-loxP,designated ΔmmcO::hyg here) of M. tuberculosis mc26230 and M. tuberculosis H37Rv were designated ML1221 and ML1223, respectively. (The gfpm+2 gene encodes a Gfp variant with enhanced folding and fluorescence, and the codon usage of the gene has been adapted for expression in mycobacteria [75]). The Cre recombinase expression vector pCreSacB1 (a kind gift from Adrie Steyn) was used to excise the loxP-flanked gfpm2+-hyg cassette from the chromosomes of the mmcO DCOs. Unmarked deletion mutants were screened for loss of gfp. Strains were cured of pCreSacB1 by growth on 7H10-OADC plates containing 2% sucrose and incubated at 37°C. Single colonies were streaked in parallel on 7H10-OADC, 7H10-OADC-Kan, and 7H10-OADC-Hyg plates to confirm the loss of hyg and pCreSacB1. The unmarked mmcO (ΔmmcO::loxP, abbreviated ΔmmcO) deletion mutants of M. tuberculosis mc26230 and M. tuberculosis H37Rv were named ML1222 and ML1224 (Table 1), respectively. Mutants were complemented with the replicative vector pML1641, pML1252, or pML1262, where indicated.
Southern blot analysis.
Chromosomal DNA was extracted from wt and mutant strains according to standard protocols (33); subsequently, 5 μg was digested with XmaI. Digested genomic DNA was separated on a 1% agarose gel; the gel was subsequently prepared and transferred according to standard protocols (34). The DNA was cross-linked to the membrane using a UV cross-linker (240,000 μJ) and prehybridized for 3 h at 42°C in Dig-Easy hybridization solution (Roche). For analysis of the wt and mutant genomic regions, a probe was generated by PCR from wt genomic DNA using the primer pair CN1702/CN1703 and labeled using the PCR DIG labeling mix (Roche). Hybridization was carried out in the presence of 250 ng of digoxigenin-labeled PCR fragment at 50°C overnight. The membrane was washed, and the hybridized digoxigenin-labeled probe was detected with a horseradish peroxidase (HRP)-conjugated antidigoxigenin antibody following the recommendations of the manufacturer (Roche). An imaging system and the software LabWorks (UVP) were used to visualize the luminescence of blots. The software Gimp2.0 was used to adjust the contrast of images.
Protein overexpression and purification and antibody production.
Protein for antibody production was overexpressed from pML1231 in E. coli strain BL21(DE3). Plasmid pML1231 expresses mmcOΔ2–35 from a T7 promoter; the TAT secretion signal and putative lipidation site were removed to avoid complications from insufficient secretion or membrane association. Cells were grown to an optical density at 600 nm (OD600) of 0.6, and expression was induced by the addition of 1 mM IPTG (isopropyl-β-d-thiogalactopyranoside) for 3 h. After lysis, protein was present and nearly pure as inclusion bodies. Protein was purified using Strep-Tag II and Strep-Tactin Sepharose (IBA Life Sciences). Polyclonal antiserum was produced in rabbits using the adjuvant Titermax (Open Biosystems).
Analysis of proteins by gel electrophoresis and Western blot.
Cell lysates of M. tuberculosis were analyzed by separation on 10% SDS-PAGE and subsequent staining with Coomassie blue G-250 stain or by transfer and Western blotting. Protein sample loading buffer contained 1% β-mercaptoethanol to ensure reduction of cysteines. MmcO was detected using polyclonal antiserum (this study), and where indicated, RNA polymerase (RNAP) was used as a loading control and detected with monoclonal antibody clone 8RB13 (Neoclone). Goat anti-rabbit and goat anti-mouse (Sigma) secondary antibodies conjugated to horseradish peroxidase were used. Blots were visualized with an enhanced chemiluminescence (ECL) Western blotting substrate (Pierce). An imaging system and the software LabWorks (UVP) were used to visualize and quantify the luminescence of blots. The software Gimp2.0 was used to adjust the contrast of images.
Induction of mmcO expression in M. tuberculosis by copper.
M. tuberculosis mc26230 was grown to an OD600 of 1.0 in liquid culture. Then, cultures were split and either left uninduced or induced with 200 μM CuSO4 for an additional 48 h. Cells were collected, washed twice with phosphate-buffered saline (PBS)–0.02% tyloxapol and resuspended in 5 μl PBS–1% SDS buffer per mg (wet weight) of cell pellet. Cells were lysed by sonication and boiled with protein sample loading buffer before SDS-PAGE and Western blot analysis as described above.
Whole-cell lysate oxidase assays.
Oxidase assays were performed as described previously, with modifications (20, 35). M. tuberculosis mc26230 and derivatives were grown in medium as described above to an OD600 of approximately 1.0. Cells were collected and washed twice with 100 mM Tris (pH 7.8)–100 mM NaCl–0.02% tyloxapol and resuspended in the same buffer at 5 μl buffer per mg (wet weight) of cell pellet. Cells were lysed by sonication, and lysate was cleared by centrifugation for 5 min at 16,000 × g. Cleared supernatant was used in oxidase assays. The oxidase reaction mix contained 50 mM sodium acetate (pH 5.5), 250 μM CuSO4, 10 μl cell lysate, and 20 mM 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) (ABTS), 2 mM dimethoxyphenol (DMP), or 2 mM para-phenylenediamine (pPD) as the substrate. Reaction mixtures (200 μl) were mixed in 96-well clear plastic, flat-bottom microplates (Costar) sealed with clear polypropylene film (USA Scientific) and monitored by microplate reader (Biotek Synergy HT). Reactions proceeded at room temperature (∼23°C) with shaking, and absorbance was read every 5 min for up to 8 h. The change in absorbance of ABTS, DMP, and pPD was monitored at 436 nm, 570 nm, and 468 nm, respectively. Ferroxidase assays were conducted as described above with 200 μM FeSO4 as the substrate. Fe(II) was injected into wells, and oxidation was monitored by the increase of absorbance at 315 nm (36) over 15 min, before precipitates formed. Reaction absorbance in all cases was corrected by subtracting the absorbance of substrate mix without lysate; samples were further normalized by the protein concentration of the lysate, which was determined by Bradford assay (Pierce). Oxidation rates of ABTS were determined using the extinction coefficient of oxidized ABTS at 436 nm, i.e., 29,300 M−1 cm−1 (20). The kinetics of Fe(II) oxidation were determined using the extinction coefficient at 315 nm, i.e., 2,200 M−1 cm−1 (36). Oxidation rates are expressed in μM min−1 mg−1 (ABTS oxidation) or mM min−1 mg−1 [Fe(II) oxidation], where mg represents the mass of total protein in the water-soluble fraction used in the oxidase assays.
Copper susceptibility of M. tuberculosis.
Drop assays to determine copper susceptibility were performed as described previously (4). Briefly, M. tuberculosis mc26230 and derivatives or M. tuberculosis H37Rv and derivatives were grown in media as described above to an OD600 of approximately 2.0, washed in PBS (pH 7.4)–0.02% tyloxapol, and serially diluted in 10-fold increments in the same buffer, and 3-μl drops were spotted onto plates (media were as described above) containing increasing concentrations of CuSO4. Plates were incubated at 37°C for 10 to 12 days until single colonies were visible at the lowest dilutions.
Subcellular fractionation of M. tuberculosis.
Protein localization was determined by subcellular fractionation as described previously (32). Briefly, M. tuberculosis mc26230 was grown in liquid culture, and cells were collected, washed in PBS, and resuspended in PBS–1 mM phenylmethylsulfonyl fluoride (PMSF) at 4 μl per mg (wet weight) of cell pellet. Samples were lysed by sonication and incubated with 1 mg/ml lysozyme plus 15 U Benzonase at 37°C to complete cell wall and chromosomal DNA digestion. Unbroken cells were removed by centrifugation at 3,200 × g. Samples were diluted 5-fold and centrifuged at 120,000 × g for 1 h, the pellet (P100.1) and supernatant (S100.1) were separated, the pellet was resuspended, and centrifugation was repeated on both samples. The final pellet (P100.2, membrane fraction) and supernatant (S100.2, cytosolic fraction) along with cleared cell lysate were analyzed by SDS-PAGE and Western blotting as described above. Antibodies against IdeR (37) and GlpX (obtained from Axel Siroy) were used as markers for cytosolic proteins, and antibody against Rv1698 (4) was used as a marker for the membrane fraction.
RESULTS
The MmcO protein is highly conserved in pathogenic mycobacteria.
MmcO (Rv0846c) was predicted as a multicopper oxidase based on conservation of active-site residues and homology to other multicopper oxidases (38). MmcO is similar to multicopper oxidases throughout the order Actinomycetales and is conserved among almost all pathogenic mycobacteria, with the notable exception of Mycobacterium leprae, which contains only a pseudogene (see Fig. S1 in the supplemental material). MmcO shares between 69 and 99% amino acid sequence identity with its homologs in other pathogenic mycobacteria, such as Mycobacterium bovis and Mycobacterium marinum. MmcO is over 40% identical to its homologs in Corynebacterium glutamicum and Corynebacterium. diphtheriae. Interestingly, no multicopper oxidases are annotated in the genomes of the nonpathogenic Mycobacterium smegmatis, Mycobacterium chelonae, and Mycobacterium terrae.
Generation of an M. tuberculosis ΔmmcO mutant.
To examine the role of MmcO in copper resistance in M. tuberculosis, we generated mmcO deletion mutants by homologous recombination. The gene mmcO does not belong to an operon, since the surrounding genes are transcribed in the opposite direction. Marked double-crossover mutants (ΔmmcO::loxP-gfpm2+-hyg-loxP, abbreviated ΔmmcO::hyg) were selected by hygromycin resistance and screened for GFP fluorescence. Mutants were subsequently unmarked, indicated by the loss of these markers, using an expression vector encoding the recombinase Cre. We generated mutants of both the virulent M. tuberculosis strain H37Rv and the avirulent strain mc26230. The avirulent mc26230 mutant contains deletions of the RD1 region as well as the pantothenate biosynthesis genes panCD (39). The avirulent strain had the same copper susceptibility as its virulent parent strain H37Rv and was used primarily for biochemical assays. Marked mutants (ΔmmcO::hyg) and unmarked mutants (ΔmmcO::loxP; referred to here as ΔmmcO) in M. tuberculosis mc26230 and M. tuberculosis H37Rv (unmarked only) were confirmed by Southern blotting (Fig. 1A and B). The deletion strategy employed replaced the mmcO gene, except for 13 bp at the 3′ end, with a single loxP site. Loss of MmcO in M. tuberculosis mc26230 was confirmed by Western blotting (Fig. 1C).
Fig 1.

Generation of an M. tuberculosis ΔmmcO mutant. (A) Depiction of genomic regions of wild-type (wt) M. tuberculosis and the ΔmmcO::hyg (marked) and ΔmmcO (unmarked) mutants. (B) Southern blot of genomic DNA from strains depicted in panel A digested with XmaI. The location of the probe is shown in panel A. Mutants were made in both the avirulent M. tuberculosis strain mc26230 and the virulent strain H37Rv. (C) Western blot of proteins in lysates of the mc26230 and ΔmmcO::hyg strains grown in cultures with (+) and without (−) 200 μM CuSO4. MmcO protein was detected with anti-MmcO antiserum. RNA polymerase (RNAP) was used as a loading control and was detected with a monoclonal antibody.
Expression of the mmcO gene is increased by copper.
Previously, quantitative RT-PCR experiments showed that mmcO transcription is increased by addition of copper (40) and is regulated by the copper-responsive RicR protein (41). To examine how MmcO protein levels changed in M. tuberculosis under copper stress, we utilized the avirulent M. tuberculosis strain mc26230 and its isogenic ΔmmcO mutant. A single band with an apparent molecular mass of 56 kDa, the predicted size of MmcO, was detected by an MmcO antiserum in wt M. tuberculosis (Fig. 1C). MmcO protein levels increased by 6-fold when 200 μM copper was added to the medium (7H9 supplemented with OADC, pantothenate, and Casamino Acids) (Fig. 1C). In addition to the dominant band with the same electrophoretic mobility as basal MmcO, a faint band with a higher apparent molecular mass was observed when M. tuberculosis was exposed to copper. MmcO contains a twin-arginine translocation (TAT) signal at the N terminus (42). Previous reports showed that the TAT secretion system is more easily saturated than the Sec secretion system, leading to incomplete transport and processing of TAT substrate proteins (43, 44). Thus, the additional band might result from unprocessed MmcO protein in copper-stressed M. tuberculosis due to incomplete translocation and processing of the TAT secretion signal when the mmcO gene is overexpressed.
MmcO is a multicopper oxidase.
We used in vitro oxidation assays to examine whether MmcO has the predicted multicopper oxidase activity. Several attempts to obtain soluble, functional MmcO from a variety of systems and conditions failed. Expression of mmcO under the control of the T7 promoter in E. coli BL21(DE3) resulted in inclusion body formation, regardless of the presence of a TAT signal sequence, the temperature during growth or induction, or addition of copper to growth medium. Expression of a fusion protein of MmcO with the maltose-binding protein at the N terminus in E. coli also resulted in an insoluble protein, possibly due to the lack of TAT signal or missing chaperones. Fusion of the E. coli CueO signal peptide (amino acid residues 1 to 28) with MmcO lacking its native TAT signal peptide (MmcO amino acid residues 36 to 504), with the gene fusion expressed under the control of the cueO native promoter, resulted in only very low levels of protein even under copper induction. The CueO-MmcO fusion protein did not complement the loss of CueO in an E. coli ΔcueO strain. Expression of mmcO using inducible expression systems in M. smegmatis (45, 46) resulted in only very small amounts of protein, which could not be purified.
Therefore, we used cleared whole-cell lysates of different strains to characterize the multicopper oxidase activity in M. tuberculosis. To this end, we monitored oxidation of 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) (ABTS), a widely used substrate for multicopper oxidases because of its rapid oxidation and easy photometric detection, by whole-cell lysates of wt mc26230 and the ΔmmcO mutant (20, 35). Wild-type lysate showed slow ABTS oxidation (2 μM min−1 mg−1 total soluble protein in assay), which was 17-fold increased when cultures were exposed to copper for 48 h before lysis (34.5 μM min−1 mg−1) (Fig. 2A). This result is consistent with the low expression of mmcO under standard growth conditions and the 6-fold-increased expression when cells were copper stressed (Fig. 1C). Lysates of the ΔmmcO mutant did not oxidize ABTS independently of copper stress during growth, demonstrating that MmcO (Rv0846c) is the only multicopper oxidase present in M. tuberculosis under the conditions tested. Wild-type whole-cell lysates also oxidized other known multicopper oxidase substrates, such as para-phenylenediamine and dimethoxyphenol (data not shown). Overexpression of mmcO in the ΔmmcO mutant restored ABTS oxidation activity, which was increased compared to that of copper-stressed wt M. tuberculosis (Fig. 2B). The rates of ABTS oxidation by MmcO were 588.2 μM min−1 mg−1 and 409.1 μM min−1 mg−1 in untreated and copper-treated samples, respectively. These results demonstrate that rv0846c encodes a copper responsive multicopper oxidase in M. tuberculosis. Therefore, we have renamed Rv0846c mycobacterial multicopper oxidase (MmcO).
Fig 2.

M. tuberculosis has multicopper oxidase activity. (A) Oxidation of 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) (ABTS) by wt M. tuberculosis mc26230 and the ΔmmcO mutant. Cultures were grown and treated with 200 μM copper for 48 h prior to lysis or left untreated. (B) ABTS oxidation by MmcO, MmcOC35A, and MmcOC486A in cell lysates of the ΔmmcO mutant. Cultures were grown in the absence of additional copper. Assays were performed in triplicate, and error bars represent standard deviations.
The putative lipidation site is dispensable for MmcO activity.
We investigated the role of the putative lipidation site, cysteine 35 (predicted by the LipoP server [47]), in MmcO oxidase activity. Mutation of cysteine 35 to alanine (MmcOC35A) left the TAT translocation signal intact and had only a minor effect on oxidase activity (Fig. 2B). ABTS was oxidized by MmcOC35A at 450.9 μM min−1 mg−1 and 236.8 μM min−1 mg−1 in lysates of M. tuberculosis untreated and copper treated, respectively, a slight reduction compared to wt MmcO. The protein levels of mutant MmcOC35A and wt MmcO in the M. tuberculosis ΔmmcO mutant were similar, indicating that the mutation did not lead to degradation (data not shown). This result showed that lipidation of MmcO is not essential for the multicopper oxidase activity of MmcO but rather stimulates its activity in M. tuberculosis.
The active-site cysteine 486 is required for MmcO activity.
Multicopper oxidases contain one cysteine residue which is critical to the coordination of the type 1 (T1) copper, the site of substrate oxidation (27). In MmcO, the T1 copper is coordinated by cysteine 486, based on the alignment with fully characterized multicopper oxidases (see Fig. S1 in the supplemental material). Mutation of the cysteine in the T1 E. coli CueO (C500S) resulted in protein which was expressed and fully folded but nonfunctional and could be crystalized under the same conditions as wt CueO (48, 49). Similarly, the MmcOC486A protein failed to restore ABTS oxidation activity in the M. tuberculosis ΔmmcO mutant (Fig. 2B). The MmcOC486A protein was detected in the ΔmmcO mutant at levels similar to those of wt MmcO by Western blot analysis (data not shown). These results show that the active-site Cys486 residue is required for substrate oxidation.
MmcO has ferroxidase activity.
Some multicopper oxidases can oxidize metals. Well-known multicopper oxidases Fet3P from Saccharomyces cerevisiae and ceruloplasmin from humans are ferrous and cuprous oxidases (50). Examples of bacterial metallo-oxidases include CueO, which oxidizes Fe(II) to Fe(III) and Cu(I) to Cu(II) (49), and the Pseudomonas aeruginosa multicopper oxidase, which also oxidizes Fe(II) to Fe(III) (51). Thus, we examined whether MmcO has ferroxidase activity similar to other multicopper oxidases (MCOs). Cultures of wt M. tuberculosis, the ΔmmcO mutant, and the complemented strains were grown in 7H9 medium with supplements (OADC, Casamino Acids, and pantothenate) and exposed to 200 μM copper prior to lysis. Oxidation of Fe(II) by whole-cell lysates was monitored by absorbance at 315 nm, which reflects the appearance of Fe(III) in solution (36). Because Fe(III) quickly forms insoluble particles in solution, the ferroxidase assay was stopped after 15 min, when such particles appeared. The parental strain M. tuberculosis mc26230 and the ΔmmcO mutant expressing mmcO or mmcOC35A had ferroxidase activity (Fig. 3). Ferroxidase activity was increased in cultures exposed to copper before lysis compared to untreated cells (data not shown). Wild-type M. tuberculosis mc26230 lysates oxidized Fe(II) at a rate of 6.6 mM min−1 mg−1. Overexpression of mmcO and mmcOC35A in the ΔmmcO strain resulted in ferroxidase rates of 21.6 and 19.9 mM min−1 mg−1, respectively. Similar to oxidation of ABTS, loss of the lipidation site cysteine 35 only slightly decreased oxidase activity compared to wt MmcO. The ΔmmcO mutant slowly oxidized Fe(II), at a rate of 3.6 mM min−1 mg−1, but oxidation activity did not reach the maximum achieved by wt lysates (Fig. 3). It is possible that other oxidases or excess copper in the whole-cell lysates of the ΔmmcO mutant resulted in slightly increased oxidation of Fe(II) over air background. Expression of mmcOC486A in the ΔmmcO mutant did not significantly alter Fe(II) oxidation compared to that obtained with the lysate of the ΔmmcO strain. These results demonstrate that MmcO has iron oxidase in addition to ABTS and phenol oxidase activity and that the active-site residue cysteine 486 is required for this catalytic activity.
Fig 3.

Fe(II) oxidation by MmcO in whole-cell lysates of M. tuberculosis. M. tuberculosis mc26230 (wt) and its derivatives were grown to an OD600 of 1.0 and exposed to 200 μM copper for 48 h prior to lysis. Oxidation of ferrous to ferric iron in cell lysates was determined by measuring the absorbance at 315 nm. The ferroxidase assay was performed in triplicate. Error bars represent standard deviations.
MmcO is required for copper resistance in M. tuberculosis.
Multicopper oxidases are required for copper resistance in several bacterial species (20–22, 24). Thus, we hypothesized that MmcO may also be required for copper resistance in M. tuberculosis. We used a serial dilution assay on Middlebrook 7H10 agar plates with OADC (for H37Rv) or 7H10 with OADC and supplemented with pantothenate and Casamino Acids (for mc26230) containing increasing amounts of copper to determine the copper susceptibility of the ΔmmcO mutants. Mutant strains did not show any growth defects on standard 7H10 medium (Fig. 4) in agreement with previous results showing that MmcO is not required for in vitro growth under standard conditions by transposon site hybridization (TraSH) (52). However, the ΔmmcO mutant was susceptible to copper at 50 μM in solid medium (Fig. 4A), while the wt grew normally at copper concentrations up to 75 μM (Fig. 4B). Overexpression of mmcO or mmcOC35A restored growth of ΔmmcO at 50 μM copper. Importantly, mmcOC486A only slightly complemented growth, demonstrating that the oxidase activity of MmcO is required for copper protection in M. tuberculosis (Fig. 4A). Additionally, we compared the ΔmmcO mutant to our previously characterized copper-susceptible Δrv1698 mutant (4). The ΔmmcO mutant was highly susceptible to 75 μM copper, while the Δrv1698 strain grew normally (Fig. 4B).
Fig 4.

MmcO is required for copper resistance in M. tuberculosis. Liquid cultures of strains were grown to an OD600 of ∼2.0, washed, normalized, and serially diluted. (A) Drops of 3 μl of 10−1 to 10−6 dilutions of M. tuberculosis mc26230 and derivatives were spotted onto Middlebrook 7H10-OADC agar plates containing Casamino Acids, pantothenate, and 6.3 μM CuSO4 or 50 μM CuSO4. The experiment was performed twice, and representative data are shown. (B) Growth of M. tuberculosis mc26230 and the ΔmmcO and Δrv1698 mutants on Middlebrook 7H10-OADC agar plates containing Casamino Acids, pantothenate, and 6.3 or 75 μM CuSO4. (C) Serial dilutions of wt M. tuberculosis H37Rv and its corresponding ΔmmcO mutant were spotted onto Middlebrook 7H10-OADC agar plates containing 6.3, 50, or 75 μM CuSO4.
To confirm that copper susceptibility was the direct result of loss of MmcO and not an unknown interaction between MmcO and components of RD1 or pantothenate biosynthesis in the avirulent M. tuberculosis strain mc26230, we repeated the experiment using virulent M. tuberculosis H37Rv and its isogenic mutant ML1224 (H37Rv ΔmmcO) (Table 1). Deletion of mmcO in H37Rv resulted in copper susceptibility, similar to results obtained in M. tuberculosis mc26230 (Fig. 4C). M. tuberculosis H37Rv and H37Rv ΔmmcO grew equally well on standard 7H10 medium with OADC, which contains 6.3 μM copper. However, ΔmmcO had a severe growth defect on plates containing 50 μM copper and did not grow on plates containing 75 μM copper. These results show that MmcO is required for copper resistance in M. tuberculosis and that this activity is dependent on a functional active site (Cys486) and independent of the putative lipidation site Cys35.
MmcO is associated with membranes.
Many multicopper oxidases are membrane associated, including eukaryotic Fet3p of S. cerevisiae, hephaestin in humans, and CotA of Bacillus subtilis (53). However, periplasmic multicopper oxidases of Gram-negative bacteria (CueO of E. coli and the multicopper oxidase of Campylobacter jejuni) are not membrane associated (24, 44). MmcO carries a potential lipidation site at Cys35 and is secreted by the TAT secretion system (42), indicating that this protein may be membrane associated in the periplasm of M. tuberculosis. We characterized the localization of MmcO by subcellular fractionation. Separation of the membrane fraction from the cytosol and soluble periplasmic fraction was monitored with the marker proteins Rv1698 (membrane) and GlpX and IdeR (cytoplasm) (Fig. 5). Seventy percent of MmcO was membrane associated (Fig. 5). The incomplete fractionation pattern may be an overexpression artifact, as was observed before (Fig. 1C).
Fig 5.
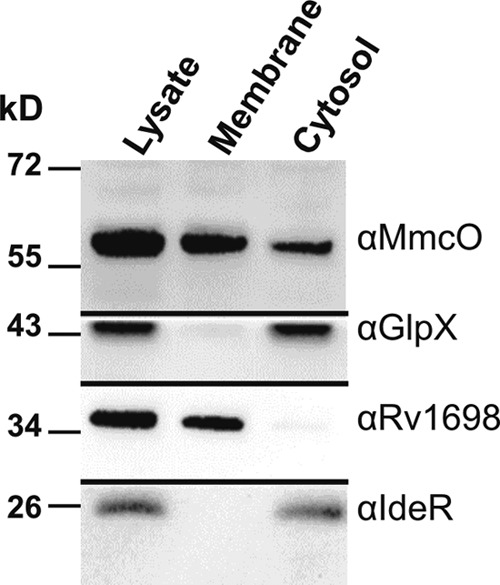
MmcO is membrane associated in M. tuberculosis. M. tuberculosis mc26230 cells were lysed and fractionated by ultracentrifugation to separate water-soluble from membrane proteins. GlpX and IdeR were used as controls for water-soluble proteins, and Rv1698 was used as a marker for membrane proteins.
DISCUSSION
MmcO is required for copper resistance in M. tuberculosis.
Deletion of the mmcO gene results in at least 10-fold-increased sensitivity to 50 μM copper in wild-type M. tuberculosis. Lack of the copper metallothionein MymT or Rv1698 increases the susceptibility of M. tuberculosis to 150 μM copper (4, 54). In contrast, the putative copper efflux pump CtpV had only a minor effect on copper resistance in M. tuberculosis (55), possibly due to redundancy with another P-type ATPase, CtpG, which is induced under copper stress in the M. tuberculosis ΔctpV mutant (55). Direct comparison revealed that the M. tuberculosis ΔmmcO mutant was more susceptible to copper than Δrv1698 mutant under the same conditions. These results indicate that MmcO is a major component of copper resistance of M. tuberculosis.
Comparison between the different mutants is difficult because the published experiments were performed under different conditions. For example, liquid culture copper exposure and subsequent CFU counts were used to determine the copper susceptibility of the M. tuberculosis ΔctpV mutant (55). We have observed that addition of copper to liquid medium, even in the presence of copper-binding albumin, causes cells to form aggregates, which makes determinations of optical density or even CFU counts difficult or error-prone (4). Further, albumin binds copper (56) and thus decreases the effective concentration of copper in the culture medium and increases the apparent MIC of copper. Finally, some experiments were performed using Middlebrook 7H11 medium, which contains enzymatically digested casein, a mixture of small peptides and amino acids which are known to sequester copper, masking the true susceptibility of mutant strains to copper (57). We found that 7H11 medium protects against copper toxicity much more than 7H10 medium supplemented with acid-hydrolyzed casein, possibly due to incomplete protein digestion of enzymatically treated casein. A further complication is that the published deletion mutants were generated using different disruption methods, i.e., by transposon mutagenesis or allelic exchange with or without leaving resistance markers in place. Clearly, direct comparison of marker-free M. tuberculosis mutants with in-frame deletions of copper homeostasis genes, under the same conditions, preferably using medium without albumin and enzymatically hydrolyzed casein, is required to ultimately quantify the contributions of individual proteins to the different mechanisms of copper homeostasis in M. tuberculosis.
MmcO is a multicopper oxidase with ferroxidase activity.
Several ferroxidases, including CueO of E. coli and McoC of C. jejuni, also have cuprous oxidase activity (58), suggesting that the physiological role of MmcO might be to oxidize toxic Cu(I). Indeed, elimination of the oxidase activity of MmcO impairs its ability to protect M. tuberculosis against copper (Fig. 4). However, siderophore oxidation has been proposed as an alternative, indirect mechanism by which bacterial MCOs could protect against copper toxicity. According to this model, a side reaction of Fe(II)-siderophore complexes is to reduce Cu(II) to the more toxic Cu(I), and thus, CueO oxidation of Fe(II)-loaded siderophores results in lower levels of Cu(I) (59). It has not been definitively determined whether CueO protects E. coli from Cu(I) toxicity by direct oxidation of Fe(II) siderophores or by a combination of both mechanisms (59).
Some multicopper oxidases, such as E. coli CueO and Pseudomonas syringae CopA, bind additional copper ions outside the active site. The extra copper binding sites of CueO are part of a methionine-rich helix which may serve to feed copper into the substrate oxidation site (48). M. tuberculosis MmcO does not contain such a methionine-rich helix (see Fig. S1 in the supplemental material). P. syringae CopA, on the other hand, acts by sequestering excess copper, either by binding it in the periplasm or by binding copper in the cytoplasm and translocating it to the periplasm (21). However, the active-site mutant MmcOC486A did not restore full copper resistance to M. tuberculosis, demonstrating that MmcO protects M. tuberculosis against copper stress mainly by oxidase activity, with possible contribution from copper sequestration. It is unclear whether MmcO oxidizes Cu(I) directly or if it acts on another substrate, which then reduces or sequesters Cu(I).
Role of lipidation of MmcO.
MmcO contains a cysteine residue downstream of the twin-arginine translocation signal sequence in a predicted lipidation motif. Acyl-transferases link fatty acids to a cysteine immediately after the cleavage site of the signal peptidase to convert the exported protein into a lipoprotein (60). Lipoproteins can be substrates of the Sec or TAT translocation systems (61). While periplasmic proteins in Gram-positive bacteria are lipoproteins (62), their counterparts in Gram-negative bacteria are often not acylated (63), probably because the outer membrane retains these proteins within the cell boundaries. Interestingly, many periplasmic proteins in mycobacteria contain lipoboxes, although mycobacteria have an outer membrane (64). For example the periplasmic binding proteins associated with ABC transporters are not acylated in Gram-negative bacteria but are lipoproteins in mycobacteria (65, 66). In this study, we show that cysteine acylation of the predicted lipoprotein MmcO of M. tuberculosis is dispensable for its function. Subcellular fractionation of MmcOC35A showed that a portion of the protein is still membrane associated (data not shown). It is unclear whether this membrane association is transient and due to the hydrophobic helix in the uncleaved TAT signal peptide. Since the TAT signal peptide as the translocation signal is not altered, it is likely that the MmcOC35A mutant is localized in the periplasm, as is wt MmcO. However, we cannot exclude the possibility that some MmcOC35A protein might be in the cytosol. Even if this were the case, it is unknown whether the usually periplasmic multicopper oxidases would be active in the cytosol. It is possible that lipidation orients the MmcO protein in the membrane or alters protein conformation such that the active site is more accessible, maybe in association with other proteins. Such a scenario would provide an explanation for the reduced oxidation activity of MmcOC35A compared to wt MmcO.
Role of protein localization in copper resistance of M. tuberculosis.
MmcO and the putative MCOs of other mycobacterial species contain a twin-arginine translocation signal sequence, which is utilized by bacteria to translocate folded proteins across the inner membrane (67). It is possible that MmcO binds copper in the cytoplasm and is then exported; however, there is evidence that MCOs are folded and exported without the full complement of cofactors (48, 68–70). Fusions of the MmcO signal peptide with β-lactamase indicate that MmcO is a periplasmic protein (42), similar to several other bacterial multicopper oxidases, including CueO (20), CopA of Pseudomonas syringae (21), and C. jejuni McoC (58). The putative localization of MmcO in the periplasm and its important role in copper resistance indicate that detoxification of periplasmic Cu(I) is the physiological role of MmcO in M. tuberculosis (Fig. 6). CtpV and MymT both protect the cytoplasm from copper stress, and hence, the absence of either one of those proteins may, at least partially, be compensated for by the other protein (Fig. 6). Locational redundancy of CtpV and MymT may explain why the ΔctpV mutant is not much more susceptible to copper than wild-type M. tuberculosis (55). The absence of Rv1698 causes copper to accumulate in M. tuberculosis (4); however, the mechanism of action of Rv1698 and its precise localization are unclear (13). Taken together, the results of this study showed that MmcO plays a key role in the copper resistance of M. tuberculosis. It is important to identify all components of copper homeostasis in M. tuberculosis (Fig. 6) and to elucidate the interplay of these mechanisms to understand how M. tuberculosis withstands the copper overload of the phagosome in macrophages (15). A better understanding of copper homeostasis in M. tuberculosis may also help to utilize copper in the development of novel anti-TB drugs, as shown recently (71).
Fig 6.

Model of copper homeostasis in M. tuberculosis. The outer membrane (OM) is an asymmetrical bilayer containing extractable lipids in both leaflets. The inner leaflet contains long-chain mycolic acids (red), which are covalently linked to the arabinogalactan-peptidoglycan polymer (72). The inner membrane (IM) is a typical plasma membrane composed of phospholipids (72). The channel proteins required for copper influx and efflux across the OM are unknown. The P-type ATPase CtpV pumps copper out of the cytoplasm and is localized in the IM (55). MymT protects the cytoplasm against copper stress by sequestering four to six copper ions (54). TatB recognizes and binds the twin-arginine (RR) motif on the N terminus of MmcO prior to translocation (73). MmcO is translocated to the periplasm by the twin-arginine translocation system (TatABC), probably as a folded protein (42). After translocation, MmcO remains membrane associated, likely through acylation (green) at cysteine 35. MmcO protects M. tuberculosis against copper stress, probably by oxidizing toxic Cu(I) to less toxic Cu(II) in the periplasm. Rv1698 is also membrane associated and protects M. tuberculosis from copper toxicity by an unknown mechanism (4).
Supplementary Material
ACKNOWLEDGMENTS
We thank Bill Jacobs for providing M. tuberculosis mc26230, Adrie Steyn for the plasmid pCreSacB1, Frank Wolschendorf for generating M. tuberculosis ML413 and the plasmids pML1641, pML1648, and pML1649, Axel Siroy for the GlpX antiserum, and members of the Mycolab for helpful discussions.
J.L.R. is supported by National Institutes of Health (NIH) training grant T32 AI7493-17. This work was supported by the NIH grant R01 AI074805 to M.N.
We have no conflicts of interest to declare.
Footnotes
Published ahead of print 14 June 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.00546-13.
REFERENCES
- 1. World Health Organization 2013. Global tuberculosis control: WHO report 2012. World Health Organization, Geneva, Switzerland [Google Scholar]
- 2. Failla ML. 2003. Trace elements and host defense: recent advances and continuing challenges. J. Nutr. 133: 1443S– 1447S [DOI] [PubMed] [Google Scholar]
- 3. Percival SS. 1998. Copper and immunity. Am. J. Clin. Nutr. 67: 1064S– 1068S [DOI] [PubMed] [Google Scholar]
- 4. Wolschendorf F, Ackart D, Shrestha TB, Hascall-Dove L, Nolan S, Lamichhane G, Wang Y, Bossmann SH, Basaraba RJ, Niederweis M. 2011. Copper resistance is essential for virulence of Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U. S. A. 108: 1621– 1626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Achard ME, Stafford SL, Bokil NJ, Chartres J, Bernhardt PV, Schembri MA, Sweet MJ, McEwan AG. 2012. Copper redistribution in murine macrophages in response to Salmonella infection. Biochem. J. 444: 51– 57 [DOI] [PubMed] [Google Scholar]
- 6. Pena MM, Lee J, Thiele DJ. 1999. A delicate balance: homeostatic control of copper uptake and distribution. J. Nutr. 129: 1251– 1260 [DOI] [PubMed] [Google Scholar]
- 7. Hong R, Kang TY, Michels CA, Gadura N. 2012. Membrane lipid peroxidation in copper alloy-mediated contact killing of Escherichia coli. Appl. Environ. Microbiol. 78: 1776– 1784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sellin S, Eriksson LE, Mannervik B. 1987. Electron paramagnetic resonance study of the active site of copper-substituted human glyoxalase I. Biochemistry 26: 6779– 6784 [DOI] [PubMed] [Google Scholar]
- 9. Hiniker A, Collet JF, Bardwell JC. 2005. Copper stress causes an in vivo requirement for the Escherichia coli disulfide isomerase DsbC. J. Biol. Chem. 280: 33785– 33791 [DOI] [PubMed] [Google Scholar]
- 10. Macomber L, Imlay JA. 2009. The iron-sulfur clusters of dehydratases are primary intracellular targets of copper toxicity. Proc. Natl. Acad. Sci. U. S. A. 106: 8344– 8349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dameron CT, Harrison MD. 1998. Mechanisms for protection against copper toxicity. Am. J. Clin. Nutr. 67: 1091S– 1097S [DOI] [PubMed] [Google Scholar]
- 12. Rosen BP. 2002. Transport and detoxification systems for transition metals, heavy metals and metalloids in eukaryotic and prokaryotic microbes. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 133: 689– 693 [DOI] [PubMed] [Google Scholar]
- 13. Rowland JL, Niederweis M. 2012. Resistance mechanisms of Mycobacterium tuberculosis against phagosomal copper overload. Tuberculosis (Edinb.) 92: 202– 210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Failla ML, Hopkins RG. 1998. Is low copper status immunosuppressive? Nutr. Rev. 56: S59– S64 [DOI] [PubMed] [Google Scholar]
- 15. White C, Lee J, Kambe T, Fritsche K, Petris MJ. 2009. A role for the ATP7A copper-transporting ATPase in macrophage bactericidal activity. J. Biol. Chem. 284: 33949– 33956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wagner D, Maser J, Lai B, Cai Z, Barry CE, III, Honer Zu Bentrup K, Russell DG, Bermudez LE. 2005. Elemental analysis of Mycobacterium avium-, Mycobacterium tuberculosis-, and Mycobacterium smegmatis-containing phagosomes indicates pathogen-induced microenvironments within the host cell's endosomal system. J. Immunol. 174: 1491– 1500 [DOI] [PubMed] [Google Scholar]
- 17. Botella H, Stadthagen G, Lugo-Villarino G, de Chastellier C, Neyrolles O. 2012. Metallobiology of host-pathogen interactions: an intoxicating new insight. Trends Microbiol. 20: 106– 112 [DOI] [PubMed] [Google Scholar]
- 18. Hodgkinson V, Petris MJ. 2012. Copper homeostasis at the host-pathogen interface. J. Biol. Chem. 287: 13549– 13555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Samanovic MI, Ding C, Thiele DJ, Darwin KH. 2012. Copper in microbial pathogenesis: meddling with the metal. Cell Host Microbe 11: 106– 115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Grass G, Rensing C. 2001. CueO is a multi-copper oxidase that confers copper tolerance in Escherichia coli. Biochem. Biophys. Res. Commun. 286: 902– 908 [DOI] [PubMed] [Google Scholar]
- 21. Cha JS, Cooksey DA. 1991. Copper resistance in Pseudomonas syringae mediated by periplasmic and outer membrane proteins. Proc. Natl. Acad. Sci. U. S. A. 88: 8915– 8919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Achard ME, Tree JJ, Holden JA, Simpfendorfer KR, Wijburg OL, Strugnell RA, Schembri MA, Sweet MJ, Jennings MP, McEwan AG. 2010. The multi-copper-ion oxidase CueO of Salmonella enterica serovar Typhimurium is required for systemic virulence. Infect. Immun. 78: 2312– 2319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lee YA, Hendson M, Panopoulos NJ, Schroth MN. 1994. Molecular cloning, chromosomal mapping, and sequence analysis of copper resistance genes from Xanthomonas campestris pv. juglandis: homology with small blue copper proteins and multicopper oxidase. J. Bacteriol. 176: 173– 188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hall SJ, Hitchcock A, Butler CS, Kelly DJ. 2008. A Multicopper oxidase (Cj1516) and a CopA homologue (Cj1161) are major components of the copper homeostasis system of Campylobacter jejuni. J. Bacteriol. 190: 8075– 8085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hsiao YM, Liu YF, Lee PY, Hsu PC, Tseng SY, Pan YC. 2011. Functional characterization of copA gene encoding multicopper oxidase in Xanthomonas campestris pv. campestris. J. Agric. Food Chem. 59: 9290– 9302 [DOI] [PubMed] [Google Scholar]
- 26. Solomon EI, Sundaram UM, Machonkin TE. 1996. Multicopper oxidases and oxygenases. Chem. Rev. 96: 2563– 2606 [DOI] [PubMed] [Google Scholar]
- 27. Quintanar L, Stoj C, Taylor AB, Hart PJ, Kosman DJ, Solomon EI. 2007. Shall we dance? How a multicopper oxidase chooses its electron transfer partner. Acc. Chem. Res. 40: 445– 452 [DOI] [PubMed] [Google Scholar]
- 28. Sambrook J, Fritsch EF, Maniatis T. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 29. Stephan J, Bender J, Wolschendorf F, Hoffmann C, Roth E, Mailänder C, Engelhardt H, Niederweis M. 2005. The growth rate of Mycobacterium smegmatis depends on sufficient porin-mediated influx of nutrients. Mol. Microbiol. 58: 714– 730 [DOI] [PubMed] [Google Scholar]
- 30. Jones CM, Niederweis M. 2011. Mycobacterium tuberculosis can utilize heme as an iron source. J. Bacteriol. 193: 1767– 1770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bi W, Stambrook PJ. 1998. Site-directed mutagenesis by combined chain reaction. Anal. Biochem. 256: 137– 140 [DOI] [PubMed] [Google Scholar]
- 32. Wells RM, Jones CM, Xi Z, Speer A, Danilchanka O, Doornbos KS, Sun P, Wu F, Tian C, Niederweis M. 2013. Discovery of a siderophore export system essential for virulence of Mycobacterium tuberculosis. PLoS Pathog. 9: e1003120. 10.1371/journal.ppat.1003120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Larsen MH, Biermann K, Tandberg S, Hsu T, Jacobs WR., Jr 2007. Genetic manipulation of Mycobacterium tuberculosis. Curr. Protoc. Microbiol. 6:10A.2.1– 10A.2.21. 10.1002/9780471729259.mc10a02s6 [DOI] [PubMed] [Google Scholar]
- 34. Ausubel FA, Brent R, Kingston RE, Moore DD, Seidmann JG, Smith JA, Struhl K. 1990. Current protocols in molecular biology. Greene Publishing and Wiley-Interscience, New York, NY [Google Scholar]
- 35. Solano F, Lucas-Elio P, Lopez-Serrano D, Fernandez E, Sanchez-Amat A. 2001. Dimethoxyphenol oxidase activity of different microbial blue multicopper proteins. FEMS Microbiol. Lett. 204: 175– 181 [DOI] [PubMed] [Google Scholar]
- 36. Kim C, Lorenz WW, Hoopes JT, Dean JF. 2001. Oxidation of phenolate siderophores by the multicopper oxidase encoded by the Escherichia coli yacK gene. J. Bacteriol. 183: 4866– 4875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dussurget O, Rodriguez M, Smith I. 1996. An ideR mutant of Mycobacterium smegmatis has derepressed siderophore production and an altered oxidative-stress response. Mol. Microbiol. 22: 535– 544 [DOI] [PubMed] [Google Scholar]
- 38. Cole ST, Brosch R, Parkhill J, Garnier T, Churcher C, Harris D, Gordon SV, Eiglmeier K, Gas S, Barry CE, III, Tekaia F, Badcock K, Basham D, Brown D, Chillingworth T, Connor R, Davies R, Devlin K, Feltwell T, Gentles S, Hamlin N, Holroyd S, Hornsby T, Jagels K, Barrell BG. 1998. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393: 537– 544 [DOI] [PubMed] [Google Scholar]
- 39. Sambandamurthy VK, Derrick SC, Hsu T, Chen B, Larsen MH, Jalapathy KV, Chen M, Kim J, Porcelli SA, Chan J, Morris SL, Jacobs WR., Jr 2006. Mycobacterium tuberculosis ΔRD1 ΔpanCD: a safe and limited replicating mutant strain that protects immunocompetent and immunocompromised mice against experimental tuberculosis. Vaccine 24: 6309– 6320 [DOI] [PubMed] [Google Scholar]
- 40. Ward SK, Hoye EA, Talaat AM. 2008. The global responses of Mycobacterium tuberculosis to physiological levels of copper. J. Bacteriol. 190: 2939– 2946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Festa RA, Jones MB, Butler-Wu S, Sinsimer D, Gerads R, Bishai WR, Peterson SN, Darwin KH. 2011. A novel copper-responsive regulon in Mycobacterium tuberculosis. Mol. Microbiol. 79: 133– 148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. McDonough JA, McCann JR, Tekippe EM, Silverman JS, Rigel NW, Braunstein M. 2008. Identification of functional Tat signal sequences in Mycobacterium tuberculosis proteins. J. Bacteriol. 190: 6428– 6438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Barrett CM, Ray N, Thomas JD, Robinson C, Bolhuis A. 2003. Quantitative export of a reporter protein, GFP, by the twin-arginine translocation pathway in Escherichia coli. Biochem. Biophys. Res. Commun. 304: 279– 284 [DOI] [PubMed] [Google Scholar]
- 44. DeLisa MP, Lee P, Palmer T, Georgiou G. 2004. Phage shock protein PspA of Escherichia coli relieves saturation of protein export via the Tat pathway. J. Bacteriol. 186: 366– 373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Posey JE, Shinnick TM, Quinn FD. 2006. Characterization of the twin-arginine translocase secretion system of Mycobacterium smegmatis. J. Bacteriol. 188: 1332– 1340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Pandey AK, Raman S, Proff R, Joshi S, Kang CM, Rubin EJ, Husson RN, Sassetti CM. 2009. Nitrile-inducible gene expression in mycobacteria. Tuberculosis (Edinb.) 89: 12– 16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Juncker AS, Willenbrock H, Von Heijne G, Brunak S, Nielsen H, Krogh A. 2003. Prediction of lipoprotein signal peptides in Gram-negative bacteria. Protein Sci. 12: 1652– 1662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Singh SK, Roberts SA, McDevitt SF, Weichsel A, Wildner GF, Grass GB, Rensing C, Montfort WR. 2011. Crystal structures of multicopper oxidase CueO bound to copper(I) and silver(I): functional role of a methionine-rich sequence. J. Biol. Chem. 286: 37849– 37857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Singh SK, Grass G, Rensing C, Montfort WR. 2004. Cuprous oxidase activity of CueO from Escherichia coli. J. Bacteriol. 186: 7815– 7817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Stoj C, Kosman DJ. 2003. Cuprous oxidase activity of yeast Fet3p and human ceruloplasmin: implication for function. FEBS Lett. 554: 422– 426 [DOI] [PubMed] [Google Scholar]
- 51. Huston WM, Jennings MP, McEwan AG. 2002. The multicopper oxidase of Pseudomonas aeruginosa is a ferroxidase with a central role in iron acquisition. Mol. Microbiol. 45: 1741– 1750 [DOI] [PubMed] [Google Scholar]
- 52. Griffin JE, Gawronski JD, Dejesus MA, Ioerger TR, Akerley BJ, Sassetti CM. 2011. High-resolution phenotypic profiling defines genes essential for mycobacterial growth and cholesterol catabolism. PLoS Pathog. 7: e1002251. 10.1371/journal.ppat.1002251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Enguita FJ, Martins LO, Henriques AO, Carrondo MA. 2003. Crystal structure of a bacterial endospore coat component. A laccase with enhanced thermostability properties. J. Biol. Chem. 278: 19416– 19425 [DOI] [PubMed] [Google Scholar]
- 54. Gold B, Deng H, Bryk R, Vargas D, Eliezer D, Roberts J, Jiang X, Nathan C. 2008. Identification of a copper-binding metallothionein in pathogenic mycobacteria. Nat. Chem. Biol. 4: 609– 616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ward SK, Abomoelak B, Hoye EA, Steinberg H, Talaat AM. 2010. CtpV: a putative copper exporter required for full virulence of Mycobacterium tuberculosis. Mol. Microbiol. 77: 1096– 1110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. de Wolf FA, Brett GM. 2000. Ligand-binding proteins: their potential for application in systems for controlled delivery and uptake of ligands. Pharmacol. Rev. 52: 207– 236 [PubMed] [Google Scholar]
- 57. Lodyga-Chruscinska E, Micera G, Szajdzinska-Pietek E, Sanna D. 1998. Copper(II) complexes of opiate-like food peptides. J. Agric. Food Chem. 46: 115– 118 [DOI] [PubMed] [Google Scholar]
- 58. Silva CS, Durao P, Fillat A, Lindley PF, Martins LO, Bento I. 2012. Crystal structure of the multicopper oxidase from the pathogenic bacterium Campylobacter jejuni CGUG11284: characterization of a metallo-oxidase. Metallomics 4: 37– 47 [DOI] [PubMed] [Google Scholar]
- 59. Grass G, Thakali K, Klebba PE, Thieme D, Muller A, Wildner GF, Rensing C. 2004. Linkage between catecholate siderophores and the multicopper oxidase CueO in Escherichia coli. J. Bacteriol. 186: 5826– 5833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Tokuda H, Matsuyama S. 2004. Sorting of lipoproteins to the outer membrane in E. coli. Biochim. Biophys. Acta 1693: 5– 13 [DOI] [PubMed] [Google Scholar]
- 61. Hutchings MI, Palmer T, Harrington DJ, Sutcliffe IC. 2009. Lipoprotein biogenesis in Gram-positive bacteria: knowing when to hold 'em, knowing when to fold 'em. Trends Microbiol. 17: 13– 21 [DOI] [PubMed] [Google Scholar]
- 62. Sutcliffe IC, Harrington DJ, Hutchings MI. 2012. A phylum level analysis reveals lipoprotein biosynthesis to be a fundamental property of bacteria. Protein Cell 3: 163– 170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Navarre WW, Schneewind O. 1999. Surface proteins of gram-positive bacteria and mechanisms of their targeting to the cell wall envelope. Microbiol. Mol. Biol. Rev. 63: 174– 229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Hoffmann C, Leis A, Niederweis M, Plitzko JM, Engelhardt H. 2008. Disclosure of the mycobacterial outer membrane: cryo-electron tomography and vitreous sections reveal the lipid bilayer structure. Proc. Natl. Acad. Sci. U. S. A. 105: 3963– 3967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Titgemeyer F, Amon J, Parche S, Mahfoud M, Bail J, Schlicht M, Rehm N, Hillmann D, Stephan J, Walter B, Burkovski A, Niederweis M. 2007. A genomic view of sugar transport in Mycobacterium smegmatis and Mycobacterium tuberculosis. J. Bacteriol. 189: 5903– 5915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Niederweis M. 2008. Nutrient acquisition by mycobacteria. Microbiology 154: 679– 692 [DOI] [PubMed] [Google Scholar]
- 67. Palmer T, Berks BC. 2012. The twin-arginine translocation (Tat) protein export pathway. Nat. Rev. Microbiol. 10: 483– 496 [DOI] [PubMed] [Google Scholar]
- 68. Ducros V, Brzozowski AM, Wilson KS, Brown SH, Ostergaard P, Schneider P, Yaver DS, Pedersen AH, Davies GJ. 1998. Crystal structure of the type-2 Cu depleted laccase from Coprinus cinereus at 2.2 A resolution. Nat. Struct. Biol. 5: 310– 316 [DOI] [PubMed] [Google Scholar]
- 69. Galli I, Musci G, Bonaccorsi di Patti MC. 2004. Sequential reconstitution of copper sites in the multicopper oxidase CueO. J. Biol. Inorg Chem. 9: 90– 95 [DOI] [PubMed] [Google Scholar]
- 70. Musci G, Di Marco S, Bellenchi GC, Calabrese L. 1996. Reconstitution of ceruloplasmin by the Cu(I)-glutathione complex. Evidence for a role of Mg2+ and ATP. J. Biol. Chem. 271: 1972– 1978 [DOI] [PubMed] [Google Scholar]
- 71. Speer A, Shrestha TB, Bossmann SH, Basaraba RJ, Harber GJ, Michalek SM, Niederweis M, Kutsch O, Wolschendorf F. 2013. Copper-boosting compounds: a novel concept for antimycobacterial drug discovery. Antimicrob. Agents Chemother. 57: 1089– 1091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Kaur D, Guerin ME, Skovierova H, Brennan PJ, Jackson M. 2009. Biogenesis of the cell wall and other glycoconjugates of Mycobacterium tuberculosis. Adv. Appl. Microbiol. 69: 23– 78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Ligon LS, Hayden JD, Braunstein M. 2012. The ins and outs of Mycobacterium tuberculosis protein export. Tuberculosis (Edinb.) 92: 121– 132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Kaps I, Ehrt S, Seeber S, Schnappinger D, Martin C, Riley LW, Niederweis M. 2001. Energy transfer between fluorescent proteins using a co-expression system in Mycobacterium smegmatis. Gene 278: 115– 124 [DOI] [PubMed] [Google Scholar]
- 75. Steinhauer K, Eschenbacher I, Radischat N, Detsch C, Niederweis M, Goroncy-Bermes P. 2010. Rapid evaluation of the mycobactericidal efficacy of disinfectants in the quantitative carrier test EN 14563 by using fluorescent Mycobacterium terrae. Appl. Environ. Microbiol. 76: 546– 554 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.