

Abstract
The expression of mepA, encoding the Staphylococcus aureus MepA multidrug efflux protein, is repressed by the MarR homologue MepR. MepR dimers bind differently to operators upstream of mepR and mepA, with affinity being greatest at the mepA operator. MepR substitution mutations may result in mepA overexpression, with A103V most common in clinical strains. Evaluation of the functional consequences of this and other MepR substitutions using a lacZ reporter gene assay revealed markedly reduced repressor activity in the presence of Q18P, F27L, G97E, and A103V substitutions. Reporter data were generally supported by susceptibility and efflux assays, and electrophoretic mobility shift assays (EMSAs) confirmed compromised affinities of MepR F27L and A103V for the mepR and mepA operators. One mutant protein contained two substitutions (T94P and T132M); T132M compensated for the functional defect incurred by T94P and also rescued that of A103V but not F27L, establishing it as a limited-range suppressor. The function of another derivative with 10 substitutions was minimally affected, and this may be an extreme example of suppression involving interactions among several residues. Structural correlations for the observed functional effects were ascertained by modeling mutations onto apo-MepR. It is likely that F27L and A103V affect the protein-DNA interaction by repositioning of DNA recognition helices. Negative functional consequences of MepR substitution mutations may result from interference with structural plasticity, alteration of helical arrangements, reduced protein-cognate DNA affinity, or possibly association of MepR protomers. Structural determinations will provide further insight into the consequences of these and other mutations that affect MepR function, especially the T132M suppressor.
INTRODUCTION
Efflux of antimicrobial agents and biocides is an important mechanism of resistance in bacteria (1). Efflux may be a primary resistance mechanism, and it has been established as such for Pseudomonas aeruginosa, but it also may predispose to the acquisition of drug target mutations resulting in high-level resistance (2). A relevant example of the latter is the development of topoisomerase mutations at a higher rate in Staphylococcus aureus with increased expression of norA compared to that in an isogenic wild-type (WT) strain (3). The ability of many efflux proteins to recognize multiple structurally diverse substrates amplifies the problem, resulting in a multidrug resistance (MDR) phenotype.
MDR-conferring efflux pumps belong to one of five protein families that are distinguished by the extent and type of secondary structures and the energy source used for substrate translocation (4). These are the ATP binding cassette (ABC), major facilitator superfamily (MFS), resistance-nodulation-division (RND), small multidrug resistance (SMR), and multidrug and toxin extrusion (MATE) families. All except ABC proteins, which derive the energy required for activity by ATP hydrolysis, utilize ion gradients as the energy source for substrate transport. Most frequently this is the proton gradient, but MATE family proteins also can utilize the sodium ion gradient (5). Bacterial MATE proteins are the least numerous of the MDR efflux proteins, with only one (MepA) encoded in the genome of S. aureus N315 (see www.membranetransport.org).
Expression of mepA is regulated by MepR, a repressor protein encoded immediately upstream of mepA (6). The structure of apo-MepR has been determined and reveals it to be a MarR family winged helix-turn-helix (wHTH) protein (7). It has six alpha helices, with the α2 through α4 helices together with the intervening beta strands comprising the DNA binding domain (see Fig. 7). Electrostatic surface mapping of apo-MepR reveals this region to be highly positive and, as such very, favorable for interaction with DNA. Detailed functional analyses of MepR have established that it binds to operator regions in the mepR and mepA promoters, with significant differences in its interaction with each site. It binds as a single dimer to the mepR operator but as two dimers to that of mepA. Binding affinity is 4-fold greater for the mepA site than that of mepR (Kd [dissociation constant] of 6.3 versus 24.3 nM, respectively). Additionally, binding to the mepA site is readily reversed by substrates, compared to a much more attenuated effect at the mepR operator (8). A seven-base-pair conserved “signature” motif (consensus sequence, 5′-GTTAGAT-3′) is found once within the mepR operator and twice within the mepA operator and clearly plays a role in the differing binding affinities of MepR for each site (Fig. 1) (7). Cooperative binding of MepR dimers to each signature motif in the mepA operator may be the basis for both the greater affinity and easier substrate induction at this site, perhaps in a manner similar to the interaction of QacR with the qacA operator (9).
Fig 1.
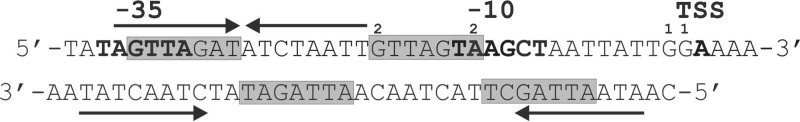
mepA operator site. 1 and 2, sites of mutations observed in the mepA-overexpressing strains SA-K4375 (G→A at −1 and −2) and SA-K4624 (A→G at −14 and G→A at −20), respectively. Inverted repeats are identified by arrows, −35 and −10 promoter motifs and the transcription start site (TSS) are indicated in bold, and the consensus GTTAGAT sequences are highlighted in gray.
Using quantitative reverse transcription-PCR (qRT-PCR) we have identified numerous clinical and laboratory strains of S. aureus that overexpress mepA (6, 10–14). Understanding the mechanism(s) of mepA overexpression may provide clues for the design of means to overcome its potential deleterious clinical consequences. One of these mechanisms is the acquisition of functionally detrimental substitution mutations in MepR. In this report, we describe the effects of various MepR mutations among clinical and laboratory strains, both previously reported and new, on repressor activity and provide a structural perspective for the observed phenotypes conferred by the mutations.
MATERIALS AND METHODS
Bacterial strains, plasmids, media, and reagents.
Clinical isolates of S. aureus (one per patient) were collected in 2005 and again in 2009 from the microbiology laboratories of the Detroit Medical Center and the John D. Dingell VA Medical Center in Detroit, MI (n = 232 and 243, respectively). The 2005 strain set has been described previously, as have in vitro-derived mutants of selected strains from this set found to overexpress mepA (10, 12). Another 446 unique clinical isolates were collected from centers in Boston, MA, Houston, TX, Omaha, NE, and Freiberg, Germany, in 2009 (14). Strains and plasmids used are listed in Table 1. Unless otherwise noted, all reagents were the highest grade available and were obtained from Sigma Chemical Co. (St. Louis, MO) or BD Biosciences (Sparks, MD).
Table 1.
Strains and plasmids used
| Strain or plasmid | Descriptiona | Source or reference |
|---|---|---|
| S. aureus strains | ||
| NCTC 8325-4 | Commonly utilized laboratory strain; rsbU deficient | 32 |
| RN4220 | Restriction-deficient derivative of NCTC 8325-4; capable of stably maintaining recombinant plasmids | 33 |
| SH1000 | NCTC 8325-4 with rsbU mutation corrected | 34 |
| SA-K2068 | mepA-overexpressing strain derived from NCTC 8325-4; mepR deficient | 6, 35 |
| SA-K4375 | Tissue isolate from San Francisco (MepR T94P+T132 M) | 14 |
| SA-K4624 | Bloodstream isolate from Germany (MepR with 10 substitution mutations) | 14 |
| SA-K2908 | SA-K2068 mepA::lacZ | 6 |
| SA-K2916 | SH1000 mepR::lacZ | 8 |
| SA-K3731 | SA-K2068 norA::erm | 17 |
| Plasmids | ||
| pALC2073 | xyl/tetO inducible promoter controlling expression of cloned genes; Cmr | 36 |
| pK434 | pALC2073 mepR (WT gene from NCTC 8325-4) | 6 |
| pK519 | pALC2073 mepR (A103V) | 10 |
| pK580 | pALC2073 mepR (Q18P) | 12 |
| pK582 | pALC2073 mepR (G97E) | 12 |
| pK712 | pALC2073 mepR (F27L) | This study |
| pK714 | pALC2073 mepR (T94P) | This study |
| pK716 | pALC2073 mepR (T132 M) | This study |
| pK756 | pALC2073 mepR (P62A) | This study |
| pK757 | pALC2073 mepR (L119F) | This study |
| pK758 | pALC2073 mepR (Q126R) | This study |
| pK793 | pALC2073 mepR (T94P+T132 M) | This study |
| pK796 | pALC2073 mepR (K4624) | This study |
| pK852 | pALC2073 mepR (A103F) | This study |
| pK854 | pALC2073 mepR (A103L) | This study |
| pK855 | pALC2073 mepR (R69K) | This study |
| pK858 | pALC2073 mepR (K23R) | This study |
| pK969 | pALC2073 mepR (A103V+T132 M) | This study |
| pK970 | pALC2073 mepR (F27L+T132 M) | This study |
Cmr, chloramphenicol resistance selection; WT, wild type. Substitutions encoded in mepR derivatives are in parentheses.
qRT-PCR.
For all strains collected in 2009, qRT-PCR was performed using a multiplex approach exactly as described previously using the Quantitect multiplex RT-PCR kit (Qiagen, Inc., Valencia, CA) and an ABI 7500 fast real-time PCR system (Applied Biosystems, Foster City, CA) (14). The comparative threshold cycle method was used to determine the relative expression of multiple chromosomal efflux pump genes, including mepA, compared to that in S. aureus SH1000, in which expression of each gene was considered to be 1.0. Values of 4.0 or greater were considered indicative of gene overexpression. The same approach was used to quantitate mepR expression in strains found to overexpress mepA in conjunction with a MepR substitution mutation (Table 2). qRT-PCR data for the 2005 strain set and selected in vitro-generated mepA-overexpressing mutants have been reported previously (10–12). For in vitro-derived mutants, the same procedures were used with the exception that gene expression was normalized to that in the appropriate parent strain. At least three independent RNA preparations were made for determination of mepA or mepR expression.
Table 2.
MepR substitution and mepA operator site mutations identified in mepA-overexpressing strains
| Substitution mutation(s) | No. of strains | Source of isolate(s) | Expression, fold increasea (range) |
mepA operator site mutationsb | |
|---|---|---|---|---|---|
| mepA | mepR | ||||
| Q18P | 1 | In vitro | 359 ± 185 (198–527) | 10.6 ± 5.3 (5–14) | None |
| F27L | 1 | Detroit, MI | 97.1 ± 52.8 (23–140) | 18.7 ± 12.9 (10–33) | None |
| K23R, P62A, R69K, L119F, Q126Rc | 1 | Germany | 6.4 ± 5.0 (3–14) | ND | A→G at −14, G→A at −20 |
| T94P+T132 Mc | 1 | San Francisco, CA | 14.8 ± 7.3 (8–25) | 5.8 ± 2.0 (4–7) | G→A at −1 and −2 |
| G97E | 1 | In vitro | 279 ± 222 (27–536) | 55.8 ± 80.4 (6–148) | None |
| A103V | 12 | Boston, MA; Detroit, MI; Houston, TX; Omaha, NE; San Francisco, CA | 17.1 ± 5.2d (7–32) | 8.7 ± 2.9d (5–13) | None |
| Premature truncation | 5 | Detroit, MI (1), in vitro (4) | 885 ± 847d (139–1,559) | 172 ± 246d (101–243) | NAe |
Mean ± standard deviation; ND, not done.
Positions of mutations relative to the mepA mRNA transcription start site.
Mutations present in one strain, each studied individually (SA-K4375 [San Francisco, CA] and SA-K4624 [Germany], which also had five additional substitutions; see text for details).
Expression data pooled if more than one strain; range data for these strain groups are mean low and high values.
NA, not applicable.
Genetic procedures.
The nucleotide sequences of the mepRA regions of all mepA-overexpressing strains were determined using an automated dideoxy chain termination method by the Applied Genomics Technology Center, Wayne State University (15).
WT mepR was amplified by PCR and cloned into pALC2073 to produce pK434, as described previously (6). MepR substitution mutations were introduced into this plasmid using the QuikChange Lightning site-directed mutagenesis kit and directions provided by the manufacturer (Agilent Technologies, Santa Clara, CA) (Table 1). Plasmids were transformed into commercially obtained Escherichia coli chemically competent cells following the methods provided by the supplier (One Shot TOP10 cells; Invitrogen, Carlsbad, CA). The presence of desired mutations was confirmed by sequencing, and plasmids were electroporated into S. aureus RN4220 and then transferred into host strains possessing mepA::lacZ (SA-K2908) or mepR::lacZ (SA-K2916) transcriptional fusions by transduction using previously described methods (16). Plasmid pK796 was constructed exactly as described for pK434, except that clinical strain SA-K4624 was used as the source for mepR. This plasmid was then transduced into appropriate S. aureus strains. Study of functional effects on both mepR and mepA expression was considered important based on the known differences in MepR stoichiometry and binding affinity at the mepR and mepA operator sites (7).
Many mepA-overexpressing strains were found to possess a MepR derivative containing an A103V substitution. In an effort to better understand the functional defect accompanying this relatively conservative change, A103L, which provides a larger branched aliphatic side chain, and A103F, which introduces a bulky aromatic side chain, were created by site-directed mutagenesis and MepR repressor function was evaluated. Based on data suggesting that the T132M substitution provides suppressor activity, site-directed mutagenesis was used to combine this substitution with A103V and F27L, and its ability to ameliorate the functional defects of these derivatives was determined.
EMSAs.
Electrophoretic mobility shift assays (EMSAs) were used to detect defects in MepR function that may not be apparent using in vivo assays. In EMSAs, the protein-to-target ratios can be adjusted to any desired value, with low ratios favoring the detection of subtle functional differences. Such differences likely would be obscured using in vivo assays, where the ratio is likely to be quite high as there is but one chromosomal copy of each MepR target.
MepR WT with a six-histidine tag on its carboxy terminus was produced and purified essentially as described previously, as were F27L, P62A, and A103V derivatives (8). Modifications included use of the Probond purification system and the native-condition purification procedure provided by the manufacturer (Invitrogen, Carlsbad, CA). EMSAs employing MepR WT and each derivative and the WT mepR (167 bp) or mepA (244 bp) operator site, established previously by DNase I protection assays, were performed at least twice essentially as described previously, modified by use of a concentration range of MepR derivatives to assess binding affinity and elimination of salmon sperm DNA as a nonspecific competitor (8). Excess (200-fold) unlabeled WT-specific target DNAs were used as specificity controls, as these sequences were WT in all strains possessing either MepR F27L or A103V. The high-end amount of input MepR used in these and subsequent experiments was chosen as the smallest amount resulting in a total or near-total shift of 1 fmol of target DNA.
The source strain for P62A possessed a MepR derivative with several additional substitutions (SA-K4624) (Tables 1 and 2). As the mepR operator sequence was WT in this strain, target DNA and specificity controls used were WT. However, the SA-K4624 mepA operator sequence was not WT (Fig. 1). As such, mepA EMSAs for P62A were performed using both WT and SA-K4624 sequences.
In addition to SA-K4624, a second mepA-overexpressing strain possessed mepA operator region mutations in conjunction with MepR substitution mutations (SA-K4375) (Tables 1 and 2 and Fig. 1). Evaluation of MepR derivatives from these strains revealed their repressor activities at the wild-type mepA operator to be well maintained (see below). The possibility that operator site mutations combined with MepR substitution mutations might affect repressor activity was investigated. Mutant mepA operators from these strains were amplified by PCR employing the same primers used to generate the 244-bp WT mepA operator target. Hexahistidine-tagged MepR from SA-K4624 was purified as described above, but that from SA-K4375 (with T94P and T132M substitutions) demonstrated solubility problems with this approach. As such, the hybrid purification procedure provided by the manufacturer of the Probond kit was used. This procedure included denaturing conditions followed by steps to renature the protein before use. EMSAs combining appropriate mepA operator mutants with a concentration range of the cognate MepR derivative were performed. The WT mepA operator and MepR WT, purified using native or hybrid conditions for comparison with MepR from SA-K4624 or SA-K4375, respectively, were also included.
The sensitivities of MepR K4375 and K4624 to substrate were determined by performing EMSAs in the presence and absence of cetrimide (50 μM). Cetrimide is a known MepA substrate and has been shown previously to mediate efficient dissociation of MepR WT from the mepA operator (8). The amount of input mutant MepR in these assays was the smallest amount resulting in a complete shift of 1 fmol of target DNA in the absence of substrate, which was 200 ng for K4375 and 25 ng for K4624. MepR WT was used in amounts matching those of the mutant proteins.
Functional evaluation of MepR substitution mutants.
In vivo assays of protein function complement in vitro assays in that they include the potential contribution of other intracellular processes to protein activity. The protein-to-target ratio in these assays is likely to be high, and as such subtle differences in protein function may be obscured. However, the data generated provide validation of in vitro assays such as EMSAs.
The in vivo efficiency of MepR derivatives in repression of chromosomal mepR or mepA expression was quantitated using a fluorescence-based β-galactosidase assay. This procedure has been described previously and utilizes triplicate cultures of test organisms growing from exponential to stationary phase over a 10-h period. At specified time intervals (every 30 min from 1 to 5 h and then at 6, 8, and 10 h), duplicate aliquots are removed for a determination of β-galactosidase activity, which correlates with expression of the gene to which lacZ is transcriptionally fused (12). All strains except those possessing substitution mutations at position A103 were grown in the absence and presence of 0.05 μg/ml of tetracycline to quantitate the effect of induction of plasmid-based mepR expression. Induction using this tetracycline concentration resulted in a growth defect for A103 substitutions, which was most pronounced for A103V, necessitating the use of a lower concentration that did not have this consequence (0.0125 μg/ml). For a proper comparison, control strains expressing MepR WT also were induced in the same manner and examined concurrently. β-Galactosidase activity was determined using a fluorescence microplate reader (FLx800; BioTek Instruments, Inc., Winooski, VT). Cumulative mepR or mepA expression was determined by integrating the areas beneath expression curves with SigmaPlot 12.0 (Systat Software, Inc., Chicago, IL). All β-galactosidase assays were replicated a minimum of three times, and data were normalized to the activity of MepR WT at the mepA or mepR operator in SA-K2908 or SA-K2916, respectively, which was considered to be 100%.
The in vivo biologic impact of MepR substitution mutations at the mepA operator was assessed by MIC and ethidium bromide (EB) efflux assays. Transduction was used to transfer empty pALC2073 and pALC2073 derivatives encoding MepR WT and all MepR mutants except K23R, R69K, L119F, and T132M into SA-K3731 (Table 1). This strain is a derivative of SA-K2068 (a mepR-deficient, mepA-overexpressing strain) in which a norA::erm mutation was introduced to eliminate potential confounding NorA activity (17). MICs of tetraphenylphosphonium bromide (TPP), a good MepA substrate, were determined a minimum of four times in the presence of chloramphenicol for maintenance of pALC2073 (10 μg/ml) without or with tetracycline induction of mepR expression (0.1 μg/ml) (18). MIC data are presented as TPP MIC ratios, which were determined by dividing the MIC in the presence by that in the absence of tetracycline. Functional MepR derivatives will repress mepA expression, resulting in reduced MepA protein production and TPP efflux and thus decreasing this ratio.
For EB efflux assays, an overnight growth of all transductants used in MIC testing, excluding those expressing MepR Q18P, G97E, A103F, and A103L, in supplemented Mueller-Hinton broth (SMHB) containing 10 μg/ml of chloramphenicol was diluted 25-fold into fresh SMHB containing chloramphenicol and tetracycline (10 and 0.05 μg/ml, respectively). Cultures were incubated at 35°C on a rotating drum (80 rpm) until the optical density at 600 nm (OD600) reached 0.7 to 0.8. Cells were pelleted and then resuspended at an OD600 of 0.8 in 0.5-ml aliquots of SMHB containing EB plus carbonyl cyanide m-chlorophenyl hydrazone (final concentrations, 25 and 100 μM, respectively). After gentle agitation for 20 min at room temperature, cells were pelleted and then resuspended in 1 ml of fresh SMHB, and 200-μl aliquots were immediately transferred into wells of opaque 96-well flat-bottom plates (Corning Inc., Corning, NY). The fluorescence of each well was monitored continuously for 5 min using the FLx800 microplate reader and excitation and emission wavelengths of 485 and 645 nm, respectively. Three independent assays were performed, and efflux was quantitated by determination of the percent reduction in fluorescence over the 5-min time course.
Strain typing.
The A103V MepR substitution was observed most frequently (Table 2). spa typing was employed to determine potential relatedness between these strains using previously described procedures (19). Assignment of spa type was made employing a web-based application at http://fortinbras.us/cgi-bin/spaTyper/spaTyper.pl, which utilizes the Ridom numbering scheme as described at http://spa.ridom.de/index.shtml.
Bioinformatics, modeling, and statistics.
CLC Main Workbench 6.8.2 (CLC bio, Cambridge, MA) was used for analyses of DNA sequences. The structures of MepR (Protein Data Bank [PDB] code 3ECO) and reduced and oxidized OhrR (PDB codes 2PEX and 2PFB, respectively) were obtained from the Research Collaboratory for Structural Bioinformatics at http://www.rcsb.org/. UCSF chimera 1.7 was used for molecular modeling, and LSQKAB was used to overlay reduced and oxidized OhrR and MepR using MepR as the reference molecule (20, 21). Quantitation of band shifts in EMSAs was done using Phoretix 1D Pro 11.8 (Nonlinear Dynamics, Newcastle upon Tyne, United Kingdom), and data are presented as percent shift. Comparisons of β-galactosidase and efflux assay data were performed using either the t test or Mann-Whitney rank sum test, as appropriate, using the statistical capabilities embedded within SigmaPlot 12.0.
RESULTS AND DISCUSSION
Mutations and gene expression quantification in mepA-overexpressing strains.
A total of 64 mepA-overexpressing strains were identified in our collection, with mutations in mepR resulting in amino acid substitutions being present in 22 (34.4%) (Table 2). Of these 22 strains, all except one also had increased mepR expression; base pair mismatching between probe and target sequences prevented obtaining mepR data from one strain (SA-K4624; Germany). The coincidence of mepA and mepR overexpression was anticipated, as the expression of both genes is under MepR control. Variability in expression between independent RNA preparations was evident based on large standard deviations, especially evident for Q18P (mepA) and G97E and premature truncation mutants (both mepA and mepR). Despite between-preparation variability, each replicate confirmed concomitant overexpression of mepA and mepR.
Of the remaining 42 mepA-overexpressing strains, 15 (23.4%) were completely WT in the mepRA region. An additional 15 had mutations resulting only in one or more MepA substitutions, and 12 possessed a variety of mutations in either the mepR (7; 10.9%) or mepA (5; 7.8%) operator region signature or nonsignature sequences. The presence of mepA overexpression in the 30 strains without mepRA mutations or only mutations resulting in MepA substitutions is consistent with the existence of as-yet-uncharacterized MepR-independent regulation of mepA expression encoded elsewhere on the chromosome.
The deleterious functional consequences of Q18P, G97E, and A103V at the mepR operator site as well as structurally based predictions as to the mechanism(s) of this have been provided previously, in part, and are expanded upon here for completeness (7, 10, 12). The A103V mutation was most commonly observed, occurring in more than half (12/22) of strains overexpressing mepA coincident with a MepR substitution mutation. This mutation was found only in clinical strains, and these strains were recovered from patients in numerous geographic locales separated by great distances. Nine of the 12 MepR A103V strains were methicillin susceptible, and 11 were spa type t008. The frequency of this spa type is consistent with the possibility of clonal relatedness among strains possessing this MepR mutation. The high frequency of this substitution may be related to the known bias in bacteria for C/G-to-T/A transitions, and the mutation in question is a C→T transition in codon 103 (GCA→GTA) (22). With the exception of G97E (G→A transition; GGA→GAA), all other substitution mutations identified involved lower-frequency transversion mutations and were found in only one strain each.
Premature truncation of the mepR coding region also was common and occurred in one clinical and four laboratory-derived mepA-overexpressing strains. The four laboratory strains have been described previously and had insertions of 1 to 4 bp (two strains) or G→T transversions (two strains) resulting in premature stop codons in the near 5′ region of the gene (12). The clinical strain was recovered from a patient in Detroit, MI, and had a 1-bp deletion in codon 74 producing a premature TGA stop codon. Previous work that we have done using SA-K2068, a strain possessing a truncated version of mepR, revealed that such mutations abrogate repressor function (6). As such, it was assumed that these mutants were inactive, and they were not studied further.
Multiple MepR mutations were present in a single bloodstream isolate from a patient in Germany, including F3Y, K23R, P62A, R69K, L119F, E121K, Q126R, M127I, A129E, and Q139K (SA-K4624) (Tables 1 and 2). Five of these residues were studied individually (K23R, P62A, R69K, L119F, and Q126R) based on their locations at potentially critical sites in the MepR monomer (see below). In apo-MepR, residues K23, L119, and Q126 lie in or near the dimerization interface and P62 and R69 are in the putative major-groove DNA binding domain.
Gene expression analyses. (i) mepR operator.
The repressor activities of the studied MepR derivatives at the mepR operator site, as determined by β-galactosidase assays, are presented in Fig. 2 (black bars), normalized to the activity of MepR WT. In all instances mepR expression peaked by 3 to 4 h and approached zero by ∼8 h. Near-total loss of repressor activity was evident for the Q18P and A103V mutants, whereas other mutants (F27L and G97E) had maintenance of low-level, but still markedly reduced, repressor function at less than 20% of the WT level. Of great interest was the T94P+T132M mutant, found in a clinical strain from San Francisco, CA. T94P in isolation resulted in ∼70% functional impairment, while T132M had no effect. Both mutations together resulted in a significant improvement in repressor activity compared to that with T94P alone, consistent with T132M compensating for the defect conferred by T94P. The possible suppressor property of T132M was studied further by combining it with F27L and A103V (discussed below).
Fig 2.

Repression of chromosomal mepR (black bars) or mepA (gray bars) expression by MepR derivatives. Values are means ± standard deviations. All data were normalized to the activity of wild-type (WT) MepR. Germany, MepR mutant with 10 substitutions; a, significantly reduced repressor activity versus WT MepR at the mepR or mepA operator; b and c, significantly increased repressor activity versus T94P or A103V MepR, respectively, at the mepR and mepA operators (P < 0.05). The expression of all derivatives except those with changes at position 103 was induced with 0.05 μg/ml tetracycline (see the text for details).
The A103F and A103L mutants had significantly impaired function, with that of A103L affected least. However, both had significantly better mepR repressor activity than that of A103V. Position 103 is completely solvent exposed, with valine and leucine both being branched-chain residues and larger than the native alanine (by two and three carbons, respectively). Phenylalanine increases the aromaticity at the position by adding a bulky phenyl group. It is intriguing that the most conservative of the studied substitutions, A103V, had the greatest functional effect. The potential reason for this is discussed below.
The MepR derivative with 10 substitution mutations recovered from a German strain was compromised modestly compared to MepR WT (∼17% reduction); the substitution(s) responsible for this phenotype did not appear to include any of the five residues studied individually (K23R, P62A, R69K, L119F, and Q126R), as each had no significant deleterious effect. It is possible that the combined effects of two or more of the 10 substitutions in this strain are responsible for the modest activity deficit observed and that other combinations of substitutions may be compensatory in nature.
Combining T132M with F27L had no beneficial effect on repressor activity. However, combining T132M with A103V significantly reversed the functional defect incurred by this substitution in isolation at the mepR operator. The data for A103V+T132M illustrated in Fig. 2 were generated using 0.0125 μg/ml tetracycline induction. However, the growth defect previously observed for A103 mutants with 0.05 μg/ml tetracycline induction did not occur in the presence of T132M, and repressor function under these conditions was 80% of the WT level (data not shown). Recovery of repressor activity at the mepR operator in the presence of this substitution in one modestly (T94P) and one severely (A103V) impaired protein indicates that it has suppressor activity for only selected deleterious MepR substitutions. Knowledge of the mechanism(s) by which this occurs awaits structural determinations of F27L, T94P, and A103V alone and in combination with T132M. It is anticipated that T132M relieves the structural defect(s) imposed on MepR by T94P and A103V but is unable to do so in the presence of F27L.
(ii) mepA operator.
Repressor activities of MepR mutants at the mepA operator site are also presented in Fig. 2 (gray bars). Similar to the case for mepR, in all instances mepA expression peaked by 3 to 4 h and approached zero by ∼8 h. Noteworthy was the fact that many mutant proteins demonstrated much better repressor function at the mepA operator than at the mepR operator (see data for T94P, T94P+T132M, A103F, A103L, and A103V+T132M). Even substitutions causing a profound loss of repressor function at the mepR operator (Q18P and A103V) had some activity at the mepA operator. This phenomenon may be related to the known higher affinity of MepR for the mepA operator site, a property that may extend to these mutants.
A marked improvement in mepA repressor function was observed for A103V+T132M versus A103V alone. With respect to T94P, the improvement observed in the presence of T132M was minor but still evident. These data support our observations at the mepR operator that T132M has suppressor activity. The limitations of this suppressor function were again demonstrated by the lack of an effect when combining it with F27L.
While statistically significant as evidenced by small standard deviations, the functional significance of minor reductions in repressor function at the mepR operator compared to WT, such as that observed for MepR K4624 (Germany), and at the mepA operator for T94P, A103L, and A103V+T132M is uncertain. The threshold in vivo concentration for effective repression by MepR at either operator is not known, and these minor reductions may or may not translate into physiologic relevance. However, these gene expression data do reveal the involvement of individual residues, however minor and regardless of whether the effect is direct or indirect, in maintenance of proper MepR function.
Figure 3 illustrates the effect of MepR derivatives on mepA expression, represented by TPP MIC ratios and EB efflux. In the absence of tetracycline, TPP MICs for all test strains were 31.3 to 62.5 μg/ml. No change in MIC in the presence of tetracycline induction, resulting in a TPP MIC ratio of 1, was observed in the absence of MepR (empty pALC2073) or with functionally compromised derivatives (Q18P, F27L, F27L+T132M, T94P, G97E, and A103V). Ratios of less than 1 indicated preserved MepR activity. This was most evident for the WT and P62A, Q126R, and K4624 derivatives, for which this ratio was 0.125 to 0.25, indicative of 4- to 8-fold MIC reductions related to good repression of mepA transcription. T94P+T132M consistently outperformed T94P in the MIC assay, and A103F, A103L, and A103V+T132M did likewise compared to A103V. A modest degree of in vivo functional compromise for all these derivatives was evident based on a lesser reduction in the TPP MIC ratio than was observed for MepR WT, but in general, TPP MIC ratios provided support for the β-galactosidase data.
Fig 3.

Tetraphenylphosphonium bromide (TPP) MIC ratios (gray bars) and ethidium bromide (EB) efflux activity (black circles) of selected MepR derivatives. MIC ratios of less than 1 indicate preserved MepR repressor activity, with 0.5, 0.25, and 0.125 equal to 2-, 4-, and 8-fold MIC reductions, respectively. Numbers within bars represent percent MepR repressor activity (compared to WT) at the mepA operator as determined in β-galactosidase assays. a, efflux activity equivalent to that of the WT; b, efflux activity equivalent to that of empty pALC2073; c, reduced efflux activity compared to that of T94P or A103V, indicative of greater MepR repression of mepA transcription in the presence of T132M (P < 0.01).
High EB efflux was consistently observed in the absence of MepR or in the presence of low-functioning derivatives and reduced efflux, indicative of inhibition of mepA expression, in the presence of derivatives with no or only modest functional compromise. Statistical analyses revealed no difference in efflux between SA-K3731 with empty pALC2073 and that expressing MepR F27L or F27L+T132M. MepR T94P and A103V both appeared to be modestly better than empty pALC2073 with respect to preserved MepR function (P = 0.004 and P = 0.02, respectively). However, both derivatives were poorly functional compared to MepR WT (P < 0.001). Additionally, the slight activity of A103V in the EB efflux assay was consistent with EMSA data showing this derivative to bind, albeit poorly, to the mepA operator (see below). MepR activity was significantly better in strains expressing T94P or A103V in combination with T132M than with each of these substitutions alone (P = 0.008 and P < 0.001, respectively), again consistent with this substitution having suppressor function. EB efflux data were consistent with TPP MIC data and also generally supportive of the β-galactosidase observations.
The variability in the magnitude of the MepR effect between β-galactosidase, MIC, and EB efflux assays is almost certainly the result of profound differences in assay conditions. In only one instance were the data truly incongruous, with MIC and EB efflux data suggesting less preservation of MepR repressor activity than that observed in the β-galactosidase assay (MepR T94P). The reason(s) for this is uncertain; other than this one instance, the three assays gave generally comparable results.
Affinity of selected MepR derivatives for the mepR and mepA operators.
Data already presented have established that the A103V substitution severely impairs or abolishes repressor activity at both the mepR and mepA operators despite this seemingly conservative substitution. EMSAs corroborated β-galactosidase, MIC, and EB efflux data by showing that binding to both operators by MepR A103V occurred but was impaired compared to that with WT protein (Fig. 4, top). A clear demarcation in the percentage of shift at the mepR and mepA operators was evident at 100 and 50 ng input protein, respectively.
Fig 4.

Electrophoretic mobility shift assays. Binding of MepR wild type (WT) versus that of MepR A103V or MepR F27L mutant protein to the WT mepR and mepA operators is shown. The amount of input protein used and percent shift of target are indicated. Binding specificity of each protein was established by including a 200-fold excess of unlabeled specific DNA (indicated by + in the final two lanes of each image).
EMSA results for F27L also were consistent with β-galactosidase, MIC, and efflux data. This derivative completely failed to interact with its cognate mepR and mepA operator sites, indicating total functional compromise (Fig. 4, bottom). MepR P62A, which was evaluated for reasons discussed below, was examined by EMSA (data not shown). This derivative had activity similar to that of MepR WT at the mepR operator. At 100, 50, and 25 ng input protein, shift percentages for WT and P62A were 81 and 67, 67 and 60, and 50 and 47%, respectively. At the mepA operator, the corresponding data were 100 and 100, 100 and 96, and 96 and 60%. The apparent defect observed at 50 ng input protein for P62A at the WT mepA operator was not observed when the cognate (K4624) mepA operator was used, where shift efficiency at 50 ng input protein was 99 and 97% for WT and P62A, respectively. The apparent modest functional defect seen at a small amount of input protein and only at the WT mepA operator was not physiologically relevant based on fully maintained repressor function in β-galactosidase, MIC, and efflux assays, which employed strains having a WT mepA operator. As discussed above, the low protein-to-target ratio in EMSAs, which for the mepA operator at 50 ng input protein was 0.4, is more likely to identify subtle functional defects that may or may not have physiologic relevance. Of significant interest was the fact that this defect was not seen with the K4624 mepA operator, suggesting that the mutations present in that region may functionally synergize with P62A to counteract a small functional defect.
Affinity for WT and cognate mepA operators and substrate responsiveness of MepR K4375 and K4624.
MepR WT shifted the WT mepA operator and that of strain K4375 with equal efficiency at all amounts of input protein tested (Fig. 5, top). Thus, the operator site mutations of K4375 do not affect binding by the native protein. However, at below 200 ng input protein, binding by MepR K4375, possessing T94P+T132M substitutions, was defective compared to that by the WT. Shift efficiency at 100, 50, and 25 ng input protein for the WT versus K4375 mepA operators was 53 and 45%, 37 and 14%, and 6 and 0%, respectively. These differences were consistent in repeat analyses. The inefficiency of binding to the mepA operator for MepR K4375 is consistent with mepA overexpression observed in the strain of origin, and worsening of this defect with the cognate operator indicates that in this case MepR substitutions combined with operator mutations can be negatively synergistic. The EMSA data for this derivative at the WT operator are not congruent with β-galactosidase assay data, in which 98% maintenance of repressor function at the mepA operator was observed (Fig. 2). However, they are consistent with MIC and efflux data that revealed MepR K4375 to be less active than MepR WT (Fig. 3). As already mentioned, profound differences in assay conditions, including protein-to-target ratios used in EMSAs, may contribute to this apparent inconsistency.
Fig 5.

Electrophoretic mobility shift assays. Binding of MepR wild type (WT) versus that of MepR K4375 (T94P+T132M) or MepR K4624 (Germany; 10 substitutions) to WT and cognate mepA DNA targets. The amount of input protein used and percent shift of target are indicated.
With respect to K4624, the MepR derivative from this strain shifted both the WT and its cognate mepA operator equally (Fig. 5, bottom). The lack of a binding defect for this derivative is consistent with all other functional data showing good maintenance of repressor activity. Of significant interest was the fact that MepR K4624 protein appeared to be more efficient than MepR WT protein at both WT and mutant operators, especially at below 50 ng input protein. The correlation of this in vitro observation to the in vivo situation is uncertain, and for reasons already discussed may not have physiologic relevance. These data also do not explain the mechanism(s) of mepA overexpression in strain K4624 (6-fold) (Table 2), as these EMSA data indicate that the operator site mutations of K4624 play no role. Increased mepA expression in this strain may be the result of MepR-independent regulatory processes for mepA expression. As mentioned above, the existence of such processes is supported by the fact that 30 of the 64 mepA-overexpressing strains in our collection had either WT mepRA sequences or only mutations in the mepA coding region resulting in amino acid substitutions. Additional work will be required to identify these processes.
Substrate responsiveness of MepR K4375 and K4624 is illustrated in Fig. 6. In the absence of a MepA substrate, all tested proteins shifted both operators completely (data not shown). Addition of cetrimide (50 μM) dissociated the majority of MepR K4375 from both operators, whereas MepR WT was unaffected. The T94P+T132M substitutions augment substrate interaction with the protein, resulting in derepression of mepA at lower ambient substrate concentrations. Toxic effects of substrates are reduced when mepA expression is increased at lower substrate concentrations, providing a selective advantage. A structural correlation to this biologic effect would be of great interest.
Fig 6.

Substrate responsiveness of MepR derivatives. Cetrimide (50 μM) was included in the mixtures for all binding reactions shown here. Experiments with K4375 MepR employed 200 ng of input protein, whereas 25 ng was used for K4624.
In contrast to MepR K4375, MepR K4624 was less responsive to substrate than MepR WT at both tested operators. The combination of improved binding efficiency and reduced substrate responsiveness would be a distinct disadvantage when organisms possessing this MepR derivative are challenged with MepA substrates. Structural analysis of this derivative compared to the WT will help to refine the cetrimide binding site precisely.
Structural consequences of MepR substitution mutations relating to function.
To understand the mechanism(s) by which certain substitution mutations affect MepR repressor function whereas others do not, we examined each in the context of the apo-MepR structure (Fig. 7) (7). Mutations that had no significant functional consequence at either operator included K23R, P62A, R69K, L119F, and Q126R. These residues were of interest based on their locations and possible involvement in MepR function by way of monomer association or DNA target site binding. With the exception of P62A, these residues will not be considered further based on their lack of effect. The homologous proline in the MepR homologue SlyA of Salmonella enterica (P61) does have functional significance in that the structural analysis of cocrystallized SlyA and cognate DNA revealed that P61 makes van der Waals contacts with nucleobases in the major groove (23, 24). Either MepR P62 does not play a similar role, or an alanine can substitute completely for any P62-base contact.
Fig 7.

MepR substitution mutations. Blue and magenta, monomers A and B of the MepR functional dimer, respectively. Helix numbers are indicated for monomer A (α1, α2, etc.), and positions of studied mutations are depicted as spheres (wild-type side chains are shown). Green, no detrimental effect at either the mepR or mepA operator; yellow, function mildly to moderately impaired; red, function markedly impaired or inactivating. (A and B) Apo-MepR face-on and rotated 180o about its central axis, respectively. The dimerization interface is indicated, as is the DNA binding domain of one monomer.
T132M also did not have any negative functional consequence, but it was evaluated to determine its contribution to the functional defect observed for MepR K4375 (T94P+T132M). The side chain of threonine at its helix 6 position is solvent exposed and not expected to have any interaction with the second MepR protomer. Methionine, a “helix-loving” residue, should be accommodated well at position 132. As such, we were not surprised that T132M had no detrimental effect. The interesting suppressor properties of this substitution, as exemplified by its restoration of activity in both T94P and A103V mutants, have already been noted.
Substitution mutations having negative functional consequences as demonstrated by β-galactosidase, MIC, and efflux assays, and for some also by EMSAs, included Q18P, F27L, T94P, T94P+T132M, G97E, and A103V. Three of these (Q18P, G97E, and A103V) have been described previously and will be briefly addressed here (7). The side chain of residue Q18 is surface exposed and centrally located on helix 1 (Fig. 7). As such, its replacement by proline would not result in steric clash but almost certainly would distort the helix and interfere with optimal dimerization of MepR (25). Such distortion is also likely to shift positioning of DNA binding motifs reducing operator site interactions.
The structural consequences of A103V are more challenging to explain. We have demonstrated that it has poor activity at both the mepR and mepA operators (Fig. 2 and 3). It also maintains some ability to bind these target sequences, but with reduced efficiency compared to the WT (Fig. 4). In an attempt to provide a structural explanation for the observed A103V phenotype, we carried out comparative analyses of MepR with the structures of induced (oxidized) and noninduced (reduced; DNA binding conformation) forms of the organic hydroperoxide resistance regulator OhrR of Xanthomonas campestris, a structural homologue of MepR (Fig. 8) (7, 26, 27). Induction of X. campestris OhrR results in a major conformational change that includes a disruption of helix 5 into two distinct helices (5A and 5B), which reorients the DNA binding domains such that they become incompatible with an interaction with cognate DNA (Fig. 8B). Given the structural homology between OhrR and MepR, it is possible that a similar helix-breaking mechanism could be induced upon drug binding by MepR. The A103V residue of MepR is located in the middle of helix 5, a position analogous to the broken midpoint of helix 5 in induced OhrR (Fig. 7 and 8). In this regard, it is worth mentioning that alanine is a helix-stabilizing residue, whereas valine is not (28–30). An X. campestris OhrR-like local deformation of MepR helix 5 may result from the substitution of valine for A103 and thus promote the induced conformation of MepR, resulting in attenuated DNA binding. The fact that the A103L and A103F substitutions, both of which provide bulkier side chains, had reduced negative consequences compared to A103V is intriguing and suggests some compensatory structural consequence that maintains the helical nature of this region. Indeed, a leucine prefers the helical conformation. However, the effect of the phenylalanine substitution remains unclear. Structural determinations of these derivatives would be most informative and are important, especially since the A103V change is the most common MepR substitution mutation found in clinical strains. Structural studies of the A103V+T132M double mutant also will reveal how T132M suppresses the negative effects of A103V.
Fig 8.

(A) Structural comparison of reduced or DNA binding-compatible Xanthomonas campestris OhrR (blue) and MepR (red). (B) Conformational differences in helix 5 between reduced (OhrR[r]; blue) and oxidized (OhrR[ox]; green) OhrR, resulting in disruption of the helix into two distinct helices (5A and 5B) and loss of DNA binding affinity. Position A103 in MepR (red) is represented by a yellow sphere. (C) Introduction of potential steric clash when proline is substituted for threonine at MepR position 94. Distances are in angstrom units. The closest approaches for both residues are illustrated; all conformers/ring puckers of the proline side chain approach the peptide backbone at isoleucine 77 more closely than any side chain rotamer of threonine.
The loss of function of the G97E substitution is easier to understand. This substitution places a bulky and negatively charged side chain into a hydrophobic environment consisting of helices 2 and 5. The likely disruption of tertiary structure caused by this substitution explains its loss of repressor function. Further support for the extreme structural defect conferred by this substitution is provided by earlier work showing a profound loss in solubility of the G97E mutant compared to MepR WT (7).
Novel MepR substitutions resulting in loss of repressor function identified since our earlier report include F27L and T94P+T132M. The side chain of F27 is located at the C-terminal end of helix 1, and the substitution of leucine here essentially abrogates DNA binding and repression by MepR (Fig. 2 to 4). Given its location away from the DNA binding domain, the defective phenotype exhibited by F27L is intriguing. However, examination of how OhrR of Bacillus subtilis, a homologue of X. campestris OhrR, interacts with cognate DNA provides a probable structural explanation for the phenotype of this derivative (31). B. subtilis OhrR employs three DNA-binding elements to interact with its cognate DNA sequences, which are also possessed by X. campestris OhrR and MepR. These include the recognition helix of the wHTH motif that interacts with the major groove, the wing region that interacts with the minor groove, and a newly recognized helix-helix motif involving the C-terminal end of helix 1, the N-terminal end of helix 2, and the intervening loop that contacts the phosphate backbone. Interestingly, F27 is near the terminus of helix 1 and thus a component of the helix-helix motif in MepR (Fig. 7). Thus, it is possible that the F27L substitution may cause a structural perturbation in its vicinity or interfere with direct interactions with DNA. Either possibility would adversely affect DNA binding by MepR. Structural studies will be necessary to understand how F27L disrupts function and why combining it with T132M has no beneficial effect.
The evaluation of T94P and T132M individually identified T94P as being responsible for the functional deficiency. T94 is the first residue of helix 5, and as mentioned above, proline in such a position is generally well tolerated, explaining the modest preservation of repressor function by this mutant (25). The side chain atoms of P94, regardless of ring pucker, approach the peptide backbone of the loop containing isoleucine at position 77, a region immediately following the α4 DNA binding helix, more closely than do any rotamers of the WT threonine side chain (proline, 2.7 to 2.9 and 3.1 to 3.7 Å; threonine, 3.7 to 3.9 and 4.5 to 4.7 Å) (Fig. 8C). This close approach would produce steric clash and result in the relocation of the loop and the adjacent helix, thereby reducing binding affinity. The ability of T132M to reverse this functional defect, as it also was able to do for A103V, provides strong evidence for its suppressor role. However, its structural mechanism of suppression remains unknown and awaits additional structural studies.
Concluding remarks.
Many substitution mutations in MepR are well tolerated, whereas others severely interfere with repressor activity. The most common change in the latter group is A103V, which is found only in clinical strains and present in the majority of such strains that we studied (12 of 16). MepR A103V binds its cognate operator sequences poorly, resulting in poor repression of both mepR and mepA expression. Our data suggested clonal relatedness among MepR A103V strains by the predominance among them of spa type t008.
The combination of operator site mutations with amino acid substitutions in MepR may synergistically compromise repressor activity, as was the case for MepR K4375 and the cognate mepA operator mutations found in the strain of origin. The opposite also appears to be true, exemplified by the improved interaction of MepR K4624 with the WT as well as its cognate operator. Combinations of substitutions also may compensate for functional defects caused by others, as clearly demonstrated by the beneficial effect of T132M on two defective proteins. The ability of T132M to function as a suppressor of the negative consequences of T94P and A103V, but not F27L, indicates its ability to reverse some but not all structural perturbations imposed by functionally compromising mutations. This is not surprising in that the structural alterations imposed by different substitutions are likely to vary from mild to severe. Those imposed by T94P and A103V may be comparable enough to allow a similar beneficial outcome by combining them with T132M, whereas that of F27L does not. A random mutagenesis approach may identify additional suppressor/compensatory mutations using functionally compromised MepR derivatives identified clinically or produced in the laboratory.
The structure-based hypotheses advanced to explain the functional consequences we observed were formulated using the known apo-MepR structure, in which the molecule was captured in an extended form with the DNA binding motifs separated too broadly to interact with DNA (7). MepR must have significant structural plasticity, allowing a shift in conformation in the presence of DNA. The ability of MepR to interact with MepA substrates, resulting in dissociation from its operator sites, is another example of structural plasticity (8). In addition to the predicted deleterious structural effects of the functionally significant substitutions we identified, these residue changes also could have detrimental effects on plasticity. Structural determinations of mutant proteins are important to identify the mechanisms of compromised function more precisely.
ACKNOWLEDGMENT
This work was supported by VA Biomedical Laboratory Research & Development grant 1IO1BX000465.
REFERENCES
- 1.Li XZ, Nikaido H. 2009. Efflux-mediated drug resistance in bacteria: an update. Drugs 69:1555–1623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lomovskaya O, Lee A, Hoshino K, Ishida H, Mistry A, Warren MS, Boyer E, Chamberland S, Lee VJ. 1999. Use of a genetic approach to evaluate the consequences of inhibition of efflux pumps in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 43:1340–1346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Markham PN, Neyfakh AA. 1996. Inhibition of the multidrug transporter NorA prevents emergence of norfloxacin resistance in Staphylococcus aureus. Antimicrob. Agents Chemother. 40:2673–2674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Borges-Walmsley MI, McKeegan KS, Walmsley AR. 2003. Structure and function of efflux pumps that confer resistance to drugs. Biochem. J. 376:313–338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Morita Y, Kataoka A, Shiota S, Mizushima T, Tsuchiya T. 2000. NorM of Vibrio parahaemolyticus is an Na(+)-driven multidrug efflux pump. J. Bacteriol. 182:6694–6697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kaatz GW, McAleese F, Seo SM. 2005. Multidrug resistance in Staphylococcus aureus due to overexpression of a novel multidrug and toxin extrusion (MATE) transport protein. Antimicrob. Agents Chemother. 49:1857–1864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kumaraswami M, Schuman JT, Seo SM, Kaatz GW, Brennan RG. 2009. Structural and biochemical characterization of MepR, a multidrug binding transcription regulator of the Staphylococcus aureus multidrug efflux pump MepA. Nucleic Acids Res. 37:1211–1224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kaatz GW, DeMarco CE, Seo SM. 2006. MepR, a repressor of the Staphylococcus aureus MATE family multidrug efflux pump MepA, is a substrate-responsive regulatory protein. Antimicrob. Agents Chemother. 50:1276–1281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schumacher MA, Miller MC, Grkovic S, Brown MH, Skurray RA, Brennan RG. 2002. Structural basis for cooperative DNA binding by two dimers of the multidrug-binding protein QacR. EMBO J. 21:1210–1218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.DeMarco CE, Cushing LA, Frempong-Manso E, Seo SM, Jaravaza TA, Kaatz GW. 2007. Efflux-related resistance to norfloxacin, dyes, and biocides in bloodstream isolates of Staphylococcus aureus. Antimicrob. Agents Chemother. 51:3235–3239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Frempong-Manso E, Raygada JL, DeMarco CE, Seo SM, Kaatz GW. 2009. Inability of a reserpine-based screen to identify strains overexpressing efflux pump genes in clinical isolates of Staphylococcus aureus. Int. J. Antimicrob. Agents 33:360–363 [DOI] [PubMed] [Google Scholar]
- 12.Huet AA, Raygada JL, Mendiratta K, Seo SM, Kaatz GW. 2008. Multidrug efflux pump overexpression in Staphylococcus aureus after single and multiple in vitro exposures to biocides and dyes. Microbiology 154:3144–3153 [DOI] [PubMed] [Google Scholar]
- 13.Kosmidis C, DeMarco CE, Frempong-Manso E, Seo SM, Kaatz GW. 2010. In silico genetic correlations of multidrug efflux pump gene expression in Staphylococcus aureus. Int. J. Antimicrob. Agents 36:222–229 [DOI] [PubMed] [Google Scholar]
- 14.Kosmidis C, Schindler BD, Jacinto PL, Patel D, Bains K, Seo SM, Kaatz GW. 2012. Expression of multidrug resistance efflux pump genes in clinical and environmental isolates of Staphylococcus aureus. Int. J. Antimicrob. Agents 40:204–209 [DOI] [PubMed] [Google Scholar]
- 15.Sanger F, Nicklen S, Coulson AR. 1977. DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. U. S. A. 74:5463–5467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Foster TJ. 1998. Molecular genetic analysis of staphylococcal virulence. Methods Microbiol. 27:433–454 [Google Scholar]
- 17.Schindler BD, Patel D, Seo SM, Kaatz GW. 2013. Mutagenesis and modeling to predict structural and functional characteristics of the Staphylococcus aureus MepA multidrug efflux pump. J. Bacteriol. 195:523–533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.CLSI 2006. Approved standard M7–A7. Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically, 7th ed Clinical and Laboratory Standards Institute, Wayne, PA [Google Scholar]
- 19.Strommenger B, Kettlitz C, Weniger T, Harmsen D, Friedrich AW, Witte W. 2006. Assignment of Staphylococcus isolates to groups by spa typing, SmaI macrorestriction analysis, and multilocus sequence typing. J. Clin. Microbiol. 44:2533–2540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kabsch W. 1976. A solution for the best rotation to relate two sets of vectors. Acta Cryatallogr. 32A:922–923 [Google Scholar]
- 21.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. 2004. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25:1605–1612 [DOI] [PubMed] [Google Scholar]
- 22.Hershberg R, Petrov DA. 2010. Evidence that mutation is universally biased towards AT in bacteria. PLoS Genet. 6:e1001115. 10.1371/journal.pgen.1001115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ellison DW, Miller VL. 2006. Regulation of virulence by members of the MarR/SlyA family. Curr. Opin. Microbiol. 9:153–159 [DOI] [PubMed] [Google Scholar]
- 24.Dolan KT, Duguid EM, He C. 2011. Crystal structures of SlyA protein, a master virulence regulator of Salmonella, in free and DNA-bound states. J. Biol. Chem. 286:22178–22185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim MK, Kang YK. 1999. Positional preference of proline in alpha-helices. Protein Sci. 8:1492–1499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Newberry KJ, Fuangthong M, Panmanee W, Mongkolsuk S, Brennan RG. 2007. Structural mechanism of organic hydroperoxide induction of the transcription regulator OhrR. Mol. Cell 28:652–664 [DOI] [PubMed] [Google Scholar]
- 27.Sukchawalit R, Loprasert S, Atichartpongkul S, Mongkolsuk S. 2001. Complex regulation of the organic hydroperoxide resistance gene (ohr) from Xanthomonas involves OhrR, a novel organic peroxide-inducible negative regulator, and posttranscriptional modifications. J. Bacteriol. 183:4405–4412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Raman SS, Vijayaraj R, Parthasarathi R, Subramanian V. 2008. Helix forming tendency of valine substituted poly-alanine: a molecular dynamics investigation. J. Phys. Chem. B 112:9100–9104 [DOI] [PubMed] [Google Scholar]
- 29.Rohl CA, Fiori W, Baldwin RL. 1999. Alanine is helix-stabilizing in both template-nucleated and standard peptide helices. Proc. Natl. Acad. Sci. U. S. A. 96:3682–3687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gregoret LM, Sauer RT. 1998. Tolerance of a protein helix to multiple alanine and valine substitutions. Fold Des. 3:119–126 [DOI] [PubMed] [Google Scholar]
- 31.Hong M, Fuangthong M, Helmann JD, Brennan RG. 2005. Structure of an OhrR-ohrA operator complex reveals the DNA binding mechanism of the MarR family. Mol. Cell 20:131–141 [DOI] [PubMed] [Google Scholar]
- 32.Novick R. 1967. Properties of a cryptic high-frequency transducing phage in Staphylococcus aureus. Virology 33:155–166 [DOI] [PubMed] [Google Scholar]
- 33.Kreiswirth BN, Lofdahl S, Betley MJ, O'Reilly M, Schlievert PM, Bergdoll MS, Novick RP. 1983. The toxic shock syndrome exotoxin structural gene is not detectably transmitted by a prophage. Nature 305:709–712 [DOI] [PubMed] [Google Scholar]
- 34.Horsburgh MJ, Aish JL, White IJ, Shaw L, Lithgow JK, Foster SJ. 2002. σB modulates virulence determinant expression and stress resistance: characterization of a functional rsbU strain derived from Staphylococcus aureus 8325-4. J. Bacteriol. 184:5457–5467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kaatz GW, Moudgal VV, Seo SM. 2002. Identification and characterization of a novel efflux-related multidrug resistance phenotype in Staphylococcus aureus. J. Antimicrob. Chemother. 50:833–838 [DOI] [PubMed] [Google Scholar]
- 36.Bateman BT, Donegan NP, Jarry TM, Palma M, Cheung AL. 2001. Evaluation of a tetracycline-inducible promoter in Staphylococcus aureus in vitro and in vivo and its application in demonstrating the role of sigB in microcolony formation. Infect. Immun. 69:7851–7857 [DOI] [PMC free article] [PubMed] [Google Scholar]