

Abstract
The periplasmic chaperone Skp has long been implicated in the assembly of outer membrane proteins (OMPs) in Escherichia coli. It has been shown to interact with unfolded OMPs, and the simultaneous loss of Skp and the main periplasmic chaperone in E. coli, SurA, results in synthetic lethality. However, a Δskp mutant displays only minor OMP assembly defects, and no OMPs have been shown to require Skp for their assembly. Here, we report a role for Skp in the assembly of the essential OMP LptD. This role may be compensated for by other OMP assembly proteins; in the absence of both Skp and FkpA or Skp and BamB, LptD assembly is impaired. Overexpression of SurA does not restore LptD levels in a Δskp ΔfkpA double mutant, nor does the overexpression of Skp or FkpA restore LptD levels in the ΔsurA mutant, suggesting that Skp acts in concert with SurA to efficiently assemble LptD in E. coli. Other OMPs, including LamB, are less affected in the Δskp ΔfkpA and Δskp bamB::kan double mutants, suggesting that Skp is specifically necessary for the assembly of certain OMPs. Analysis of an OMP with a domain structure similar to that of LptD, FhuA, suggests that common structural features may determine which OMPs require Skp for their assembly.
INTRODUCTION
The cell envelope of the Gram-negative bacterium Escherichia coli includes three subcellular compartments: the inner membrane (IM), the outer membrane (OM), and the aqueous space between them, known as the periplasm. The outer membrane is unique, as it is an asymmetric bilayer with an outer leaflet composed of lipopolysaccharide (LPS) and an inner leaflet of phospholipids. The OM also contains two major classes of proteins: lipoproteins, most of which are essentially soluble periplasmic proteins that are attached to the inner leaflet of the OM by a lipidated N terminus, and integral β-barrel proteins known as outer membrane proteins (OMPs) (1).
There are two essential OMPs in the outer membrane (2–4). One is BamA, which along with its four associated lipoproteins, BamBCDE, is responsible for inserting itself and other β-barrel proteins into the OM (2, 5, 6). The other is LptD, which, along with its associated lipoprotein LptE, is responsible for inserting LPS into the outer leaflet of the OM (7, 8). LptD is an especially complicated substrate of the OMP assembly machinery. The C terminus of this protein forms its integral β-barrel, while the N terminus is a soluble periplasmic domain that is homologous to the periplasmic LPS transport protein LptA (4, 7, 8). The N- and C-terminal domains of LptD possess two cysteines each, which form two nonconsecutive disulfide bonds, each of which joins the N terminus to the barrel domain (9). In order for these disulfide bonds to be correctly formed and, thus, for the cell to be viable, LptD must first be inserted into the OM by the Bam complex, which also requires an interaction with its accessory lipoprotein, LptE (9, 10). The periplasmic oxidase DsbA has also been shown to play an important, although not essential, role in the formation of LptD's disulfide bonds (9).
Because the periplasm is an aqueous, oxidizing environment, LptD and other OMPs must be escorted by chaperones as they travel from the Sec translocon in the IM to the Bam complex in the OM (11). The main periplasmic chaperone in E. coli is SurA, a protein that also possesses peptidyl-prolyl cis-trans isomerase activity (11, 12). This protein is responsible for the assembly of the bulk mass of OMPs and is especially important for the biogenesis of LptD (9, 12). Although lptD transcription increases in the absence of surA (13), due to the induction of the σE stress response, the levels of LptD in this mutant are decreased dramatically, because the misassembled LptD that accumulates is rapidly degraded in the periplasm (9, 12).
Although the SurA pathway is the principal periplasmic chaperone pathway for OMPs in E. coli, there is also a secondary pathway made up of two proteins: the chaperone Skp and the chaperone and protease DegP (11, 14). As long as one of these two pathways remains intact, the cell is viable. However, if both the main SurA pathway and the backup Skp/DegP pathway are compromised (as in either an surA skp or surA degP double mutant), this synthetic interaction results in cell death (11, 14). It has also been demonstrated that the levels of virtually all OMPs decrease in the absence of both of these pathways (15).
Skp was initially reported to be a histone-like protein that binds DNA (16); it was later shown to bind OMPs and periplasmic proteins (17, 18). It exists in solution as a trimer and is structurally similar to the eukaryotic cytoplasmic chaperone prefoldin (19). Skp has been shown to interact with phospholipid membranes (20) and to facilitate the release of OMPs from spheroplasts (21) and, along with LPS, to insert unfolded OmpA into phospholipid membranes (22). More recently, experiments have provided evidence that β-barrels may be protected from aggregation within the cavity of the Skp trimer (23). Skp has also been shown to interact with the passenger domain of the autotransporter EspP at a different, earlier assembly step than SurA (24, 25). Despite this wealth of evidence supporting a role in OM biogenesis, an skp mutant displays only minor OM permeability and OMP assembly phenotypes in vivo (11), and no OMPs appear to depend on the Skp/DegP pathway for their assembly in E. coli (11, 15). However, in other organisms, Skp appears to play a more important physiological role (26). For example, it has been reported that Skp is essential for the folding and surface presentation of the α-domain (although not the barrel) of the autotransporter IcsA in Shigella flexneri (27).
In addition to SurA, the E. coli periplasm contains three additional peptidyl-prolyl cis-trans isomerases, FkpA, PpiA, and PpiD (which is anchored to the IM) (28), as well as additional chaperones. Mutants lacking any of these three proteins, or even all three simultaneously, exhibit only minor defects in vivo with respect to OM biogenesis and OMP assembly (28).
FkpA has long been known to act as a chaperone for nonnative or mutant E. coli proteins (29–31), but until recently, its physiological role was largely unclear. It has been shown that FkpA is necessary for colicin M toxicity in vivo (32, 33) and that its PPIase activity is required for this function (34). It has also been shown that FkpA interacts with high affinity with the passenger domain of the autotransporter EspP (35), and, along with DsbC, FkpA has been implicated in the folding of the nonnative passenger domain of a hybrid autotransporter protein (36).
Here, we show that Skp does play a role in OMP assembly, but this role can be performed by FkpA and is thus masked when FkpA is present in the cell. This role appears to be important only in the assembly of certain OMPs and can be compensated for by the overexpression of an additional periplasmic chaperone, Spy.
MATERIALS AND METHODS
Bacterial strains and plasmids.
Bacterial strains were constructed by P1 transduction and plasmid transformation as previously described (37). Strains and plasmids used in this work are described in Table 1. The deletion alleles used are from the Keio collection (38). Alleles from the Keio collection were cured using the pCP20 plasmid as previously described (39). The bamB::kan insertion allele is from the Blattner collection from the E. coli Genome Project at the University of Wisconsin-Madison. The degPS210A allele (11) was transduced using a linked yadC::Tn10 allele (40), and the presence of the degPS210A mutation was confirmed by sequencing. The pTrc99A::cam plasmid was constructed by amplifying the cat gene using primers with PvuI restriction sites (underlined) ( 5′-TTCGATCGTTGTAGGCTGGAGCTGCT-3′ and 5′-TTCGATCGGCATATGAATATCCTCCT-3′ ). This PCR product and the pTrc99A vector (41) were digested with PvuI and ligated together. The resulting pTrc99A::cam plasmid was used as the vector control, and candidate genes were cloned into it. The skp gene was amplified by PCR to contain EcoRI and HindIII sites (underlined) (primers 5′-GGAGAATTCGGTAAGGAGTTTATTATGAAAAAGTGG-3′ and 5′-CATGCAAAGCTTATCCAACTGCTGCGCTAAAT-3′ ) and cloned into pTrc99A::cam. The forward primer changed the naturally occurring GTG start codon to ATG (underlined). The fkpA gene was cloned using the PstI and HindIII sites (primers 5′-CATGCACTGCAGGTTAACCCTGGGGTGAGATG-3′ and 5′-CATGCAAAGCTTTTCCGCTTTCCAGCACTAAT-3′ ). The dsbA overexpression plasmids were constructed by amplifying the dsbA gene to contain EcoRI and XbaI sites (underlined) (primers 5′-CATGCTGAATTCCCCTTTGCAATTAACACCTATG-3′ and 5′-GAGCATTCTAGATTCACG GGCTTTATGTAATTT-3′ ). The pAER1 (14) (pdsbAlow) and pBAD18 (pdsbAhigh) plasmids were digested with EcoRI and XbaI enzymes and ligated with the dsbA PCR product. Construction of the psurA plasmid (pAER1) has been previously described (14). The pspy plasmid was reconstructed as previously described (42).
Table 1.
Strains and plasmids used in this study
| Strain or plasmid | Description | Reference/source |
|---|---|---|
| Escherichia coli strains | ||
| MC4100 | F− araD139 Δ(arg-lac)U169 rpsL150 relA1 flbB5301 deoC1 ptsF25 thi | 53 |
| JAS412 | MC4100 Ara+/r ΔfkpA | This study |
| JAS416 | MC4100 Ara+/r | This study |
| JAS417 | MC4100 Ara+/r Δskp | This study |
| JAS420 | MC4100 Ara+/r Δskp ΔfkpA | This study |
| JAS431 | MC4100 Ara+/r Δskp ΔfkpA yadC::Tn10 degPS210A | This study |
| JAS458 | MC4100 Ara+/r ΔfkpA yadC::Tn10 degPS210A | This study |
| JAS459 | MC4100 Ara+/r yadC::Tn10 degPS210A | This study |
| JAS460 | MC4100 Ara+/r Δskp yadC::Tn10 degPS210A | This study |
| JAS80 | MC4100 Δskp | This study |
| JAS195 | MC4100 ΔfkpA | This study |
| JAS475 | MC4100 Δskp ΔfkpA | This study |
| JAS16 | MC4100 ΔsurA | 54 |
| JAS502 | MC4100 Ara+/r psurA | This study |
| JAS501 | MC4100 Ara+/r pACYC177 | This study |
| JAS505 | MC4100 Ara+/r Δskp ΔfkpA pACYC177 | This study |
| JAS506 | MC4100 Ara+/r Δskp ΔfkpA psurA | This study |
| JAS497 | MC4100 Δskp ΔfkpA ΔlamB | This study |
| JAS523 | MC4100 Δskp ΔfkpA ΔlamB ΔompA | This study |
| JAS535 | MC4100 Δskp ΔfkpA ΔlamB ΔompA ΔompC::kan | This study |
| JAS215 | MC4100 ΔsurA pfkpA | This study |
| JAS509 | MC4100 ΔsurA pTrc99A::cam | This study |
| JAS510 | MC4100 ΔsurA pskpATG | This study |
| JAS866 | MC4100 Ara+/r pdsbAlow | This study |
| JAS867 | MC4100 Ara+/r pBAD18 | This study |
| JAS868 | MC4100 Ara+/r pdsbAhigh | This study |
| JAS874 | MC4100 Ara+/r Δskp ΔfkpA pdsbAlow | This study |
| JAS875 | MC4100 Ara+/r Δskp ΔfkpA pBAD18 | This study |
| JAS876 | MC4100 Ara+/r Δskp ΔfkpA pdsbAhigh | This study |
| JAS185 | MC4100 bamB::kan | 54 |
| JAS205 | MC4100 ΔfkpA bamB::kan | This study |
| JAS187 | MC4100 Δskp bamB::kan | This study |
| GS67 | MC4100 Δskp ΔfkpA baeSP255L | This study |
| GS95 | MC4100 Δskp ΔfkpA baeSP255L Δspy::kan ynjB::Tn10 | This study |
| GS100 | MC4100 Δskp ΔfkpA pTrc99A | This study |
| GS99 | MC4100 Δskp ΔfkpA pspy | This study |
| GS108 | MC4100 ΔsurA pTrc99A | This study |
| GS109 | MC4100 ΔsurA pspy | This study |
| GS321 | MC4100 baeSP255L ΔyegL::kan | This study |
| Plasmids | ||
| psurA | surA cloned into pBAD18, then araC-PBAD-surA fragment subcloned into pACYC177 | 14 |
| pTrc99A::cam | pTrc99A with cam cassette cloned into bla gene | This study |
| pfkpA | fkpA cloned into pTrc99A::cam | This study |
| pskpATG | skp with start codon mutated to ATG cloned into pTrc99A::cam | This study |
| pdsbAlow | dsbA subcloned into the psurA plasmid | This study |
| pdsbAhigh | dsbA cloned into pBAD18 | This study |
| pTrc99A | High-copy-number vector with inducible lac promoter and amp resistance | 41 |
| pspy | spy cloned into pTrc99A | This study |
Growth conditions.
Strains were grown in Luria-Bertani (LB) medium at 37°C unless otherwise noted. Where appropriate, the LB medium was supplemented with antibiotics at the following concentrations: ampicillin, 125 μg/ml; kanamycin, 25 μg/ml; tetracycline, 25 μg/ml; and chloramphenicol, 20 μg/ml. Also, where indicated, l-arabinose was added to the medium at a concentration of 0.2% to induce the psurA and pdsbA plasmids and isopropyl-β-d-thiogalactopyranoside (IPTG) at a concentration of 50 μM to induce the pspy plasmid.
Efficiency-of-plating assays.
Efficiency-of-plating assays were performed by growing an overnight culture in the appropriate medium and at the permissive temperature. The overnight cultures were added to the first row of wells of a 96-well plate (200 μl/well) and serially diluted 10-fold in the subsequent wells (20 μl of the preceding well into 180 μl of fresh LB medium). The overnight culture and serial dilutions were spotted onto LB agar plates containing the indicated antibiotics using a 48-pin replicator. Spots were allowed to dry, and the plates were incubated overnight at 30°C and 37°C unless otherwise indicated.
Western blot analysis.
One-milliliter samples of strains were pelleted (16,000 × g, 1 min) and resuspended at a volume equal to the optical density at 600 nm (OD600)/14 (for LptD and FhuA blots), OD600/40 (for Spy blots), or OD600/7 (all other blots). Reducing and nonreducing LptD blots were performed as previously described (9). FhuA blots were performed using a polyvinylidene difluoride (PVDF) membrane (Bio-Rad, Hercules, CA), and all others were performed with a nitrocellulose (Whatman GmbH, Dassel, Germany) membrane. Immunoblotting was performed using the following antibodies at the indicated dilutions: anti-LamB antibody (which cross-reacts with OmpA), 1:30,000; anti-BamA antibody, 1:20,000; anti-LptD antibody, 1:5,000; anti-FhuA antibody, 1:2,500; anti-TolC antibody, 1:30,000; anti-LptE antibody, 1:20,000; anti-BamB antibody, 1:7,000; anti-BamD antibody, 1:5,000; anti-BamC antibody, 1:20,000; anti-BamE antibody, 1:20,000; anti-SurA antibody, 1:8,000; anti-Skp antibody, 1:10,000; and anti-Spy antibody, 1:25,000. For FhuA blots, goat anti-mouse secondary antibody conjugated to horseradish peroxidase (HRP) was used at a dilution of 1:5,000 (Bio-Rad). For all other blots, donkey anti-rabbit secondary antibody conjugated to horseradish peroxidase was used at a dilution of 1:8,000 (GE Healthcare). Immunoblots were visualized using Luminata Classico Western HRP substrate (EMD Millipore Corporation, Massachusetts). The anti-TolC antibody was a gift from R. Misra; the anti-BamB, anti-BamD, and anti-LptE antibodies were a gift from D. Kahne; the anti-SurA antibody was a gift from R. Kolter; and the anti-FhuA antibody was a gift from J. Coulton.
Coomassie staining.
Samples resuspended in SDS-PAGE sample buffer at a volume equal to OD600/14 were boiled for 10 min and analyzed by SDS-PAGE. Gels were then incubated overnight in Coomassie brilliant blue staining solution (43) and destained the next day with Coomassie destaining solution (43).
Suppressor selection.
Suppressor mutants were selected by plating 10 μl of an overnight culture of the Δskp ΔfkpA strain on agar plates containing 55 mg/liter vancomycin. After overnight incubation at 37°C, colonies were isolated and analyzed.
RESULTS
Genetic interactions exist between skp and fkpA.
The existence of two OMP chaperone pathways in E. coli, SurA and Skp/DegP, was first established by the discovery of synthetic phenotypes in double-mutant strains. The simultaneous loss of surA and skp or surA and degP causes synthetic lethality (11, 14). In order to further investigate the chaperone network in the E. coli periplasm, we constructed strains lacking combinations of the known periplasmic chaperones and fkpA (38). We then assayed the growth of these mutant strains. None of the single-mutant strains exhibited a growth defect at 37°C, although the Δskp ΔdegP double mutant did exhibit slightly slowed growth at this increased temperature. Strikingly, we found that although the Δskp ΔdegP ΔfkpA::kan triple mutant grows normally at 30°C, this strain grows poorly at 37°C. This temperature sensitivity suggests that in the absence of skp and degP, FkpA becomes critically important.
DegP possesses both chaperone and protease functions, and we investigated which of these is more important in the absence of skp and fkpA. It is known that the loss of degP results in temperature sensitivity at 42°C (44). Strains expressing a proteolytically inactive allele known as degPS210A are temperature sensitive at 42°C unless the DegPS210A protein is overexpressed; this suggests that the loss of the protease function of DegP is primarily responsible for the temperature-sensitive phenotype of the degP mutant (45). We hypothesized that the loss of fkpA and skp lowers the temperature at which the degP mutation becomes lethal, resulting in the observed temperature sensitivity of the triple mutant at 37°C. In order to examine this more closely, we constructed strains lacking skp, fkpA, or both in the degPS210A mutant background and assayed their growth by an efficiency-of-plating assay. None of the strains examined exhibit a growth defect at the permissive temperature, 30°C (Fig. 1A). However, the Δskp ΔfkpA degPS210A triple mutant is temperature sensitive; like the Δskp ΔdegP ΔfkpA::kan strain, it grows very poorly at 37°C (Fig. 1A). The temperature sensitivity of this strain can be complemented by the expression of wild-type degP in trans, which provides further evidence of protein misfolding in the Δskp ΔfkpA double mutant. In the absence of both skp and fkpA, the protease activity of DegP is required to degrade misfolded proteins that accumulate in the periplasm.
Fig 1.

The Δskp ΔfkpA double mutant exhibits phenotypes consistent with OMP assembly defects. (A) Overnight cultures were grown at the permissive temperature of 30°C. These cultures were then serially diluted in fresh LB medium, spotted onto LB agar, and incubated overnight at 30°C and 37°C. The degPS210A allele was moved using the linked yadC::Tn10 marker, so this allele is also present in all of the degPS210A strains, and all of the strains shown are Ara+/r. The Δskp degPS210A ΔfkpA strain exhibits a defect of approximately 3 to 4 logs of growth at 37°C compared to that of the single and double mutants. (B) Growth of the wild-type, Δskp, ΔfkpA, and Δskp ΔfkpA strains was assayed on LB agar supplemented with 65 mg/liter vancomycin by efficiency-of-plating assay. The Δskp ΔfkpA double mutant exhibits a defect of approximately 5 to 6 logs of growth on 65 mg/liter vancomycin.
Loss of Skp and FkpA causes defects in OM biogenesis.
Mutant strains that are defective in OM biogenesis also exhibit increased permeability to antibiotics and other small molecules (3). Thus, if the Δskp ΔfkpA mutant is defective in the assembly of OMPs, it should exhibit increased OM permeability. We tested this possibility by performing efficiency-of-plating assays with the single- and double-mutant strains at 37°C on LB agar supplemented with 65 mg/liter vancomycin. Indeed, the Δskp ΔfkpA double mutant is much more sensitive to this concentration of vancomycin than either single mutant (Fig. 1B).
To further investigate the potential OMP assembly defect in the Δskp ΔfkpA strain, we examined OMP levels in this strain by Western blotting. The OMPs LamB, OmpA, and TolC have been previously used as model proteins. Because the LamB and OmpA proteins are rapidly degraded in the periplasm when they fall off-pathway (11), their levels in whole-cell lysates are representative of their levels in the OM, and decreased levels of these proteins are indicative of a general OMP assembly defect (11, 12). Conversely, TolC levels are known to increase in certain strains that exhibit an OMP assembly defect (46). Neither LamB nor OmpA is present at reduced levels in the Δskp ΔfkpA mutant strain, and levels of TolC are not increased (Fig. 2A). This suggests that the OMP assembly defect in the Δskp ΔfkpA mutant strain is not general. The ΔsurA strain, which exhibits clear defects in the assembly of LamB, is shown here for comparison (Fig. 2A).
Fig 2.
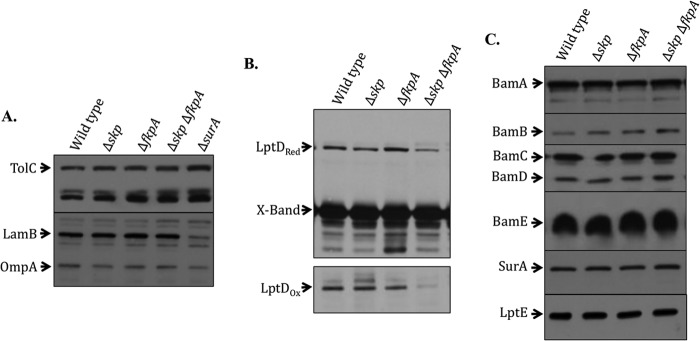
The Δskp ΔfkpA double mutant exhibits an LptD assembly defect that is not due to loss of other LptD assembly factors. Cultures were grown to an OD600 of approximately 0.7 to 0.8. Whole-cell lysates were analyzed by SDS-PAGE and immunoblotted with the appropriate antibody. (A) A ΔsurA strain is included for comparison. Levels of TolC, LamB, and OmpA are unaffected by the loss of skp and fkpA. (B) Samples were analyzed by SDS-PAGE in sample buffer with (LptDRed) and without (LptDOx) 2-mercaptoethanol. “X-Band” is a cross-reacting band of approximately 55 kDa, shown here as a loading control. Levels of both oxidized and reduced LptD are decreased in the absence of Skp and FkpA. (C) The levels of proteins known to be important for LptD assembly are unchanged in the absence of Skp and FkpA.
Loss of Skp and FkpA affects LptD levels.
In the absence of a general OMP assembly defect in the Δskp ΔfkpA strain, we investigated whether the observed OM permeability of this strain could be due to an LPS assembly defect resulting from decreased levels of LptD in the OM. Thus, we examined the levels of LptD by Western blotting. Because LptD is not fully oxidized until it has interacted with its accessory lipoprotein, LptE, and been inserted into the OM by the Bam complex, nonreducing SDS-PAGE provides us with an assay to determine the levels of correctly assembled LptD in the cell (8, 9). While the Δskp and ΔfkpA single mutants exhibited no apparent reduction in oxidized or reduced LptD levels, a Δskp ΔfkpA double mutant showed a considerable reduction in the levels of both oxidized and reduced LptD (Fig. 2B). These results suggest that in the absence of Skp and FkpA, there is a defect in the assembly of LptD.
If the loss of Skp and FkpA affects the levels of the Bam complex members, SurA, or the accessory lipoprotein LptE, it could cause the LptD assembly defect that we observed, because these proteins are necessary for the correct assembly of LptD. However, the levels of all of these proteins remained unchanged in the Δskp ΔfkpA double mutant (Fig. 2C), leading us to conclude that Skp and FkpA participate directly in the assembly of LptD.
The role of Skp and FkpA in LptD assembly is unique.
We considered the possibility that the decreased LptD levels in the Δskp ΔfkpA double mutant might be caused by overloading the SurA pathway with substrate. If Skp and FkpA function as OMP chaperones, it is possible that the loss of both might saturate the main OMP assembly pathway, titrating SurA and causing some LptD to fall off-pathway and be degraded by periplasmic proteases (such as DegP). If this were indeed the case, increasing the levels of SurA in the Δskp ΔfkpA double-mutant strain should restore LptD levels. Accordingly, we overexpressed SurA in a Δskp ΔfkpA double mutant and examined the LptD levels in this strain by Western blotting. Overexpression of SurA did not increase the levels of LptD in the Δskp ΔfkpA double mutant (Fig. 3A). Further, the deletion of a number of the most highly expressed OMPs (LamB, OmpA, and OmpC), which should decrease the load on the SurA pathway, did not restore levels of LptD in the Δskp ΔfkpA double mutant (Fig. 3B).
Fig 3.

Skp/FkpA and SurA have distinct roles in LptD assembly. Cultures were grown to an OD600 of about 0.7 to 0.8 in LB medium. Whole-cell lysates were analyzed by SDS-PAGE and immunoblotted with the appropriate antibody. “X-Band” is a cross-reacting band of approximately 55 kDa, shown here as a loading control. (A) Medium was supplemented with 0.2% l-arabinose to induce the psurA plasmid. Overexpression of SurA in the Δskp ΔfkpA strain has no effect on LptD levels. (B) The deletion of major OMPs has no effect on LptD levels in the absence of skp and fkpA. (C) Overexpression of Skp or FkpA does not increase LptD levels in the absence of surA. (D) Medium was supplemented with 0.2% l-arabinose to induce the plasmids. DsbA levels were visualized using Coomassie brilliant blue staining after SDS-PAGE analysis. Overexpression of DsbA in the Δskp ΔfkpA strain does not affect LptD levels.
We then wondered whether either Skp or FkpA could substitute for SurA in the assembly of LptD. In order to test this, we overexpressed Skp and FkpA in a ΔsurA background and examined the levels of LptD in these strains by Western blotting. Overexpression of Skp or FkpA did not restore LptD levels (Fig. 3C). It is clear that SurA cannot perform the role of Skp and FkpA in LptD assembly, nor can Skp or FkpA compensate for the role of SurA in this process.
Another protein folding factor that is known to be required for the proper and efficient assembly of LptD is DsbA, which catalyzes the formation of the disulfide bonds in LptD (9). If the loss of Skp and FkpA leads to a defect in the formation of these disulfide bonds, LptD assembly would be impaired in a way that could possibly be compensated for by the overexpression of DsbA. However, the overexpression of DsbA from both low- and high-copy-number plasmids failed to restore LptD levels in the Δskp ΔfkpA double mutant (Fig. 3D). Therefore, none of the periplasmic protein folding factors that are known to be important for LptD assembly can substitute for Skp and FkpA.
Skp/FkpA and BamB exhibit a functional relationship.
The nonessential Bam complex lipoprotein BamB contributes to the assembly of a large number of OMPs. In most cases, OMPs that require SurA also require BamB (2, 6, 11, 46, 47). LptD, however, is an exception. In the absence of BamB, LptD levels remain relatively unaffected (2). However, when LptD assembly is impaired, as is the case in an lptD4213 mutant, the loss of bamB causes synthetic lethality (2). Thus, bamB and skp mutants exhibit similar phenotypes; each has little effect on LptD assembly unless an additional defect exists.
We hypothesized that LptD is assembled normally in a bamB mutant strain because of the role played by Skp and FkpA. In order to address this possibility, we examined LptD levels in Δskp bamB::kan and ΔfkpA bamB::kan double-mutant strains by Western blotting. In the Δskp bamB::kan double-mutant strain, LptD levels are much lower than in either of the single mutants, and the ΔfkpA bamB::kan strain exhibits a slight LptD assembly defect (Fig. 4). LptD levels are largely unaffected by the loss of any of these individual proteins, but the loss of two of these proteins in combination impairs LptD assembly.
Fig 4.
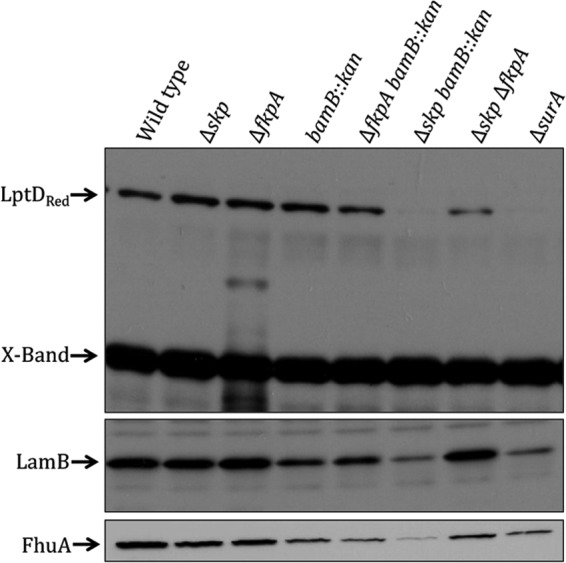
Loss of Skp and BamB results in synthetic LptD, LamB, and FhuA assembly defects. Cultures were grown to an OD600 of about 0.75 to 0.9 in LB medium. Samples were analyzed by SDS-PAGE and immunoblotted with anti-LptD, anti-LamB, and anti-FhuA antibodies. “X-Band” is a cross-reacting band of approximately 55 kDa, shown here as a loading control. The ΔsurA strain is shown here as a control. The Δskp bamB::kan and ΔfkpA bamB::kan strains exhibit decreased LptD and FhuA levels compared to those of the single-mutant strains.
We also examined the assembly of LamB in these mutants. LamB represents an OMP that is strongly affected by the loss of BamB and SurA, but its assembly is unaffected by the loss of skp and fkpA (Fig. 2A). Although the Δskp bamB::kan double-mutant strain exhibits impaired LamB assembly, this defect is not as severe as the observed LptD assembly defect in this double mutant, especially compared to that in the bamB single mutant (Fig. 4). The ΔfkpA bamB::kan strain exhibits LamB levels that are essentially equivalent to those of the bamB::kan single-mutant strain (Fig. 4).
FhuA and LptD are similarly affected in double-mutant strains.
The TonB-dependent siderophore transport protein FhuA (48) is, like LptD, one of the few OMPs whose assembly is strongly dependent on SurA (12). In fact, in a surA mutant, LptD and FhuA are the only known OMPs present at decreased levels that do not exhibit a concomitant decrease in transcript levels (12). FhuA is also structurally similar to LptD; it possesses a soluble N-terminal domain and a C-terminal barrel domain. Although we had already shown that the loss of skp and fkpA does not affect a number of model OMPs (LamB, OmpA, and TolC), the similarity of FhuA to LptD led us to investigate whether FhuA assembly is affected in the Δskp ΔfkpA strain.
Indeed, like LptD, while FhuA levels are unaffected in the Δskp and ΔfkpA single-mutant strains, there is an assembly defect in the Δskp ΔfkpA double mutant (Fig. 4). FhuA also exhibits assembly defects that are similar to those of LptD in the absence of bamB. Although FhuA assembly is only moderately affected by the loss of BamB, its assembly is dramatically reduced in the Δskp bamB::kan double-mutant strain (Fig. 4). This result suggests that FhuA and LptD are targeted to the Bam complex by similar chaperone assembly pathways.
A novel baeS* allele suppresses the defects of a Δskp ΔfkpA double mutant.
In order to learn more about the role of Skp and FkpA in the assembly of LptD, we selected suppressors of the Δskp ΔfkpA strain by plating on vancomycin. One vancomycin-resistant suppressor also restored growth of the temperature-sensitive Δskp ΔfkpA ΔdegP::kan strain at 37°C, in addition to restoring levels of LptD in the Δskp ΔfkpA double mutant (Fig. 5A). This suppressor mapped to the baeSR locus, and sequencing revealed that amino acid 255 in BaeS had been changed from proline to leucine (baeSP255L).
Fig 5.

Spy overexpression suppresses the LptD assembly defect of a Δskp ΔfkpA strain but not a ΔsurA strain. Cultures were grown to stationary phase in LB medium. Where indicated, medium was supplemented with 50 μM IPTG. Samples were analyzed by SDS-PAGE and immunoblotted with anti-LptD and Spy antibodies. (A) The baeS* suppressor (baeSP255L) and Spy overexpression suppress the LptD assembly defect of a Δskp ΔfkpA strain. (B) Spy is induced approximately 1,000-fold in the baeS* and pspy strains. The X-band is too diluted to be visible in the 1:100 or 1:1,000 dilutions. (C) Spy is induced similarly when the baeS* suppressor is transduced into a wild-type (MC4100) background. (D) Spy overexpression does not suppress the LptD defect of a ΔsurA strain.
The BaeS protein is the sensor kinase of a two-component system; its cognate response regulator is BaeR (49). This two-component system is known to regulate several genes in response to a variety of stimuli, including spheroplast formation, PapG overexpression, and indole exposure (49). Among the genes regulated by this system are those specifying multidrug efflux pump components (mdtABCD and acrD) and the periplasmic chaperone spy (50). Previously isolated baeS alleles, which dramatically upregulate the production of Spy, mapped to nearby amino acids (E264 and D268) (42). It has also been shown that Spy can protect substrate proteins from aggregation in vitro and that overexpression of Spy can stabilize an unstable variant of the colicin E7 immunity protein (Im7) in vivo (42). Thus, it seemed likely that baeSP255L is a gain-of-function allele and that the LptD defect is suppressed due to the overexpression of Spy. Similar mutants in the Cpx two-component system have previously been called cpx* mutants (51), and thus we will refer to this mutation as a baeS* mutant.
The levels of Spy in this suppressor confirm that it is a baeS* allele; in the Δskp ΔfkpA strain containing the baeSP255L suppressor, there is approximately 1,000-fold more Spy than in a wild-type strain (Fig. 5B). Spy levels are similarly induced when the suppressor allele is introduced into a wild-type background (Fig. 5C). When a Δspy::kan allele is introduced into the Δskp ΔfkpA baeSP255L strain, levels of LptD return to the levels observed in the Δskp ΔfkpA double mutant. When Spy alone is overexpressed in the Δskp ΔfkpA strain, LptD levels are restored to nearly the levels observed in a wild-type strain (Fig. 5A). Thus, Spy is both necessary and sufficient to suppress the LptD assembly defect in the Δskp ΔfkpA strain.
It seemed likely that the restoration of LptD levels in the Δskp ΔfkpA strain when Spy is overexpressed is due to the general chaperone activity of the Spy protein. We investigated whether Spy overexpression is capable of suppressing a broader range of LptD assembly defects by overexpressing Spy in a ΔsurA mutant. As shown in Fig. 5D, LptD levels remain unchanged even at high levels of Spy overexpression. This result reinforces the specificity of the roles played by Skp/FkpA and SurA in the LptD assembly process. Although Spy is capable of compensating for the role played by Skp and FkpA, it is not capable of compensating for the role played by SurA.
DISCUSSION
Several studies have shown that the periplasmic chaperone Skp can interact directly with OMPs (17, 23), and genetic analysis has revealed a role for this protein in the backup OMP assembly pathway in E. coli (11, 14). However, no substrates specifically dependent on Skp for their assembly have ever been identified. We have discovered a role for Skp in the assembly of LptD. This function was not previously identified, because it can be performed by another periplasmic chaperone, FkpA. The loss of both Skp and FkpA substantially reduces the levels of correctly assembled LptD.
Previous work has shown that LptD assembly is much more complicated than the assembly of a typical OMP. After translocation through the IM by the Sec machinery, LptD is escorted across the periplasm by SurA in a manner that no other chaperone can replicate (9, 12). Once LptD reaches the Bam complex, it must interact with its partner lipoprotein LptE in order for its β-barrel domain to be properly assembled (9, 10). Finally, oxidation, a process catalyzed by DsbA, must occur to form the disulfide bonds that connect the amino-terminal periplasmic domain to the carboxy-terminal β-barrel (9). Our results demonstrate that Skp or FkpA must act in concert with the main periplasmic chaperone SurA to efficiently assemble LptD but that SurA and Skp or FkpA perform distinct roles in this process. Overexpression of SurA or DsbA does not restore levels of LptD in a Δskp ΔfkpA strain, nor can overexpressing Skp or FkpA restore LptD levels in a ΔsurA strain. The loss of SurA, or of both Skp and FkpA, causes a profound defect in LptD assembly that cannot be compensated for by the other proteins.
The baeS* suppressor isolated here provides further evidence of a novel role for Skp or FkpA in LptD assembly. This suppressor restores LptD assembly in mutants lacking Skp and FkpA, and the overproduction of Spy that occurs in this mutant is both necessary and sufficient for this restoration. However, Spy overexpression fails to restore LptD levels in the absence of SurA. We conclude that LptD assembly requires the participation of at least two different periplasmic chaperones, each of which must perform distinct functions. Spy is capable only of substituting for one of these two functions.
The loss of Skp, FkpA, or BamB has little effect on LptD assembly. However, unlike the loss of Skp or FkpA, the absence of BamB does cause defects in the assembly of many other OMPs, such as LamB and OmpA, and when LptD assembly is impaired, BamB becomes essential (2). Although LptD assembly is not measurably compromised in strains lacking either Skp or BamB, assembly of this protein is decreased significantly in strains lacking both Skp and BamB. The synthetic phenotypes exhibited by these double mutants could indicate that Skp and BamB possess a redundant function in LptD assembly. Alternatively, there could be two different LptD assembly pathways: one that requires Skp (or FkpA) and another that uses BamB. At present, we cannot distinguish between these possibilities.
Strikingly, although other model OMPs, including LamB, are unaffected or less severely affected than LptD in mutant strains that lack Skp and FkpA, at least one other OMP, FhuA, exhibits assembly defects that are similar to those of LptD. Both LptD and FhuA are strongly dependent on SurA for their assembly, and both also require Skp or FkpA. It is tempting to speculate that Skp and FkpA aid in the assembly of these two proteins because of their structural similarities. One somewhat unique feature shared by LptD and FhuA is the presence of a large, periplasmic, N-terminal domain, which is the first to emerge into the periplasm during translocation. Skp has been shown to play a role in the folding and assembly of the N-terminal, soluble passenger domain of the autotransporter IcsA in S. flexneri (27), and both Skp and FkpA have been shown to interact with the N-terminal passenger domain of EspP in E. coli (24, 25, 35). It is possible that Skp and FkpA play a role in assembling proteins with these soluble N-terminal domains. Another possibility is that Skp and FkpA are involved at an early step in the assembly process and that only some OMPs require interaction with these chaperones at this step. Skp has been shown to associate with OMPs at the inner membrane (52) and to interact with EspP at an earlier step in its assembly than SurA (24) does.
There may be additional OMPs in E. coli that share the chaperone requirements of FhuA and LptD. Further study of these proteins and their structural characteristics could provide insight into the functional roles of Skp and FkpA. Moreover, because this is the first time a physiological substrate of the Spy protein has been identified, further study of this interaction may provide insight into the function of Spy in the cell.
ACKNOWLEDGMENTS
J.S., T.F.M., and G.R.S. were supported by the Genetics and Molecular Biology Training Grant from the NIH GM07388, and T.J.S. is supported by the National Institute of General Medical Sciences grant GM34821.
We thank the members of the Silhavy lab for their helpful comments and discussions and Jennifer Munko for the assistance in preparing the manuscript. We also thank Natividad Ruiz (The Ohio State University) for technical assistance and helpful discussions.
Footnotes
Published ahead of print 14 June 2013
REFERENCES
- 1. Silhavy TJ, Kahne D, Walker S. 2010. The bacterial cell envelope. Cold Spring Harb. Perspect. Biol. 2:a000414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ruiz N, Falcone B, Kahne D, Silhavy TJ. 2005. Chemical conditionality: a genetic strategy to probe organelle assembly. Cell 121:307–317 [DOI] [PubMed] [Google Scholar]
- 3. Sampson BA, Misra R, Benson SA. 1989. Identification and characterization of a new gene of Escherichia coli K-12 involved in outer membrane permeability. Genetics 122:491–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Braun M, Silhavy TJ. 2002. Imp/OstA is required for cell envelope biogenesis in Escherichia coli. Mol. Microbiol. 45:1289–1302 [DOI] [PubMed] [Google Scholar]
- 5. Wu T, Malinverni J, Ruiz N, Kim S, Silhavy TJ, Kahne D. 2005. Identification of a multicomponent complex required for outer membrane biogenesis in Escherichia coli. Cell 121:235–245 [DOI] [PubMed] [Google Scholar]
- 6. Sklar JG, Wu T, Gronenberg LS, Malinverni JC, Kahne D, Silhavy TJ. 2007. Lipoprotein SmpA is a component of the YaeT complex that assembles outer membrane proteins in Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 104:6400–6405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wu T, McCandlish AC, Gronenberg LS, Chng S-S, Silhavy TJ, Kahne D. 2006. Identification of a protein complex that assembles lipopolysaccharide in the outer membrane of Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 103:11754–11759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chng S-S, Ruiz N, Chimalakonda G, Silhavy TJ, Kahne D. 2010. Characterization of the two-protein complex in Escherichia coli responsible for lipopolysaccharide assembly at the outer membrane. Proc. Natl. Acad. Sci. U. S. A. 107:5363–5368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ruiz N, Chng S-S, Hiniker A, Kahne D, Silhavy TJ. 2010. Nonconsecutive disulfide bond formation in an essential integral outer membrane protein. Proc. Natl. Acad. Sci. U. S. A. 107:12245–12250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chimalakonda G, Ruiz N, Chng S-S, Garner RA, Kahne D, Silhavy TJ. 2011. Lipoprotein LptE is required for the assembly of LptD by the β-barrel assembly machine in the outer membrane of Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 108:2492–2497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sklar JG, Wu T, Kahne D, Silhavy TJ. 2007. Defining the roles of the periplasmic chaperones SurA, Skp, and DegP in Escherichia coli. Genes Dev. 21:2473–2484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Vertommen D, Ruiz N, Leverrier P, Silhavy TJ, Collet J. 2009. Characterization of the role of the Escherichia coli periplasmic chaperone SurA using differential proteomics. Proteomics 9:2432–2443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dartigalongue C, Missiakas D, Raina S. 2001. Characterization of the Escherichia coli sigma E regulon. J. Biol. Chem. 276:20866–20875 [DOI] [PubMed] [Google Scholar]
- 14. Rizzitello AE, Harper JR, Silhavy TJ. 2001. Genetic evidence for parallel pathways of chaperone activity in the periplasm of Escherichia coli. J. Bacteriol. 183:6794–6800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Denoncin K, Schwalm J, Vertommen D, Silhavy TJ, Collet J-F. 2012. Dissecting the Escherichia coli periplasmic chaperone network using differential proteomics. Proteomics 12:1391–1401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Holck A, Lossius I, Aasland R, Kleppe K. 1987. Purification and characterization of the 17 K protein, a DNA-binding protein from Escherichia coli. Biochim. Biophys. Acta 914:49–54 [DOI] [PubMed] [Google Scholar]
- 17. Chen R, Henning U. 1996. A periplasmic protein (Skp) of Escherichia coli selectively binds a class of outer membrane proteins. Mol. Microbiol. 19:1287–1294 [DOI] [PubMed] [Google Scholar]
- 18. Jarchow S, Lück C, Görg A, Skerra A. 2008. Identification of potential substrate proteins for the periplasmic Escherichia coli chaperone Skp. Proteomics 8:4987–4994 [DOI] [PubMed] [Google Scholar]
- 19. Walton TA, Sousa MC. 2004. Crystal structure of Skp, a prefoldin-like chaperone that protects soluble and membrane proteins from aggregation. Mol. Cell 15:367–374 [DOI] [PubMed] [Google Scholar]
- 20. De Cock H, Schäfer U, Potgeter M, Demel R, Müller M, Tommassen J. 1999. Affinity of the periplasmic chaperone Skp of Escherichia coli for phospholipids, lipopolysaccharides and non-native outer membrane proteins. Role of Skp in the biogenesis of outer membrane protein. Eur. J. Biochem. 259:96–103 [DOI] [PubMed] [Google Scholar]
- 21. Schäfer U, Beck K, Müller M. 1999. Skp, a molecular chaperone of Gram-negative bacteria, is required for the formation of soluble periplasmic intermediates of outer membrane proteins. J. Biol. Chem. 274:24567–24574 [DOI] [PubMed] [Google Scholar]
- 22. Bulieris PV, Behrens S, Holst O, Kleinschmidt JH. 2003. Folding and insertion of the outer membrane protein OmpA is assisted by the chaperone Skp and by lipopolysaccharide. J. Biol. Chem. 278:9092–9099 [DOI] [PubMed] [Google Scholar]
- 23. Walton TA, Sandoval CM, Fowler CA, Pardi A, Sousa MC. 2009. The cavity-chaperone Skp protects its substrate from aggregation but allows independent folding of substrate domains. Proc. Natl. Acad. Sci. U. S. A. 106:1772–1777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ieva R, Tian P, Peterson JH, Bernstein HD. 2011. Sequential and spatially restricted interactions of assembly factors with an autotransporter beta domain. Proc. Natl. Acad. Sci. U. S. A. 108:E383–E391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ieva R, Bernstein HD. 2009. Interaction of an autotransporter passenger domain with BamA during its translocation across the bacterial outer membrane. Proc. Natl. Acad. Sci. U. S. A. 106:19120–19125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Volokhina EB, Grijpstra J, Stork M, Schilders I, Tommassen J, Bos MP. 2011. Role of the periplasmic chaperones Skp, SurA, and DegQ in outer membrane protein biogenesis in Neisseria meningitidis. J. Bacteriol. 193:1612–1621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wagner JK, Heindl JE, Gray AN, Jain S, Goldberg MB. 2009. Contribution of the periplasmic chaperone Skp to efficient presentation of the autotransporter IcsA on the surface of Shigella flexneri. J. Bacteriol. 191:815–821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Justice SS, Hunstad DA, Harper JR, Duguay AR, Pinkner JS, Bann J, Frieden C, Silhavy TJ, Hultgren SJ. 2005. Periplasmic peptidyl prolyl cis-trans isomerases are not essential for viability, but SurA is required for pilus biogenesis in Escherichia coli. J. Bacteriol. 187:7680–7686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ow DS-W, Lim DY-X, Nissom PM, Camattari A, Wong VV-T. 2010. Co-expression of Skp and FkpA chaperones improves cell viability and alters the global expression of stress response genes during scFvD1.3 production. Microb. Cell Fact. 9:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Saul FA, Arié J-P, Vulliez-le Normand B, Kahn R, Betton J-M, Bentley GA. 2004. Structural and functional studies of FkpA from Escherichia coli, a cis/trans peptidyl-prolyl isomerase with chaperone activity. J. Mol. Biol. 335:595–608 [DOI] [PubMed] [Google Scholar]
- 31. Zhang Z, Song L, Fang M, Wang F, He D, Zhao R, Liu J, Zhou Z, Yin C, Lin Q, Huang H. 2003. Production of soluble and functional engineered antibodies in Escherichia coli improved by FkpA. Biotechniques 35:1032–1038, 1041–1042 [DOI] [PubMed] [Google Scholar]
- 32. Hullmann J, Patzer SI, Römer C, Hantke K, Braun V. 2008. Periplasmic chaperone FkpA is essential for imported colicin M toxicity. Mol. Microbiol. 69:926–937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Barnéoud-Arnoulet A, Barreteau H, Touzé T, Mengin-Lecreulx D, Lloubès R, Duché D. 2010. Toxicity of the colicin M catalytic domain exported to the periplasm is FkpA independent. J. Bacteriol. 192:5212–5219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Helbig S, Patzer SI, Schiene-Fischer C, Zeth K, Braun V. 2011. Activation of colicin M by the FkpA prolyl cis-trans isomerase/chaperone. J. Biol. Chem. 286:6280–6290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ruiz-Perez F, Henderson IR, Nataro JP. 2010. Interaction of FkpA, a peptidyl-prolyl cis/trans isomerase with EspP autotransporter protein. Gut Microbes 1:339–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Veiga E, de Lorenzo V, Fernández LA. 2004. Structural tolerance of bacterial autotransporters for folded passenger protein domains. Mol. Microbiol. 52:1069–1080 [DOI] [PubMed] [Google Scholar]
- 37. Silhavy TJ, Berman ML, Enquist LW. 1984. Experiments with gene fusions. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 38. Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H. 2006. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol. Syst. Biol. 2:2006.0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Cherepanov PP, Wackernagel W. 1995. Gene disruption in Escherichia coli: TcR and KmR cassettes with the option of Flp-catalyzed excision of the antibiotic-resistance determinant. Gene 158:9–14 [DOI] [PubMed] [Google Scholar]
- 40. Singer M, Baker TA, Schnitzler G, Deischel SM, Goel M, Dove W, Jaacks KJ, Grossman AD, Erickson JW, Gross CA. 1989. A collection of strains containing genetically linked alternating antibiotic resistance elements for genetic mapping of Escherichia coli. Microbiol. Rev. 53:1–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Amann E, Ochs B, Abel KJ. 1988. Tightly regulated tac promoter vectors useful for the expression of unfused and fused proteins in Escherichia coli. Gene 69:301–315 [DOI] [PubMed] [Google Scholar]
- 42. Quan S, Koldewey P, Tapley T, Kirsch N, Ruane KM, Pfizenmaier J, Shi R, Hofmann S, Foit L, Ren G, Jakob U, Xu Z, Cygler M, Bardwell JCA. 2011. Genetic selection designed to stabilize proteins uncovers a chaperone called Spy. Nat. Struct. Mol. Biol. 18:262–269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Simpson RJ. 2007. Staining proteins in gels with Coomassie blue. Cold Spring Harb. Protoc. 2007:pdb.prot4719. [DOI] [PubMed] [Google Scholar]
- 44. Lipinska B, Fayet O, Baird L, Georgopoulos C. 1989. Identification, characterization, and mapping of the Escherichia coli htrA gene, whose product is essential for bacterial growth only at elevated temperatures. J. Bacteriol. 171:1574–1584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Spiess C, Beil A, Ehrmann M. 1999. A temperature-dependent switch from chaperone to protease in a widely conserved heat shock protein. Cell 97:339–347 [DOI] [PubMed] [Google Scholar]
- 46. Charlson ES, Werner JN, Misra R. 2006. Differential effects of yfgL mutation on Escherichia coli outer membrane proteins and lipopolysaccharide. J. Bacteriol. 188:7186–7194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ureta AR, Endres RG, Wingreen NS, Silhavy TJ. 2007. Kinetic analysis of the assembly of the outer membrane protein LamB in Escherichia coli mutants each lacking a secretion or targeting factor in a different cellular compartment. J. Bacteriol. 189:446–454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sansom MS. 1999. Membrane proteins: a tale of barrels and corks. Curr. Biol. 9:R254–R257 [DOI] [PubMed] [Google Scholar]
- 49. Raffa RG, Raivio TL. 2002. A third envelope stress signal transduction pathway in Escherichia coli. Mol. Microbiol. 45:1599–1611 [DOI] [PubMed] [Google Scholar]
- 50. Leblanc SKD, Oates CW, Raivio TL. 2011. Characterization of the induction and cellular role of the BaeSR two-component envelope stress response of Escherichia coli. J. Bacteriol. 193:3367–3375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Raivio TL, Silhavy TJ. 1997. Transduction of envelope stress in Escherichia coli by the Cpx two-component system. J. Bacteriol. 179:7724–7733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Harms N, Koningstein G, Dontje W, Muller M, Oudega B, Luirink J, de Cock H. 2001. The early interaction of the outer membrane protein Phoe with the periplasmic chaperone Skp occurs at the cytoplasmic membrane. J. Biol. Chem. 276:18804–18811 [DOI] [PubMed] [Google Scholar]
- 53. Casadaban MJ. 1976. Transposition and fusion of the lac genes to selected promoters in Escherichia coli using bacteriophage lambda and mu. J. Mol. Biol. 104:541–555 [DOI] [PubMed] [Google Scholar]
- 54. Rigel NW, Schwalm J, Ricci DP, Silhavy TJ. 2012. BamE modulates the Escherichia coli beta-barrel assembly machine component BamA. J. Bacteriol. 194:1002–1008 [DOI] [PMC free article] [PubMed] [Google Scholar]