

Abstract
Several two-component regulatory systems are known to be involved in the signal transduction pathway of the ethanol oxidation system in Pseudomonas aeruginosa ATCC 17933. These sensor kinases and response regulators are organized in a hierarchical manner. In addition, a cytoplasmic putative iron-containing alcohol dehydrogenase (Fe-ADH) encoded by ercA (PA1991) has been identified to play an essential role in this regulatory network. The gene ercA (PA1991) is located next to ercS, which encodes a sensor kinase. Inactivation of ercA (PA1991) by insertion of a kanamycin resistance cassette created mutant NH1. NH1 showed poor growth on various alcohols. On ethanol, NH1 grew only with an extremely extended lag phase. During the induction period on ethanol, transcription of structural genes exa and pqqABCDEH, encoding components of initial ethanol oxidation in P. aeruginosa, was drastically reduced in NH1, which indicates the regulatory function of ercA (PA1991). However, transcription in the extremely delayed logarithmic growth phase was comparable to that in the wild type. To date, the involvement of an Fe-ADH in signal transduction processes has not been reported.
INTRODUCTION
Upon aerobic growth on ethanol, Pseudomonas aeruginosa ATCC 17933 expresses a periplasmic, soluble quinoprotein, ethanol dehydrogenase (QEDH), with pyrroloquinoline quinone (PQQ) as a cofactor (1). QEDH transfers electrons to soluble cytochrome c550 (2). After phenotypic characterization and complementation of chemical mutants that were unable to grow on ethanol, it was concluded that six or seven different genes might be involved in regulating ethanol oxidation (3, 4). These genes controlling the ethanol oxidation system in Pseudomonas aeruginosa are organized in a hierarchical manner (see Fig. 2, below). The response regulator ErbR controls transcription of structural genes encoding components necessary for ethanol oxidation (5). exaB codes for cytochrome c550, exaC codes for an NAD+-dependent acetaldehyde dehydrogenase (6), the operon pqqABCDEH encodes enzymes for PQQ biosynthesis (7), and eraSR code for a two-component regulatory system. EraSR activate transcription of exaA encoding quinoprotein ethanol dehydrogenase (QEDH) (3). In addition, two sensor kinases, ErcS (encoded by PA1992) and ErcS′ (encoded by PA1976) and the global response regulator ErdR (encoded by PA3604) have been identified as regulatory components of the ethanol oxidation system in P. aeruginosa (8).
Fig 2.
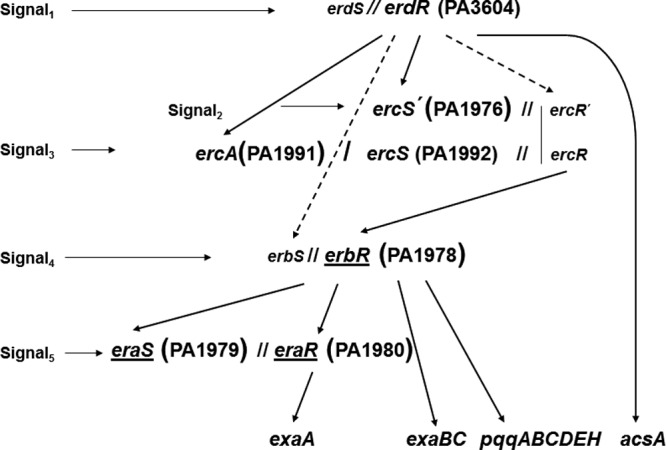
Hypothetical model of the complex regulatory network of the quinoprotein ethanol oxidation system in P. aeruginosa. In the present study, gene ercA (PA1991), which encodes a Fe-ADH, was identified to be essential for expression of the ethanol oxidation system. The hierarchical arrangement of sensor kinases and response regulators control the expression of QEDH, cytochrome c550, and PQQ biosynthetic enzymes (8). Solid arrows indicate demonstrated positive control of transcription, and broken arrows indicate assumed positive control of transcription. The identified genes encoding sensor kinases S and response regulators R are shown in a larger font size with the respective gene numbers. The proposed corresponding two-component system genes not yet identified are shown in a smaller font size. Two slashes indicate response regulator-sensor kinase pairing. This figure is a modified version of Fig. 3 from our previous publication (8).
In the present study, we tried to elucidate the function of ercA (PA1991), which is located adjacent to the sensor kinase ErcS (PA1992). PA1991 codes for a putative iron-containing NAD-dependent alcohol dehydrogenase (Fe-ADH). PA1146 and PA5186 are also annotated to code for putative Fe-ADHs in the Pseudomonas Genome Database (9, 10). Fe-ADHs form family III of the NAD(P)-dependent alcohol dehydrogenases (11). The three putative members of the Fe-ADH family found in the P. aeruginosa genome (encoded by PA1146, PA1991, and PA5186) show high sequence similarities to the 1,3-propanediol dehydrogenase of Klebsiella pneumoniae (12). While the majority of characterized Fe-ADH enzymes are involved in fermentation processes, a minority have been identified to play a major oxidative role: methanol dehydrogenase of Bacillus methanolicus and methanol:N,N-dimethyl-4-nitrosoaniline oxidoreductase from Amycolatopsis methanolica and Mycobacterium gastri, which contain a tightly bound NAD(P) cofactor (13), are active in oxidative pathways.
In this study, the putative Fe-ADH, which is encoded by PA1991, was identified as a regulatory component of the ethanol oxidation system in P. aeruginosa ATCC 17933. So far, the involvement of an Fe-ADH in signal transduction processes has not been reported. Thus, this study may contribute to further elucidate the complex regulation of the ethanol oxidation system in P. aeruginosa.
MATERIALS AND METHODS
Strains and culture conditions.
The strains and plasmids used in this work are listed in Table 1.
Table 1.
Strains and plasmids used in this study
| Strain or plasmid | Relevant genotype or phenotype | Reference |
|---|---|---|
| P. aeruginosa strains | ||
| ATCC 17933 | Wild type | 29 |
| MD1 | ATCC 17933 derivative, ΔPA3604::Kmr | 8 |
| NH1 | ATCC 17933 derivative, PA1991::Kmr | This study |
| NH2 | SH1 derivative, PA1991::Kmr ΔPA1992::Gmr | This study |
| NH3 | ATCC 17933 derivative, ΔPA2572::Kmr | This study |
| NH5 | ATCC 17933 derivative, PA5186::Kmr | This study |
| SH1 | ATCC 17933 derivative, ΔPA1992::Gmr | 8 |
| E. coli strains | ||
| DH5α | supE44 ΔlacU169(ϕ80lacZΔM15) hsdR17 recA1 endA1 gyrA96 thi-1 relA1 | 30 |
| HB101 | supE44 hsdS20(rB− mB−) recA13 ara-14 proA2 lacY1 galK2 rpsL20 xyl-5 mtl-1 | 31 |
| Plasmids | ||
| pACΩGm | Gmr; cloning vector containing Gmr cassette and Ω fragments | 32 |
| pEDY305 | Tcr; lacZ promoter-probe vector | 22 |
| pEX18Ap | Apr; oriT+ sacB+, gene replacement vector with multiple cloning site (MCS) from pUC18 | 20 |
| pLAFR3 | Tcr; broad-host-range cosmid | 33 |
| pQF50 | Apr; lacZ promoter-probe vector | 21 |
| pRK2013 | Kmr; helper plasmid for triparental mating | 34 |
| pUC18, 19 | Apr; cloning and expression vector | 35 |
| pUCP20T | Apr; broad-host-range vector | 36 |
| pTB3001 | Tcr; ∼25-kbp genomic DNA partially digested with Sau3AI from P. aeruginosa cloned into BamHI site of pLAFR3 | 6 |
| pTB3070 | Apr; 3.2-kbp BamHI-PstI fragment from pTB3001 cloned into BamHI-PstI sites of pUC18 | 6 |
| pTB3112 | Apr; 6.2-kbp PstI-PstI fragment from pTB3001 cloned into PstI site of pUCP20T | 3 |
| pTB3131 | Apr Kmr; 1.0-kbp PCR product with promoter and Kmr cassette of Tn5 cloned into EcoRI-BamHI sites of pUC19 | 16 |
| pTB3135 | Apr; 0.89-kbp HindIII-SalI-fragment from pTB3070 cloned into HindIII-SalI sites of pUC19 | 3 |
| pTB3138 | Tcr; P_exaA′-lacZ; 0.89-kbp PstI-SalI fragment from pTB3070 cloned into PstI-XhoI sites of pEDY305 | 3 |
| pTB3139 | Tcr; P_exaB′-lacZ; 0.89-kbp PstI-XbaI fragment from pTB3135 cloned into PstI-XbaI sites of pEDY305 | 3 |
| pTB3144 | Apr; 3-kbp MluI-SmaI fragment of pTB3112 removed, ends filled in, and vector religated | 3 |
| pTB7023 | Tcr; P_pqqAB́-lacZ; 2.5-kbp XhoI-BamHI fragment of pTB3070 cloned into XhoI-BglII sites of pEDY305 | 5 |
| pTB7060 | Apr; 1.67-kbp PCR product of erbR cloned into PstI-BamHI sites of pUCP20T | 5 |
| pTB7074 | Apr; P_eraSR(PA1979/1980)′-lacZ; 1.16-kbp promoter region of exaDE cloned into BglII-XbaI sites of pQF50 | 5 |
| pTB7082 | Apr; P_ercA(PA1991)′-lacZ; 0.53-kbp PCR product containing promoter region of PA1991 cloned into XbaI-HindIII sites of pQF50(pPA1991) | 8 |
| pTB7101 | Apr; 1.98-kbp PCR product of PA1992 cloned into SphRI-EcoRI sites of pUCP20T | 8 |
| pTB7107 | Apr; P_erdR(PA3604)′-lacZ; 280-bp promoter region of PA3604 cloned into XbaI-HindIII sites of pQF50 | 8 |
| pTB7108 | Apr; P_erbR(PA3604)′-lacZ; 1.0-kbp promoter region of agmR cloned into KpnI-HindIII sites of pQF50 | 8 |
| pTB7200 | Apr; 3.3-kbp PCR product containing ercA-ercS and its promoter cloned into EcoRI-HindIIII sites of pUCP20T | This study |
| pTB7212 | Apr; 1.2-kbp PCR product of PA1991 cloned into EcoRI-HindIII sites of pUCP20T | This study |
| pTB7219 | Apr; 1.2-kbp PCR product of PA1146 cloned into EcoRI-HindIII sites of pEX18Ap | This study |
| pTB7220 | Apr; 1.2-kbp PCR product of PA1991 cloned into EcoRI-HindIIII sites of pEX18Ap | This study |
| pTB7222 | Apr; 1.3-kbp PCR product of PA2572 cloned into EcoRI-HindIII sites of pEX18Ap | This study |
| pTB7224 | Apr; 1.2-kbp PCR product of PA5186 cloned into EcoRI-KpnI sites of pEX18Ap | This study |
| pTB7225 | Apr Kmr; 380-bp SmaI-SmaI fragment of pTB7219 removed and filled with PCR product of Kmr cassette | This study |
| pTB7226 | Apr Kmr; 1.0-kbp PCR product of Kmr cassette cloned into SmaI site of pTB7220 | This study |
| pTB7227 | Apr Kmr; 500-bp PstI-PstI fragment of pTB7222 removed and filled with PCR product of Kmr cassette | This study |
| pTB7229 | Apr Kmr; 1.0-kbp PCR product of Kmr cassette cloned into blunted NcoI site of pTB7224 | This study |
| pTB7234 | Apr; P_PA1146́-lacZ; 500-bp PCR product containing promoter region of PA1146 cloned into NcoI-HindIIII sites of pQF50(pPA1146) | This study |
| pTB7235 | Apr; P_PA2572′-lacZ; 500-bp PCR product containing promoter region of PA2572 cloned into NcoI-HindIIII sites of pQF50 | This study |
| pTB7237 | Apr; P_PA5186′-lacZ; 500-bp PCR product of promoter region of PA5186 cloned into NcoI-HindIIII sites of pQF50(pPA5186) | This study |
Escherichia coli was cultivated in Luria-Bertani (LB) medium, and P. aeruginosa was cultivated in LB or minimal medium (1) containing different carbon sources. Alcohols were added at 0.5% (vol/vol). Glucose and succinate were used at 40 mM, and acetate was used at 20 mM. Antibiotics were added as follows: tetracycline at 20 μg/ml, carbenicillin at 100 μg/ml, kanamycin at 50 μg/ml, and gentamicin at 50 μg/ml.
General genetic techniques and PCR.
Routine recombinant DNA work was performed according to the protocols described previously by Sambrook et al. and Ausubel et al. (14, 15). Triparental matings were performed as described by Kretzschmar et al. (16). Electrotransformation of P. aeruginosa ATCC 17933 was performed as described by Smith and Iglewski (17).
Genomic DNA was isolated from P. aeruginosa ATCC 17933 as the template, and Pfu DNA polymerase (Promega) was used for PCR. For primer design, the sequence of P. aeruginosa strain PAO1 was used, since the nucleotide identity between PAO1 and ATCC 17933 has been reported to be 99% (18, 19). For amplification of most genes, oligonucleotides with restriction sites for EcoRI and HindIII, indicated in bold below, were used. For amplification of PA1146, the forward primer 5′-GCGAATTCATGAGCGACCTGCATTACTGGA-3′ and the reverse primer 5′-TATAAGCTTTCAGGCGAGAGTCCCGGCCAC-3′ were used. The 1.2-kbp PCR product containing PA1146 was cloned into EcoRI-HindIII sites of pEX18Ap, resulting in plasmid pTB7219. For PCR amplification of PA1991, the forward primer was 5′-CATGAATTCCAGATGAGCCACGACCTCAG-3′ and the corresponding reverse primer was 5′-AATAAGCTTTCAGAGGGCCTCGCCATAGAC-3′. The 1.2-kbp PCR product containing PA1991 was cloned into EcoRI-HindIII sites of pEX18Ap, resulting in plasmid pTB7220. For the PCR amplification of PA2572, the forward primer was 5′-GAGAATTCATGAACGATAGCGCACCTCCTT-3′ and the corresponding reverse primer was 5′-TGTAAGCTTCTAGGTCGTCGACTCCGGGAG-3′. The 1.3-kbp PCR product containing PA2572 was cloned into EcoRI-HindIII sites of pEX18Ap, resulting in plasmid pTB7222.
For amplification of the promoter region of PA1991 plus PA1991 and PA1992, the forward primer was 5′-CAGAATTCCCAGTCTCTATGGGGTCAG-3′ and the reverse primer was 5′-ATCAAGCTTCCGCTCGATGTTCCTCTTC-3′. The 3.3-kbp PCR product containing the 445-bp region upstream of the start codon of PA1991 plus PA1991 and PA1992 with a possible terminator region was cloned into EcoRI-HindIII sites of pUCP20T, resulting in plasmid pTB7200. PA5186 was amplified using oligonucleotides with restriction sites for EcoRI and KpnI (shown in bold). The forward primer was 5′-CTGAATTCATGCAAGCTTTCAGTTTCGCCA-3′, and the corresponding reverse primer was 5′-CTGGTACCTCAGTATGCCGCGCGATAGATC-3′. The 1.2-kbp PCR product containing PA5186 was cloned into the EcoRI-KpnI sites of pEX18Ap, resulting in plasmid pTB7224.
Gene inactivation.
For inactivation of genes by a kanamycin resistance cassette, the sacB-based strategy with the suicide vector pEX18Ap (20) was employed. Sucrose-resistant colonies were obtained by streaking P. aeruginosa merodiploids on LB plates supplemented with 5% sucrose. For generation of mutants, the kanamycin resistance cassette of pTB3131 was amplified using primers with restriction sites (indicated in bold) for SmaI. The forward primer was 5′-ATCCCGGGGCAAAGAGAAAGCAGGTAGC-3′, and the corresponding reverse primer was 5′-CATCCCGGGCTCAGAACTCGTCAAGAA-3′. For inactivation of PA1146, the SmaI-digested kanamycin resistance cassette was cloned into the SmaI site of pTB7219, resulting in pTB7225. PA1991 was inactivated by cloning the SmaI-digested kanamycin resistance cassette into the SmaI site of pTB7220, resulting in pTB7226. For inactivation of PA2572, the PstI-PstI fragment of the vector pTB7222 was removed, the ends were filled, and the SmaI-digested resistance cassette was cloned into blunted ends of pTB7222, resulting in pTB7227. PA5186 was inactivated by ligating the SmaI-digested kanamycin resistance cassette into the blunted NcoI site of pTB7224, resulting in pTB7229. The same orientation for the Kmr gene and interrupted gene was verified. The suicide vectors were introduced into P. aeruginosa by triparental mating, and after two independent homologous recombinations, potential site-directed double-crossover mutants with a Kmr (Gmr) Cbs Sucr phenotype were selected.
Construction of promoter-probe vectors.
Promoter-probe vectors were constructed to study the transcriptional regulation of PA1146, PA5186, and PA2572. Putative promoter regions (0.5 kbp upstream of the start codon) were amplified by PCR by using primers with restriction sites for NcoI and HindIII, indicated in bold. The PCR products were cloned into the NcoI-HindIII sites of the promoter-probe vector pQF50 upstream of the lacZ gene (21). For amplification of the promoter region of PA1146, the forward primer 5′-GCGAATTCATGAGCGACCTGCATTACTGGA-3′ and the corresponding reverse primer 5′-TATAAGCTTTCAGGCGAGAGT-CCCGGCCAC-3′ were used, and the promoter region of PA5186 was amplified using the forward PCR primer 5′-ACTACCATGGACCAATGGCATCCAGGCGCT-3′ and the corresponding reverse primer 5′-TGAAAGCTTGCATATCGGTCTCCTTGGGCG-3′. The forward PCR primer 5′-ACTACCATGGTCCCTGCAAAGGCAGGCCGA-3′ and the corresponding reverse primer 5′-ACGAAGCTTCCATCTCCGTCTCGTTGGAAG-3′ were used. The corresponding promoter probe vectors pTB7234 (PA1146), pTB7237 (PA5186), and pTB7235 (PA2572) were constructed.
As previously described, the promoter-probe vectors for exaA (pTB3138), exaBC (pTB3139) (3), and pqqABCDEH (pTB7023) (5) are derivatives of pEDY305 (22). The promoter-probe vector for PA1991 (pTB7082) (8) is a derivative of pQF50 (21).
Demonstration of a common mRNA for ercA and ercS.
Total RNA of wild-type cells was extracted using the RNeasy minikit (Qiagen). The resulting solution was incubated with RNase-free DNase (MBI Fermentas) according to the manufacturer's instructions. To generate cDNA, the RNA was incubated with reverse transcriptase (Hoffman-La Roche) according to instructions provided by the supplier. For amplification of a region spanning parts of both genes, which begins 150 bp upstream of and ends 150 bp downstream of the start codon of ercS (PA1992), the forward PCR primer 5′-CCTTCAAGCACGCCGTGGGTTTCCACGAGA-3′ and the corresponding reverse primer 5′-GCTCGAACAGCCATTTGTAGCGGTTGCGCT-3′ were used. The resulting DNA was visualized by agarose gel electrophoresis and ethidium bromide staining. The DNA product showed the expected length of 300 bp.
Enzyme assays and protein determination.
The activity of β-galactosidase was determined with toluene-treated cells according to the procedure of Miller (23). Determination of β-galactosidase activity in NH1 was performed after induction on ethanol, as described by Schobert and Görisch (3) and after growth for 50 h to an optical density at 620 nm (OD620) of 0.6. The empty promoter-probe vector pQF50 or pEDY305 without any promoter cloned into them was used as a baseline condition for measuring promoter activity. The presented values of promoter activities are the calculated mean values of the probes minus the baseline mean values.
Enzyme activity of the NAD+-dependent Fe-ADH was determined with cell extracts. The method described for 1,3-propanediol oxidoreductase of K. pneumoniae was used (12). Cells were collected at an OD620 of 0.6 and disrupted by sonication, and cell debris was removed by centrifugation. Substrate-dependent NADH formation was determined spectrophotometrically at 340 nm. The assay mixtures contained potassium bicarbonate buffer (0.1 M; pH 9.0), ammonium sulfate (30 mM), NAD+ (0.6 mM), various alcohols (0.01 M), and cell extract; 1,3-propanediol at a concentration of 0.1 M was used. Protein concentrations were determined using the method described by Groves et al. (24).
Bioinformatics tools.
The Pseudomonas aeruginosa Genome Database (9, 10) was used to obtain DNA sequences of PAO1. Primers were designed using the Primer3 bioinformatics tool (25). Similarities between amino acid sequences of PA1991 and other protein sequences available in GenBank were assessed from BLAST searches (26). For determination of possible interactions between proteins, the program STRING (27) was used.
RESULTS AND DISCUSSION
The operon ercA-ercS and other genes possibly involved in regulating ethanol utilization.
In P. aeruginosa PAO1, ercA (PA1991) and ercS (PA1992) are located downstream of and close to genes encoding components of the ethanol oxidation system (9, 10). This arrangement of genes is also found in P. aeruginosa ATCC 17933, the organism studied in the present report (Fig. 1).
Fig 1.
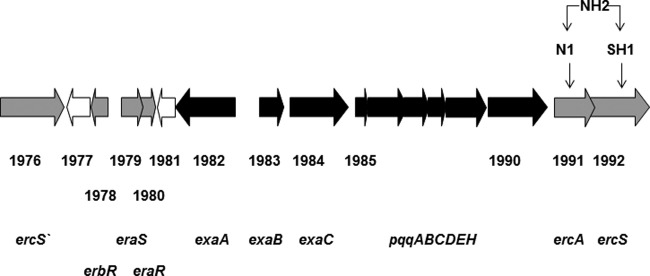
Arrangement of genes of the aerobic ethanol oxidation system and its regulation in P. aeruginosa ATCC 17933. Structural genes exaA, exaBC, and genes of the pqq operon are shown in black. Genes coding for regulatory components of the ethanol oxidation system, eraSR, erbR, ercS, and ercS′, as well as ercA, which encodes a putative Fe-ADH, are shown in gray. Downstream of ercS are genes encoding a probable major facilitator superfamily (MSF) transporter, two hypothetical proteins, and a peptidyl-prolyl cis-trans isomerase (PA1996). All these genes are transcribed in the opposite direction with respect to ercS. The gene erdR (PA3604) is not shown. Mutants discussed in the text are indicated by their names.
The gene ercS overlaps with the upstream gene PA1991 by 17 bp. From transcriptional studies with different constructs of PA1991, encoding a putative Fe-ADH (9, 10), and ercS, it has been concluded that both genes are organized in an operon (8). In this study, we confirmed a common mRNA for the two genes by generating a 300-bp cDNA that spanned the end of PA1991 and the beginning of ercS (data not shown).
PA1991 shows high sequence similarity to the 1,3-propanediol dehydrogenase of Klebsiella pneumoniae (12). The NCBI BLAST analysis of the PA2572 nucleotide sequence showed high sequence similarity to a response regulator in Pseudomonas sp. M18 (complete CDS identity, 99%). The interaction between a putative response regulator encoded by PA2572 and the sensor kinase ErcS was predicted based on the program STRING (27) with a relatively low confidence (k = 0.6). In contrast, interaction of the putative Fe-ADH encoded by PA1991 with the sensor kinase ErcS was predicted, surprisingly, with high confidence (k = 0.9). Two other genes of P. aeruginosa, PA1146 and PA5186, also encode putative Fe-ADHs (9, 10) with significant sequence similarities to the 1,3-propanediol dehydrogenase of K. pneumoniae. For the latter two Fe-ADHs, no interaction was predicted with the sensor kinase ErcS (k < 0.15).
Generation of putative regulatory mutants.
PA2572, encoding a putative response regulator, was inactivated by combined deletion of 500 bp and insertion of a Kmr cassette (pTB7227), resulting in the mutant NH3. PA1991, encoding a putative Fe-ADH, was interrupted by insertion of a Kmr cassette (pTB7226), resulting in the mutant NH1. Double mutant NH2, with both genes of the operon PA1991-ercS inactivated, was generated by interrupting PA1991 in the mutant SH1 (8) by insertion of a Kmr cassette (pTB7226). PA1146(pTB7225) and PA5186(pTB7229), both encoding additional putative Fe-ADHs, were inactivated by insertion of a Kmr cassette. The mutant NH5 with an interrupted PA5186 was isolated, but we were unable to isolate a mutant with an inactivated PA1146. Presumably, such a defect is lethal. The correct insertion of resistance cassettes and construction of mutants was verified by PCR product analysis (data not shown). The mutants and their genotypes are listed in Table 1.
Growth of putative regulatory mutants.
NH1 is unable to grow on 1,3-propanediol. Compared to the wild type, NH1 showed poor growth on butanol, with a significantly increased generation time. On ethanol, 1-propanol, and 1,2-propanediol, it showed an 8-fold-prolonged lag phase (Table 2). However, after the extended lag phase, NH1 grew on the three alcohols with the same generation time as the wild type did. To exclude reversion of NH1 during cultivation, it was reinoculated in fresh minimal medium with ethanol. This culture showed again the extreme 8-fold-prolonged lag phase. No differences in duration of lag phase or in generation time were found on glycerol, acetate, glucose, or succinate (Table 2). Growth of NH1 on the substrates tested was comparable to growth of SH1 with the inactivated sensor kinase ErcS (8). NH2, with both genes of operon PA1991-ercS inactivated, showed an identical growth pattern as NH1 (Table 2). Growth on ethanol was restored in NH1 by plasmid pTB7212, carrying PA1991, in SH1 by plasmid pTB7101, carrying PA1992, and in NH2 by plasmid pTB7200, carrying operon PA1991-ercS (Table 3). NH3 with interrupted PA2572 and NH5 with interrupted PA5186 showed wild-type behavior on the substrates tested (Table 2). We assume that the products of both genes are not directly involved in aerobic ethanol oxidation or in its regulation.
Table 2.
Growth parameters of different P aeruginosa mutants on various substrates
| Carbon source(s) | Growth of mutanta |
|||||||
|---|---|---|---|---|---|---|---|---|
| NH1 [ercA (PA1991)::Kmr], Fe-ADH gene inactivated |
NH2 [ercA (PA1991)::Kmr ΔercS (PA1992)::Gmr], Fe-ADH and SK genes inactivated |
NH3 [PA2572::Kmr], RR gene inactivated |
NH5 [PA5186::Kmr], Fe-ADH gene inactivated |
|||||
| Gen | Lag | Gen | Lag | Gen | Lag | Gen | Lag | |
| Ethanol, 1-propanol | wt | ≫ | wt | ≫ | wt | wt | wt | wt |
| 1-Butanol | ≫ | wt | ≫ | wt | ND | ND | wt | wt |
| 1,2-Propanediol | wt | ≫ | wt | ≫ | ND | ND | wt | wt |
| 1,3-Propanediol | NG | NG | NG | NG | ND | ND | wt | wt |
| Acetate | wt | wt | wt | wt | wt | wt | wt | wt |
| Glucose | wt | wt | wt | wt | ND | ND | Wt | wt |
| Succinate | wt | wt | wt | wt | wt | wt | Wt | wt |
Gen, generation time; Lag, lag phase; SK, two-component regulatory system sensor kinase; RR, response regulator; wt, response similar to wild type; ≫, generation time or lag phase greater than 5 times the wild-type value; NG, no growth for at least 72 h; ND, not determined. Judgments of differences in generation times and lag phases were based on means ± standard deviations from at least three independent experiments.
Table 3.
Growth comparison of P. aeruginosa complemented mutants versus the wild type on ethanol
| Complementation of P. aeruginosa mutant | Growth of mutanta |
|||||
|---|---|---|---|---|---|---|
| NH1 (PA1991::Km) |
SH1 (ΔPA1992::Gmr) |
NH2 (PA1991::Kmr ΔPA1992::Gmr) |
||||
| Gen | Lag | Gen | Lag | Gen | Lag | |
| pUCP20T (empty vector) | wt | ≫ | wt | > | wt | ≫ |
| pTB7212 (PA1991) | wt | wt | wt | > | wt | > |
| pTB7101 (PA1992) | wt | ≫ | wt | wt | wt | ≫ |
| pTB7200 (PA1991, PA1992) | wt | wt | wt | wt | wt | wt |
Gen, generation time; Lag, lag phase; wt, response similar to wild type; >, lag phase greater than 2 times that of the wild type; ≫, lag phase greater than 5 times the wild-type value. Judgments of differences in generation times and lag phases were based on means ± standard deviations of at least three independent experiments.
Transcription of genes coding for the response regulator PA2572 and putative Fe-ADHs.
To test promoter activity of the putative response regulator PA2572 and the different putative Fe-ADHs, promoter-probe vectors were generated. The respective promoter regions were cloned into the vector pQF50, resulting in plasmids pTB7235 (P_PA2572), pTB7234 (P_PA1146), and pTB7237 (P_PA5186). Transcription of the respective genes in wild-type cells was determined after growth on different carbon sources: ethanol, succinate, 1,2-propanediol, 1,3-propanediol, acetate, glycerol, and glucose. Transcription of PA1991 was determined using the promoter-probe vector pTB7082 (8). On ethanol or succinate, promoter activities of PA2572 of 94 ± 12 Miller units (MU) and 42 ± 6 MU were determined, respectively (means ± standard deviations). On ethanol or 1,2-propanediol, the highest promoter activity of PA1991, 703 ± 31 MU, was measured, while on glucose or succinate only 15% of this level was detected. On ethanol, promoter activities of PA1146 and PA5186 reached only 11 to 13% of the promoter activity of PA1991. PA1146 showed the highest activity, with 143 ± 16 MU on acetate, while on ethanol, 1,2-propanediol, or 1,3-propanediol, the activity was 74 ± 6 MU. PA5186 showed the highest activity, with 92 ± 6 MU on glycerol, while on ethanol and acetate the activity was 77 ± 7 MU. Upon growth on ethanol, the putative Fe-ADH encoded by PA1991 appeared to be the most prominent enzyme.
Promoter activities of operons exa and pqq in NH1.
To determine transcriptional levels of the essential structural genes encoding QEDH (exaA), cytochrome c550 (exaB), and PQQ biosynthetic enzymes (pqqABCDEH), promoter-probe vectors pTB3138, pTB3139, and pTB7023 (3, 5) were transferred into wild-type P. aeruginosa or NH1 by triparental mating. The β-galactosidase activities were determined after induction of growth on minimal medium with ethanol as the carbon source.
Promoter activities of exaA and the operon exaBC in NH1 were reduced after induction by a factor of more than 100, compared to that of the wild type. Promoter activity of the operon pqqABCDEH was reduced by a factor of about 30 (Table 4). After complementation of NH1 by vector pTB7212, carrying PA1991, wild-type-like activities were detected again (Table 4). The results indicated that the putative Fe-ADH encoded by PA1991 is involved in the signal transduction process of the ethanol oxidation system. Because of this involvement, and the fact that PA1991 forms an operon with ercS, we named it ercA.
Table 4.
Promoter activities of operons exaA, exaBC, and pqqABCDEH in P. aeruginosa wild type, mutant NH1, and NH1 complemented by vector pTB7212 (PA1991)a
| Reporter gene construct in P. aeruginosa strain and condition | Promoter activity (kMU) |
|||
|---|---|---|---|---|
| P_exaA (pTB3138) | P_exaBC (pTB3139) | P_pqq (pTB7023) | Empty vector (pEDY305) | |
| WT, 6-h growth | 45.5 ± 6.4 | 37.7 ± 5.3 | 26.7 ± 4.1 | 0.06 ± 0.02 |
| WT(pTB7212) (PA1991), 6-h growth | 43.3 ± 5.9 | 36.9 ± 5.1 | 27.7 ± 3.9 | 0.07 ± 0.02 |
| NH1, 6-h induction | 0.3 ± 0.03 | 0.3 ± 0.04 | 0.8 ± 0.07 | ND |
| NH1, 50-h growth | 24.8 ± 3.4 | 23.6 ± 4.4 | 20.5 ± 3.1 | 0.05 ± 0.02 |
| NH1(pTB7212) (PA1991), 6-h growth | 39.7 ± 4.4 | 36.7 ± 4.3 | 27.3 ± 3.2 | 0.06 ± 0.02 |
The reporter gene constructs are derivatives of pEDY305. Activities with the wild type and complemented NH1 mutant were determined after growth on ethanol to an OD600 of 0.6 for about 6 h. Activities with NH1 were determined after induction on ethanol for 6 h and growth to an OD600 of 0.6 for about 50 h. The data represent means and standard deviations of three independent experiments. ND, not determined.
After an extremely extended lag phase, NH1 started to grow on ethanol with a generation time comparable to that of the wild type. The promoter activities of exaA, exaBC, and pqqABCDEH in NH1 reached values of about 50 to 75% of that of the wild type (Table 4). Since growth of NH1, after an extended lag phase, showed a generation time comparable to that of the wild type, the putative Fe-ADH encoded by ercA apparently does not catalyze a critical enzymatic step in the log phase of aerobic ethanol metabolism.
Activity of the NAD+-dependent Fe-ADH.
Wild-type P. aeruginosa ATCC 17933 was grown on ethanol for 6 h, while NH1 was grown for 50 h or was induced on ethanol for 6 h. In cell extracts, no NAD-dependent alcohol dehydrogenase activity could be detected in the absence of Mn2+ ions. By using the test described for 1,3-propanediol:NAD-oxidoreductase (12), extracts with or without membrane fragments showed identical enzyme activities for NAD+-dependent Fe-ADH. The results indicated that the enzyme is soluble. After growth on ethanol, cell extracts of the wild type showed low Fe-ADH activities of about 18 MU/mg. Enzyme activity was also detected with propanol and 1,2-propanediol. The highest activity (82 to 92 MU/mg) was found with 100 mM 1,3-propanediol as the substrate. In extracts of wild-type cells grown on succinate, Fe-ADH activity with ethanol was below the detection limit, 5 MU/mg. Moreover, QEDH, the enzyme responsible for aerobic ethanol oxidation in P. aeruginosa, was not detected. After induction of NH1 for 6 h on ethanol, the activity of Fe-ADH was also below the detection limit, and again no QEDH activity was found. It appears that a functional Fe-ADH encoded by ercA (PA1991) is required for effective expression of QEDH. The Fe-ADH encoded by ercA (PA1991) might generate a signal for the sensor kinase ErcS. After the extremely long lag phase on ethanol, NH1 showed in the following logarithmic growth phase Fe-ADH activity of about 15 MU/mg and QEDH activity similar to the wild type. Under these conditions, the lost function of ErcA was probably compensated by another gene product that generated the signal needed for QEDH expression.
Transcription of eraSR, erbR, and erdR in NH1.
Transcription of the two-component regulatory genes eraSR, erbR, and erdR was determined in NH1 in order to investigate the role of the ercA gene product in controlling the quinoprotein ethanol regulon (Fig. 2). The promoter-probe vectors pTB7074 (5), pTB7107, and pTB7108 (8) were used. After induction on ethanol for 6 h, transcription of the regulatory components EraSR and ErbR was reduced in NH1 to below 5% that of the wild type, indicating a direct influence of the ercA gene product (Table 5). Transcription of erdR was only slightly reduced, to about 75%, in NH1. These results were essentially the same as those found with SH1, in which ercS is inactivated (8). After 50 h, when NH1 resumed growth after the extremely long lag phase, transcription of the regulatory genes erbR and erdR reached the wild-type levels, while that of the eraSR operon reached only 25% (Table 5).
Table 5.
Activities of eraSR, erbR, and erdR promoters in P. aeruginosa wild-type and NH1 mutant strainsa
| P. aeruginosa strain and condition | Gene inactivated | Promoter activity (MU) |
|||
|---|---|---|---|---|---|
| P_eraSR PA1979, PA1980 (pTB7074) | P_erbR PA1978 (pTB7108) | P_erdR PA3604 (pTB7107) | Empty vector (pQF50) | ||
| WT, 6-h growth | 1,417 ± 153 | 1,649 ± 168 | 210 ± 4 | 23 ± 7 | |
| NH1, 6-h induction | PA1991 (Fe-ADH) | 37 ± 5 | 55 ± 7 | 166 ± 7 | 25 ± 8 |
| NH1, 50-h growth | PA1991 (Fe-ADH) | 334 ± 29 | 1,409 ± 160 | 221 ± 14 | 27 ± 9 |
The reporter gene constructs are derivatives of pQF50. Activities were determined after growth on ethanol to an OD620 of 0.6 for about 6 h with the wild type and for about 50 h with NH1. Promoter activities were also determined in NH1 after induction on ethanol for 6 h. The data represent means and standard deviations from three independent experiments.
The data demonstrate transcriptional control of the quinoprotein ethanol regulon by the ercA gene product and lend further support for naming the gene ercA. Both gene products of the ercA-ercS operon are required for effective expression of the ethanol oxidation system. Presumably, the Fe-ADH encoded by ercA generates a signal that activates the sensor kinase ErcS. However, this signal remains unknown. Experiments to restore wild-type behavior of NH1 on ethanol by exogenous acetaldehyde were unsuccessful (data not shown).
Constitutive expression of the operon ercA-ercS does not restore expression of the ethanol oxidation system in MD1.
The response regulator ErdR represents the highest level of the proposed signal cascade that regulates ethanol oxidation in P. aeruginosa (8). The cascade encompasses 4 levels of two-component systems (Fig. 2). This regulatory system is hypothetical and complex, but it is the simplest one to explain all of our former experimental results, without making further complicated and unlikely assumptions. ErdR regulates transcription of the one-level-lower operon ercA-ercS. In MD1, with an inactivated erdR, the promoter activity of the operon ercA-ercS was reduced to about 12%, and the promoter activity of the next-level-lower response regulator gene erbR was reduced to below 5% compared to transcription in the wild type. Constitutive expression of ercS in MD1 did not result in the expression of the exa and pqq operons, which encode the components of the ethanol oxidation system in P. aeruginosa (8). Furthermore, constitutive expression of erbR restored expression of the ethanol oxidation system in the mutant SH2, with an inactivated ercS (8). To study if transcription of the exa and pqq operons could be restored by constitutive expression of the complete ercA-ercS operon, the vector pTB7200 was constructed. This vector carried the operon ercA-ercS under the control of the lac promoter, which is recognized in P. aeruginosa and leads to constitutive expression of both genes. Vector pTB7200 was transferred into MD1. After induction on ethanol, expression levels of exaA and operons exaBC and pqqABCDEH were determined with promoter-probe vectors pTB3138, pTB3139, and pTB7023. Under these conditions, the transcription levels of exaA, exaBC, and pqqABCDEH were below 5% that of the wild type and comparable to that in MD1 without the vector pTB7200 (data not shown). This result is in agreement with our previously proposed regulatory scheme (8). In MD1, transcription of the so-far-unknown sensor kinase ErbS, corresponding to the response regulator ErbR, is assumed to depend also on the response regulator ErdR, which is missing in MD1 (Fig. 2).
Constitutive expression of EraSR and ErbR partially restores expression of the ethanol oxidation system in NH1 and SH1.
The response regulator ErbR controls transcription of the exaBC, the pqq and the eraSR operons (5). The two-component regulatory system EraSR controls transcription of exaA, which encodes QEDH (3). We previously showed that transcription of erbR depends on the sensor kinase ErcS (8) and, as shown in the present report, it also depends on the gene product of ercA. As shown in Fig. 2, in NH1, with a defective ercA, and in SH1, with a defective ercS, it is to be expected that constitutive expression of the one-level-lower ErbR will lead to the expression of the exa and pqq operons. Furthermore, constitutive expression of the next-level-lower EraSR will lead to the expression of exaA, but not of exaBC and pqq. The vector pTB7060 and vector pTB3144 were used for constitutive expression of erbR and the two-component system eraSR, respectively. Both vectors carry the respective genes under the control of a lac promoter, which is recognized in P. aeruginosa. The vectors were transferred into NH1 and SH1. Constitutive expression of ErbR led to induction of operons exaA, exaBC, and pqq (Table 6). The relative expression levels were about 25 to 40% in NH1 and about 25 to 50% in SH1 compared to that in the wild type. As expected, constitutive expression of EraSR in both mutants did not induce the exaBC or pqq operons. However, in both mutants, exaA, encoding QEDH, was induced, but to a rather low level of 5 to 9% of the wild-type level (Table 6). The reason for this low expression level is not clear. The hypothetical model shown in Fig. 2 does explain the basic experimental results, but it may need to be modified to explain the low expression levels described.
Table 6.
Promoter activities of exaA, exaBC, and pqqABCDEH operons in the wild type and mutants NH1 and SH1a
| P. aeruginosa construct: strain/vector/gene(s) | Gene inactivated | Promoter activity (kMU) |
Conditionb | |||
|---|---|---|---|---|---|---|
| P_exaA (pTB3138) | P_exaBC (pTB3139) | P_pqqAB (pTB7023) | Vector (pEDY305) | |||
| Wild type/pUCP20T | 41.4 ± 5.7 | 37.7 ± 5.3 | 26.7 ± 4.1 | 0.06 ± 0.02 | G | |
| NH1/pTB7060/erbR | ercA (PA1991) | 12.9 ± 2.3 | 10.0 ± 1.4 | 11.6 ± 2.3 | 0.07 ± 0.02 | I |
| NH1/pTB3144/eraSR | ercA (PA1991) | 1.8 ± 0.2 | 0.42 ± 0.15 | 0.9 ± 0.15 | 0.06 ± 0.02 | I |
| NH1/pUCP20T | ercA (PA1991) | 0.28 ± 0.03 | 0.32 ± 0.04 | 0.8 ± 0.08 | ND | I |
| SH1/pTB7060/erbR | ercS (PA1992) | 10.5 ± 1.6 | 9.4 ± 1.4 | 14.3 ± 1.7 | 0.06 ± 0.02 | I |
| SH1/pTB3144/eraSR | ercS (PA1992) | 3.7 ± 0.5 | 0.46 ± 0.1 | 1.3 ± 0.2 | 0.05 ± 0.02 | I |
| SH1/pUCP20T | ercS (PA1992) | 0.52 ± 0.14 | 0.9 ± 0.19 | 1.5 ± 0.17 | ND | I |
Activities were determined in mutants with vector pTB7060 constitutively expressing the response regulator ErbR and vector pTB3144 constitutively expressing the two-component system EraSR. Reporter constructs of pEDY305 were used. Activities were determined after induction of mutants for 6 h on ethanol or after growth of the wild type to an OD620 of 0.6 for about 6 h. Results represent means ± standard deviations of at least three independent experiments. ND, not determined.
G, after growth; I, after induction.
Conclusions.
In the present study, we confirmed that PA1991 and ercS (PA1992) form an operon. Inactivation of the respective genes generated mutants NH1 and SH1, with similar properties (Table 2). Both genes are essential for effective expression of the ethanol oxidation system in P. aeruginosa. Therefore, we named PA1991, which forms an operon with ercS, ercA. The ercA-ercS operon regulates transcription of the response regulator ErbR, which controls the operons eraSR, exaBC, and pqq. The results generated with NH1 provided further support to the hierarchical complex regulatory network (Fig. 2), comprising possibly five different two-component systems that regulate ethanol oxidation in P. aeruginosa. It has been recently reported that the global response regulator ErdR is required for expression of the gene acsA, which encodes acetyl coenzyme A synthetase (28). Thus, ErdR controls, in addition to ethanol, acetate metabolism in P. aeruginosa. This observation is incorporated in Fig. 2.
ACKNOWLEDGMENT
Alexandra Hogel is acknowledged for excellent technical assistance.
Footnotes
Published ahead of print 28 June 2013
REFERENCES
- 1.Rupp M, Görisch H. 1988. Purification, crystallization and characterization of quinoprotein ethanol dehydrogenase from Pseudomonas aeruginosa. Biol. Chem. Hoppe-Seyler 369:431–439 [DOI] [PubMed] [Google Scholar]
- 2.Mennenga B, Kay CWM, Görisch H. 2009. Quinoprotein ethanol dehydrogenase from Pseudomonas aeruginosa: the unusual disulfide ring formed by adjacent cysteine residues is essential for efficient electron transfer to cytochrome c550. Arch. Microbiol. 191:361–367 [DOI] [PubMed] [Google Scholar]
- 3.Schobert M, Görisch H. 2001. A soluble two-component regulatory system controls expression of quinoprotein ethanol dehydrogenase (QEDH) but not expression of cytochrome c550 of the ethanol-oxidation system in Pseudomonas aeruginosa. Microbiology 147:363–372 [DOI] [PubMed] [Google Scholar]
- 4.Görisch H. 2003. The ethanol oxidation system and its regulation in Pseudomonas aeruginosa. Biochim. Biophys. Acta 1647:98–102 [DOI] [PubMed] [Google Scholar]
- 5.Gliese N, Khodaverdi V, Schobert M, Görisch H. 2004. AgmR controls transcription of a regulon with several operons essential for ethanol oxidation in Pseudomonas aeruginosa ATCC 17933. Microbiology 150:1851–1857 [DOI] [PubMed] [Google Scholar]
- 6.Schobert M, Görisch H. 1999. Cytochrome c550 is an essential component of the quinoprotein ethanol oxidation system in Pseudomonas aeruginosa: cloning and sequencing of the genes encoding cytochrome c550 and an adjacent acetaldehyde dehydrogenase. Microbiology 145:471–481 [DOI] [PubMed] [Google Scholar]
- 7.Gliese N, Khodaverdi V, Görisch H. 2010. The PQQ biosynthetic operons and their transcriptional regulation in Pseudomonas aeruginosa. Arch. Microbiol. 192:1–14 [DOI] [PubMed] [Google Scholar]
- 8.Mern DS, Ha SW, Khodaverdi V, Gliese N, Görisch H. 2010. A complex regulatory network controls aerobic ethanol oxidation in Pseudomonas aeruginosa: indication of four levels of sensor kinases and response regulators. Microbiology 156:1505–1516 [DOI] [PubMed] [Google Scholar]
- 9.Winsor GL, Lo R, Sui SJ, Ung KS, Huang S, Cheng D, Ching WK, Hancock RE, Brinkman FS. 2005. Pseudomonas aeruginosa Genome Database and PseudoCAP: facilitating community-based, continually updated, genome annotation. Nucleic Acids Res. 33:338–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Winsor GL, Lam DK, Fleming L, Lo R, Whiteside MD, Yu NY, Hancock RE, Brinkman FS. 2011. Pseudomonas Genome Database: improved comparative analysis and population genomics capability for Pseudomonas genomes. Nucleic Acids Res. 39:D596–D600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Reid MF, Fewson CA. 1994. Molecular characterization of microbial alcohol dehydrogenases. Crit. Rev. Microbiol. 20:13–56 [DOI] [PubMed] [Google Scholar]
- 12.Johnson EA, Lin EC. 1987. Klebsiella pneumoniae 1,3-propanediol:NAD+ oxidoreductase. J. Bacteriol. 169:2050–2054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bystrykh LV, Vonck J, van Bruggen EFJ, van Beeumen J, Samyn B, Govorukhina NI, Arfman N, Duine JA, Dijkhuizen L. 1993. Electron microscopic analysis and structural characterization of novel NADP(H)-containing methanol:N,N′-dimethyl-4-nitrosoaniline oxidoreductases from the gram-positive methylotrophic bacteria Amycolatopsis methanolica and Mycobacterium gastri MB19. J. Bacteriol. 175:1814–1822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sambrook J, Fritsch EF, Maniatis T. 1989. Molecular cloning: a laboratory manual, 2nd ed Cold Spring Harbor Laboratory, Cold Spring Harbor, NY [Google Scholar]
- 15.Ausubel FA, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K. 2002. Current protocols in molecular biology. Wiley, New York, NY [Google Scholar]
- 16.Kretzschmar U, Schobert M, Görisch H. 2001. The Pseudomonas aeruginosa acsA gene, encoding an acetyl-CoA synthetase, is essential for growth on ethanol. Microbiology 147:2671–2677 [DOI] [PubMed] [Google Scholar]
- 17.Smith AW, Iglewski BH. 1989. Transformation of Pseudomonas aeruginosa by electroporation. Nucleic Acids Res. 17:10509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schobert M. 1999. Molekulargenetische Untersuchungen zum Ethanol-oxidierenden System in Pseudomonas aeruginosa. Ph.D thesis Technische Universität Berlin, Berlin, Germany [Google Scholar]
- 19.Stover CK, Pham XQ, Erwin AL, Mizoguchi SD, Warrener P, Hickey MJ, Brinkman FS, Hufnagle WO, Kowalik DJ, Lagrou M, Garber RL, Goltry L, Tolentino E, Westbrock-Wadman S, Yuan Y, Brody LL, Coulter SN, Folger KR, Kas A, Larbig K, Lim R, Smith K, Spencer D, Wong GK, Wu Z, Paulsen IT, Reizer J, Saier MH, Hancock RE, Lory S, Olson MV. 2000. Complete genome sequence of Pseudomonas aeruginosa PAO1, an opportunistic pathogen. Nature 406:959–964 [DOI] [PubMed] [Google Scholar]
- 20.Hoang TT, Karkhoff-Schweizer RR, Kutchma AJ, Schweizer HP. 1998. A broad-host-range Flp-FRT recombination system for site-specific excision of chromosomally-located DNA sequences: application for isolation of unmarked Pseudomonas aeruginosa mutants. Gene 212:77–86 [DOI] [PubMed] [Google Scholar]
- 21.Farinha MA, Kropinski AM. 1990. Construction of broad-host-range plasmid vectors for easy visible selection and analysis of promoters. J. Bacteriol. 172:3496–3499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schwartz E, Gerischer U, Friedrich B. 1998. Transcriptional regulation of Alcaligenes eutrophus hydrogenase genes. J. Bacteriol. 180:3197–3204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Miller JM. 1992. A short course in bacterial genetics: a laboratory manual and handbook for Escherichia coli and related bacteria. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY [Google Scholar]
- 24.Groves WE, Davis FC, Sells BH. 1968. Spectrophotometric determination of microgram quantities of protein without nucleic acid interference. Anal. Biochem. 22:195–210 [DOI] [PubMed] [Google Scholar]
- 25.Rozen S, Skaletsky HJ. 2000. Primer3 on the WWW for general users and for biologist programmers. Methods Mol. Biol. 132:365–386 [DOI] [PubMed] [Google Scholar]
- 26.Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. 1997. Gapped Blast and psi-Blast: a new generation of protein database search programs. Nucleic Acids Res. 25:3389–3402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Szklarczyk D, Franceschini A, Kuhn M, Simonovic M, Roth A, Minguez P, Doerks T, Stark M, Muller J, Bork P, Jensen LJ, Von Merking C. 2011. The STRING database in 2011: functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res. 39:D561–D568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kretzschmar U, Khodaverdi V, Adrian L. 2010. Transcriptional regulation of the acetyl-CoA synthetase gene acsA in Pseudomonas aeruginosa. Arch. Microbiol. 192:685–690 [DOI] [PubMed] [Google Scholar]
- 29.Cetin ET, Toereci K, Ang O. 1965. Encapsulated Pseudomonas aeruginosa (Pseudomonas mucosus) strains. J. Bacteriol. 89:1432–1433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hanahan D. 1983. Studies on transformation of Escherichia coli with plasmids. J. Mol. Biol. 166:557–580 [DOI] [PubMed] [Google Scholar]
- 31.Boyer HW, Roulland-Dussoix D. 1969. A complementation analysis of the restriction and modification of DNA in Escherichia coli. J. Mol. Biol. 14:459–472 [DOI] [PubMed] [Google Scholar]
- 32.Schweizer HD. 1993. Small broad-host-range gentamycin resistance cassettes for site-specific insertion and deletion mutagenesis. Biotechniques 15:831–834 [PubMed] [Google Scholar]
- 33.Staskawicz B, Dahlbeck D, Keen N, Napoli C. 1987. Molecular characterization of cloned avirulence genes from race 0 to race 1 of Pseudomonas syringae pv. glycinea. J. Bacteriol. 169:5789–5794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Figurski DH, Helinski DR. 1979. Replication of an origin-containing derivative of plasmid RK2 dependent on a plasmid function provided in trans. Proc. Natl. Acad. Sci. U. S. A. 76:1648–1652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yanisch-Perron C, Vieira J, Messing J. 1985. Improved M13 phage cloning vectors and host strains: nucleotide sequence of the M13mp18 and pUC19 vectors. Gene 33:103–119 [DOI] [PubMed] [Google Scholar]
- 36.Schweizer HP, Klassen TR, Hoang T. 1996. Improved methods for gene analysis in Pseudomonas, p 229–237 In Nakazawa T, Furukawa K, Haas D, Silver S. (ed), Molecular biology of pseudomonads. American Society for Microbiology, Washington, DC [Google Scholar]