

Abstract
Pyridine-2,6-bis(thiocarboxylate) (PDTC), produced by certain pseudomonads, is a sulfur-containing siderophore that binds iron, as well as a wide range of transition metals, and it affects the net hydrolysis of the environmental contaminant carbon tetrachloride. The pathway of PDTC biosynthesis has not been defined. Here, we performed a transposon screen of Pseudomonas putida DSM 3601 to identify genes necessary for PDTC production (Pdt phenotype). Transposon insertions within genes for sulfate assimilation (cysD, cysNC, and cysG [cobA2]) dominated the collection of Pdt mutations. In addition, two insertions were within the gene for the LysR-type transcriptional activator FinR (PP1637). Phenotypic characterization indicated that finR mutants were cysteine bradytrophs. The Pdt phenotype of finR mutants could be complemented by the known target of FinR regulation, fprA (encoding ferredoxin:NADP+ oxidoreductase), or by Escherichia coli cysJI (encoding sulfite reductase). These data indicate that fprA is necessary for effective sulfate assimilation by P. putida and that the effect of finR mutation on PDTC production was due to deficient expression of fprA and sulfite reduction. fprA expression in both P. putida and P. aeruginosa was found to be regulated by FinR, but in a manner dependent upon reduced sulfur sources, implicating FinR in sulfur regulatory physiology. The genes and phenotypes identified in this study indicated a strong dependence upon intracellular reduced sulfur/cysteine for PDTC biosynthesis and that pseudomonads utilize sulfite reduction enzymology distinct from that of E. coli and possibly similar to that of chloroplasts and other proteobacteria.
INTRODUCTION
Pyridine-2,6-bis(thiocarboxylate) (PDTC) is a novel siderophore produced by certain strains of bacteria of the genus Pseudomonas (1, 2). Its novelty lies in the ability to form stable complexes with a wide range of transition metals in addition to iron (3), a role in zinc nutrition (4), and unique reactivity with a toxic pollutant (carbon tetrachloride [CCl4]) (5). Its novel characteristics are imparted by the constituent ligands thiocarboxylate sulfur and pyridine nitrogen atoms, which coordinate both hard and soft metal ions. Evidence that PDTC is a siderophore includes the high stability constant of the ferric complex (6), iron-repressible production (7, 8), and receptor-mediated uptake of the ferric-PDTC complex (9). This has yielded insights into how PDTC production is regulated (10), but questions remain as to its biosynthesis and how that may relate to global regulatory circuits.
Individual organisms may be capable of producing more than one siderophore (11, 12). How an organism makes the metabolic “decision” to produce one siderophore as opposed to an alternative in its repertoire is not completely understood. Regulatory features reflecting the unique demands of specific siderophore biosynthetic pathways may also be present. Siderophore biosynthetic genes may thus have been selected for appropriate regulation by global regulators that prevent the export of a limiting nutrient. There are few data to support this assumption in the current literature on siderophore regulation, however. Studies have identified global regulatory systems that affect siderophore production such as quorum sensing (13–15). Studies that have identified nutritional cues other than iron are limited. Farmer and Thomas (16) identified a connection between sulfur assimilatory processes and production of a sulfur-containing siderophore. In that case, Burkholderia cenocepacia was found to curtail pyochelin production when sulfate starvation conditions were imposed. More recently, Matthijs et al. (17) have shown that production of the only other characterized thiocarboxylate siderophore, thioquinolobactin, requires effective sulfate assimilation or provision of a suitable sulfur source. It is not known how sulfur sources affect production of the respective secondary metabolites; regulation could be somewhat passive, e.g., due to parameters such as intracellular cysteine concentrations and the relative affinities of primary metabolic enzymes versus secondary metabolism, or more active such as via transcriptional or posttranscriptional regulatory processes.
Although the PDTC biosynthetic pathway has not yet been established biochemically, genetic sequence data and limited isotopic tracer studies have informed speculation as to how it may proceed (8, 18). Genes necessary for PDTC production by P. stutzeri KC and P. putida DSM 3601 (pdt gene clusters) have been described (9, 18) (GenBank accession no, AY319946). Homology of some of the respective gene products with proteins known to function in forming protein thiocarboxylates indicated that cysteine desulfurase, sulfur transferase, and acyl-adenylate ligase activities are involved (Fig. 1) (19–21). An obvious candidate for the carboxylic acid substrate is dipicolinate (DPA) (Fig. 1). Isotopically labeled DPA ([3-2H]DPA) was found to be incorporated into PDTC when provided to cells of P. putida DSM 3601 (22). Based on the above-mentioned homology the precursor molecule providing sulfur for PDTC biosynthesis is presumed to be cysteine (Fig. 1). It should be pointed out that a pathway for PDTC biosynthesis that includes a sulfenic acid as an intermediate has been proposed (23). That pathway predicts a cleavage (apparently monooxygenase-dependent) of a thioester intermediate, and reduction of the resulting sulfenic acid to form the thiocarboxylate. That pathway would require genes (e.g., for a monooxygenase system) that are not encoded within the described pdt gene clusters.
Fig 1.
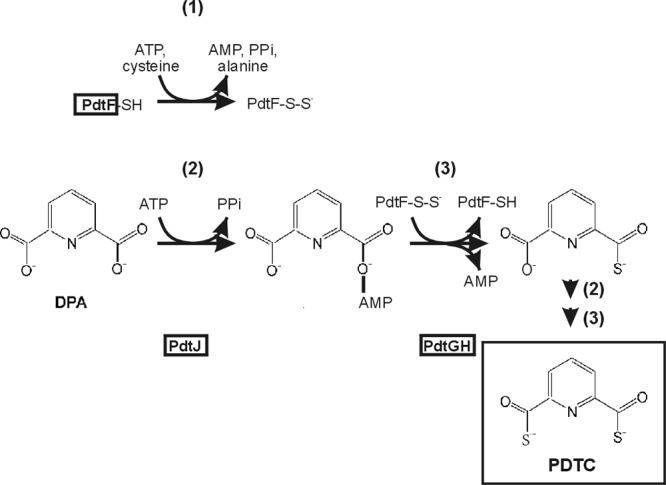
Predicted sulfur transfer steps in PDTC biosynthesis. The three steps depicted are (i) cysteine desulfurylase activity attributed to PdtF, an autosulfurylation forming persulfide-containing, modified PdtF; (ii) acyl activation; and (iii) transulfurylation activities, giving 6-(monothiocarboxylic acid)-picolinic acid. A second cycle of activation/transulfurylation of that product would give PDTC. Gene products (PdtF, PdtJ, PdtG, and PdtH) catalyzing the respective steps are denoted by boxes within or below each reaction.
The hypothetical PDTC biosynthetic pathway described in Fig. 1 predicts PDTC production to be limited by factors affecting intracellular cysteine levels, as has been seen for pyochelin and thioquinolobactin (16, 17, 24). The biochemistry and genetics of cysteine biosynthesis has been extensively studied in Gram-negative bacteria in which it constitutes a major regulon (25). That system displays finely coordinated activities of serine activation and sulfur incorporation. Coordinate regulation is achieved through transcriptional, and posttranslational mechanisms. CysB is the master regulator of sulfur assimilation and is a LysR-type transcriptional activator. In the presence of its co-effector, N-acetylserine (NAS), CysB binds regulatory elements within promoters of sulfur assimilatory genes to allow their maximal expression. Production of inducer (NAS) is dependent upon the serine transacetylase activity of CysE. CysE is in turn (negatively) allosterically regulated by intracellular cysteine. Several gene products catalyzing steps of sulfate assimilation (e.g., CysD and CysN) display rapid turnover, requiring active transcription to maintain steady intracellular levels (25). The result is balance between the demand for reduced sulfur and the abundance of sulfur assimilatory enzymes. Although PDTC production is an example of secondary metabolism rather than assimilatory metabolism, its regulation may involve some overlap of regulatory elements in order to optimally allocate intracellular sulfur.
To more fully address questions of how PDTC production is integrated into central metabolism, a more comprehensive analysis of functions associated with PDTC production, and encoded outside the pdt cluster was undertaken. We used a genetic approach to search for potential accessory functions within the genome of a PDTC-producing pseudomonad. We used the genomic background of P. putida DSM 3601, owing to the advantages afforded by the availability of a published genome sequence for that species (26), and a well-characterized alternative siderophore system (pyoverdine). Using a pyoverdine-deficient genetic background, a transposon insertion library was screened for strains with altered PDTC production by exploiting the iron-containing dye chrome azurol S (CAS) (12, 27). That screening procedure yielded only a single pdt insertion (pdtI) that has been described elsewhere (24). In the present study, we describe other insertions that led to the complete loss of or to a reduction in PDTC production. Identification of the affected genes and characterization of the resulting phenotypes yielded insights into sulfate assimilation by pseudomonads and support a role for cysteine as the immediate sulfur donor for PDTC biosynthesis.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
The strains used in the present study are listed in Table 1. P. putida strains were routinely maintained on tryptic soy medium. Minimal media used for studies of siderophore production in Pseudomonas were PM and M9. PM is a PIPES-buffered minimal medium and was prepared as described previously (24). PIPES (>99.0%) was from Research Organics (Cleveland, OH). M9 is a phosphate-buffered minimal medium and was prepared as described by Maniatis et al. (24a). M9 was made without sulfur by replacing ammonium sulfate with ammonium chloride and magnesium sulfate with magnesium chloride to give an equivalent amount of ammonium and magnesium. M9 minimal medium with 0.2% sodium citrate was used for selection of Pseudomonas transconjugants. CAS plates were prepared by the method of Schwyn and Neilands (27). P. aeruginosa and E. coli strains were maintained on Luria-Bertani (LB) medium. Antibiotics were used at the following concentrations: kanamycin (Km), 50 μg/ml; tetracycline (Tc), 15 μg/ml; gentamicin (Gm), 15 μg/ml for E. coli and 30 μg/ml for Pseudomonas; ampicillin (Ap), 100 μg/ml; and chloramphenicol (Cm), 25 μg/ml. P. putida cultures were grown at 30°C, and P. aeruginosa and E. coli cultures were grown at 37°C. Cultures used for siderophore quantitation were 5 ml in 17-by-125-mm test tubes grown with constant shaking (P. putida) or 25 ml in 125-ml baffled flasks with constant shaking (P. aeruginosa). For the growth and luminescence measurements, 96-well plates were used with a final medium volume of 250 μl per well using a BioTek Synergy H4 plate reader and constant agitation. Optical density (OD) readings were adjusted to a 1-cm path-length value. Clear plates (Thermo/Nunc, catalog no. 266120) were used when growth was measured alone, and white plates with clear optical bottoms (Thermo/Nunc, catalog no. 165306) were used whenever luminescence measurement was included (200 μl of medium volume per well). Luminescence was measured using the extended range setting of the instrument.
Table 1.
Bacterial strains and plasmids examined in this study
| Strain or plasmid | Description or genotypea | Source or referenceb |
|---|---|---|
| Strains | ||
| P. putida | ||
| DSM 3601 | Wild-type PDTC producer | DSMZ (2) |
| BK8 | DSM 3601 pfrI::Tcr; Pvd− | Laboratory collection (24) |
| SO3B9 | BK8 finR::mini-Tn5xy/E Kmr | This work |
| TA690 | BK8 ΔfinR::Gmr (+ orientation) | This study |
| TA691 | BK8 ΔfinR::Gmr (− orientation) | This study |
| P. aeruginosa | ||
| MPA01 | Wild type | UWGSD |
| UWID#1789 | MPA01 PA4130::TnphoA | UWGSD (54) |
| UWID#1631 | MPA01 PA4130::TnphoA | UWGSD (54) |
| UWID#33115 | MPA01 PA4513::TnphoA | UWGSD (54) |
| UWID#33735 | MPA01 PA4513::TnphoA | UWGSD (54) |
| Δ3398 | PA01 ΔfinR | This study |
| TA791 | MPA01 ΔpvdF | This study |
| TA975 | MPA01 ΔpvdF ΔfinR | This study |
| E. coli | ||
| JM109 | recA endA host | Promega, Madison, WI |
| DH5α λpir | pir+ recA endA host | K. Mintz, University of Vermont |
| BW20767 | pir+ mob+ donor | 32 |
| Plasmids | ||
| pUTKm mini-Tn5xylE | Mini-transposon vehicle | 28 |
| pGEM-Teasy | Apr; oricolE1 TA cloning vector used for direct cloning of PCR products | Promega |
| pBluescript SK(−) | Apr; oricolE1 cloning vector | Stratagene, La Jolla, CA |
| pBsdelSal | pBluescript SK(−) with XhoI-SalI deletion | This study |
| pUCGM | Source of Gmr cassette | 55 |
| pJB3Tc20 | Tcr broad-host-range vector | 56 |
| pJB3Km1 | Kmr broad-host-range vector | 56 |
| pJB3TcGm | pJB3Tc20 with Gmr cassette inserted at BamHI | This study |
| pJB3cysDNC | pJB3TcGm with P. putida DSM 3601 cysDNC and flanking DNA inserted at EcoRI | This study |
| pJB3PP1637 | pJB3TcGm with P. putida mt-2 finR gene and flanking DNA inserted at EcoRI | This study |
| pVT1460 | oriR6K, Apr, mobRP4 suicide vector; Kmr derivative of pGP704 | K. Mintz (57) |
| pJBKm1 | Kmr broad-host-range vector | 56 |
| pJBKmfinR | pJB3Km1 with P. putida DSM 3601 finR | This study |
| pJBKmfprA | pJB3Km1 with P. putida DSM 3601 fprA | This study |
| pJN105 | GmR broad-host-range expression vector with ParaBAD promoter | 58 |
| pJN105GW | pJN105 with Gateway recombination site inserted at SmaI | This study |
| pJN105GW::fpr2-3 | pJN105GW with MPA01 fprA | This study |
| pJN105GW::cysJI | pJN105GW with E. coli cysJI | This study |
| pEX18T | ori ColE1 Apr, sacB suicide vector | 59 |
| pEX18T::Δ3398 | pEX18T with PA finR deletion allele, Gmr cassette | This study |
| pEX18T::ΔpvdF | pEX18T with PA pvdF deletion allele, Gmr cassette | This study |
| pMF54 | Apr/Cbr broad-host-range vector | 60 |
| pMF418 | pMF54 with PA finR | This study |
| pUC18-mini-Tn7T-Gm-lux | Mini-Tn7lux vector | 31 |
| pAG4lux | pUC18-mini-Tn7T-Gm-lux with a 1048-1114 deletion and a 2036-2503 deletion for reduced-background lux expression | A. Glassing and T. A. Lewis, unpublished data |
| pAG4lux::PfprA | pAG4lux with P. aeruginosa fprA promoter | This study |
Gmr, gentamicin resistance; Tetr, tetracycline resistance; Apr, ampicillin resistance; Cbr, carbenicillin resistance; Kmr, kanamycin resistance.
DSMZ, Deutsche Sammlung von Mikroorganismen und Zellkulturen, Braunschweig, Germany; UWGSD, University of Washington, Genome Sciences Department.
Transposon mutagenesis and insertion characterization.
P. putida strain BK8 was mutagenized with Mini-TnKmxylE using the filter-mating technique (28). Several independent matings were performed on each of two separate occasions. Kanamycin-resistant clones were replicated onto CAS Km plates with 0.3% Casamino Acids (first screen) or without Casamino Acids (second screen). Sites of transposon insertion were determined by sequencing. Strains LL1, LL3, PP1, TL1, LLBr1, and SEMBr1 were characterized by inverse PCR using a nested set of primers (xylE with KMR, followed by KmlacZRV with KmlacZFW). The remaining transposon insertions were characterized using arbitrary PCR with primers ARB6 and ARB2 (29) in combination with individual, nested transposon-derived primers given above.
Strains obtained from the University of Washington Transposon Mutant Collection were screened by PCR and sequencing as described by Bailey and Manoil (30).
P. putida finR deletion construction.
P. putida deletion alleles were constructed by individually amplifying upstream and downstream finR-flanking sequences using the primer pairs dgkFWSal/dgkRVXba, and fprAFWXba/fprARVSal, respectively. The two products were assembled using engineered XbaI sites and ligated products were amplified using the corresponding outside primers to give the fused deletion allele. That product was cloned into pGEM-T Easy and prepared for insertion of the gentamicin resistance cassette from pUCGM by digestion with XbaI. The aacC1 gene was removed from pUCGM by digestion with XbaI and gel purification. Alleles with both orientations of aacC1 were identified by PCR with combinations of either primer GmF1311 or GmRV1269 in combination with primers dgkFWSal or fprARVSal. Each respective allele was then ligated into the vector pVT1460 using the SalI site and the resulting plasmid vehicles were used in binary matings from E. coli host BW20767 into P. putida BK8. Transconjugants that underwent double recombination events, resulting in replacement of the wild-type allele were selected on M9 citrate with Gm (TA690) or tryptic soy agar with Tc and Gm (TA691). Replacements were resolved from cointegrants by PCR with primers PP1635FW2 and fprARV805 in combination with either GmF1311 or GmRV1269. The resulting products obtained from strains TA690 and TA691 were cloned and sequenced to verify that no other mutations were introduced in the affected locus.
P. aeruginosa deletion alleles.
Overlap-extension PCR was used to generate P. aeruginosa deletion alleles as described by Choi and Schweizer (31). Gm-F and Gm-R primers designed by those authors were used to produce the excisable gentamicin resistance cassette. The primers PA3398-EcoRI3′Dn, PA3398-DnF-Gm, PA3398-UpR-Gm, and PA3398-EcoRI5′Up were used to generate the Δ3398 strain, and pvdF-UpF2-GWL, pvdF-UpR2-Gm, pvdF-DnF2-Gm, and pvdF-DnR2-GWR were used for the ΔpvdF strain. Products were cloned into the EcoRI site of pEX18T and transferred into PA01 by conjugation. Genomic replacements were selected on LB medium lacking sodium chloride and containing 10% sucrose. The gentamicin resistance cassette was removed by FLP-mediated recombination as described by Choi and Schweizer (31). Replacements were verified by PCR. For the Δ3398 strain, these were PA33985′Up and PA33983′Dn. For ΔpvdF primers that annealed outside the cloned region were used in combination with primers annealing within the Gmr cassette; pvdFUpdiag, pvdFDndiag, GmRV1269, and GmF1311.
P. aeruginosa transposon mutants.
Strains were obtained from the University of Washington Genomes Sciences Department P. aeruginosa two-allele library and positions of the transposon insertions verified by the methods of Bailey and Manoil (30).
Functional gene and promoter cloning.
Cloning for complementation testing was carried out by designing PCR primers from the P. putida KT2440 genome, the DSM 3601 region encompassing finR-fprA region, the P. aeruginosa PA01 genome, or the E. coli W3110 genome. The corresponding genes were amplified using genomic DNA or colony suspension as a template. PCR products for CysDNCDSM3601, and finRKT2440, were ligated into the pGEM-T Easy vector (Promega, Madison, WI) according to the manufacturer's protocol. Cloned inserts were then excised and recloned into broad-host-range vectors with appropriate selective markers (vectors are listed in Table 1). For complementation of SO3B9 by E. coli cysJI, and P. aeruginosa fprA, the vector pJN105GW was used. The respective genes amplified with att sequence-containing primers and cloned into pDONR221 (Invitrogen, Carlsbad, CA) and the pDONR clone used for cloning into the destination vector as recommended by the supplier. Clones were mobilized from E. coli BW20767 into Pseudomonas strains by binary matings (32). Primer sequences are given in Table 2 and were used in the following combinations: for cysDNC amplification (mt-2 genomic DNA as a template), cysDF2 and cysNCRv2; for finR (mt-2 genomic DNA as a template), PP1637F and PP1637R; for finR (DSM 3601), PP1637FWXba and 1637RVXba; for fprA (DSM 3601) 1637W4Xba and fprARV810Xba; for P. aeruginosa finR, PA3398 NcoI 5′, PA3398 Xba 3′; for P. aeruginosa fprA, PA fprFW GWL, PAfprRV GWR; for E. coli cysJI, Ec cysJFW GWL2, Ec cysJRV GWR; and for the P. aeruginosa fprA promoter, Pfpr Bam L2, Pfpr EcoR.
Table 2.
PCR primers used in this study
| Primer | Sequence (5′-3′)a |
|---|---|
| cysDF2 | ATTCTAGACACCTGTTCATCGATTGCC |
| cysNCRv2 | GGATCCTTACTGACGCAGTACGTCCAACAC |
| PP1637F | ATTCTAGAAAAATGCCAAGGACATGGG |
| PP1637R | ATGGATCCGAACTGACCGTTCTCGAAGC |
| dgkFWSal | ATAGCTGTCGACATGACATCGCCATTCAAGG |
| dgkRVXba | TCTAGATCGCTTAAAGCAGGATCACC |
| fprAFWXba | TCTAGAGATATTGTCGCTGCCCCTAA |
| fprARVSal | ATAGCTGTCGACATGTCGCTGAACAGCTTGC |
| GmF1311 | GGCTCAAGTATGGGCATCAT |
| GmRV1269 | CAAGCGCGATGAATGTCTTA |
| PP1635FW2 | GCTGCTGGACTTGAACATGC |
| fprARV805 | TCGCCTTCTTCGTACCTACC |
| PP1637FWXba | ACCATCTAGAGATGGGGCTCCTGAAGAAA |
| 1637RVXba | ACCATCTAGAGTCGAAAAACGCCAAGGAC |
| 1637W4Xba | ACCATCTAGAACGAAGACTTGCAGTTGACG |
| fprARV810Xba | ACCATCTAGATACCTGCGCCTTATTTCTCG |
| KMR | TCAGCAACACCTTCTTCAG |
| KmlacZFW | GCCGCACTTGTGTATAAG |
| KmlacZRV | GGCCAGATCTGATCAAGA |
| PAfprFW GWL | TACAAAAAAGCAGGCTCCGCGTTTTCCTAGGAGTCT |
| PAfprRV GWR | TACAAGAAAGCTGGGTGGGCCGGAAAGCAGAAAG |
| Ec cysJFW GWL2 | TACAAAAAAGCAGGCTAACATAACGACGCATGACGA |
| Ec cysJIRV GWR | TACAAGAAAGCTGGGTCGCGTTCTTATCAGGCCTAC |
| PA3398-EcoRI3′Dn | GAATTCTACATGGCCGGCTACAGCTGG |
| PA3398-DnF-Gm | AGGAACTTCAAGATCCCCAATTCGTGACTCAGTTGGCCAGGGACAGGT |
| PA3398-UpR-Gm | TCAGACGCTTTTGAAGCTAATTCGCATCCAGGCTTCCTCGTCTAGAGC |
| PA3398-EcoRI5′Up | GAATTCAGGTGCTGCAGGCGCGAGGTC |
| Pfpr Bam L2 | ATGGATCCTGAATTTCATCCAGGCTTCC |
| Pfpr EcoR | ATGAATTCACACCAACAGCAGCAGAC |
| pvdF-UpF2-GWL | TACAAAAAAGCAGGCTCGCTTGGGATTGGTCATAGT |
| pvdF-UpR2-Gm | TCAGAGCGCTTTTGAAGCTGCGACACCTCTTCCTGATCT |
| pvdF-DnF2-Gm | AGGAACTTCAAGATCCCCAATTCGCTCCGGCCTTCTTCATTCT |
| pvdF-DnR2-GWR | TACAAGAAAGCTGGGTAAGACCGGCAAACGCTAC |
| pvdFUpdiag | GAGTGCAAGGCGTTGTTGAT |
| pvdFDndiag | GGTATGCGTCGACTACAACG |
| PA3398 NcoI 5′ | CTCCATGGAATTCACCCTCCGCCAGCTCG |
| PA3398 Xba 3′ | CGTCTAGACCGGATCGCCGGTGGCGCCG |
| PAO1fprfor1 | CCTGGAGTTCTTCAGCATCAA |
| PAO1fprrev1 | CTCGTAGCGCTCGTAGGTTTC |
| dsm3601fprfor | CTGAAGGAAGGCGATGAGA |
| dsm3601fprrev | CAGGTGCTCGGTGATGAA |
Underlined letters indicate engineered restriction sites.
Analytical procedures.
PDTC was assayed from culture supernatants as described previously (24). The detection limit for PDTC in culture supernatants was ∼2.5 μM. CAS medium was used to assess pyochelin production.
Northern analysis.
To measure fpr expression in response to sulfur sources by P. putida strains, overnight cultures grown in M9 succinate medium with 0.5 mM l-cystine were used to inoculate fresh cultures in the same medium and grown to an optical density at 600 nm (OD600) of 0.8. The cultures were washed in M9 succinate medium without sulfur and resuspended in 10 ml of either M9 succinate 0.5 mM l-cystine or M9 succinate 1 mM sulfate to an OD600 of 0.2 to 0.3. Cells were incubated in 50-ml baffle flasks with shaking for 2 h before harvesting for RNA extraction. For comparison of gene expression in response to paraquat, cells were grown in M9 succinate 1 mM sulfate, resuspended in 10 ml of the same medium to an OD600 of 0.3 (P. aeruginosa) or 0.1 to 0.3 (P. putida), and incubated in 50-ml flasks with shaking until an OD600 of 0.6 to 0.8 was reached. Those cultures were then washed once in M9 succinate without sulfur, before resuspending to an OD600 of 0.10 (P. aeruginosa) or 0.15 (P. putida) in 10-ml aliquots of M9 succinate 1 mM sulfate to which either no additives were included (control), or 0.5 mM l-cysteine, l-cystine, or d-cystine, or the same treatments plus 1 mM paraquat (N,N′-dimethyl-4,4′-bipyridinium dichloride; Sigma-Aldrich, Milwaukee, WI). Cells were incubated under those conditions for 30 min (P. putida) or for 10 min and 30 min (P. aeruginosa) before RNA extraction. RNA was extracted using the hot phenol method. Northern analysis was carried out using RNA (amounts of indicated in figure legends) run on 1% agarose morpholinepropanesulfonic acid denaturing gels (6.5% formaldehyde). Probes were generated by PCR using primers dsm3601fprfor, and dsm3601fprrev for fprAPp (P. putida DSM 3601 template) or PAO1fprfor1 and PAO1fprrev1 for fprAPa (P. aeruginosa MPA01 template). Probes were labeled using the BrightStar Psoralen-Biotin kit from Ambion (Austin, TX) and detected using the BrightStar BioDetect kit (Ambion) and a Kodak IS440 Imaging system.
RESULTS
Mutations generating Pdt phenotypes.
The approach taken to obtain mutants with altered PDTC production relied on PDTC-dependent bleaching of the iron-containing dye CAS (27) to produce “halos” surrounding siderophore-producing colonies. Strain BK8, defective in production of pyoverdine, was used for random transposon mutagenesis. Two screening media were used, one minimal medium formulation and one utilizing Casamino Acid supplementation. Approximately 9,000 transposon mutants were screened. Strains lacking halos were further characterized by quantitation of PDTC in a liquid minimal test medium (PM) that allowed normalization to growth yields. Two phenotypic categories were distinguished: class I mutants (nine isolates) gave no detectable PDTC, and class II mutants (two isolates) showed PDTC production significantly lower than the parental strain (Table 3). The locations of transposon insertions were determined by inverse PCR and sequencing of DNA flanking the various transposon insertion sites and are shown in Fig. 2. To confirm that the PDTC production phenotypes could be ascribed to the respective insertions, complementation experiments were undertaken with each class of mutant. These used either single genes or intact operons (Fig. 2) from DSM 3601 or P. putida mt-2, provided in multicopy. PDTC production data indicated successful trans complementation and therefore that the Pdt phenotypes observed were due to loss of the indicated gene products (Table 3).
Table 3.
PDTC production by wild type, Pdt mutants obtained by CAS screening, and trans complements on PM medium
| Strain | Genotype | Phenotypea | Avg PDTC production (μM/OD600) ± SDb |
|---|---|---|---|
| Wild type | |||
| DSM 3601 | Wild type | Pdt+ | 47.3 ± 9.0 |
| DSM 3601/pJB3Tc20 | Wild type/vector | Tcr control | 19.0 ± 0.9 |
| BK8 | pfrI (pvdS) | Pvd− | 40.8 ± 14.9 |
| Class I mutants | |||
| SEM1 | pfrI pdtI | Pvd−, Pdt− | BD |
| LL1 | pfrI cysG (cobA2) | Pvd−, Pdt−, Cys− | BD |
| SEM10D10 | pfrI cysD | Pvd−, Pdt−, Cys− (aux) | BD |
| LL3 | pfrI cysNC | Pvd−, Pdt−, Cys− | BD |
| LL3/pJB3TcGm | LL3/vector control | Pvd−, Pdt−, Cys− | BD |
| LL3/pJB3TcGm::cysDNC | LL3/cysDNC | Pvd− | 27.4 ± 1.6 |
| TL1 | pfrI mutL | Pvd−, Pdt−, Cys− | BD |
| Class II mutants | |||
| SO3B9 | pfrI finR | Pvd−, Pdt−, Cys (brad) | 4.7 ± 0.5 |
| SO3B9/pJB3TcGm | SO3B9/vector control | Pvd−, Pdt−, Cys (brad) | 4.4 ± 2.9 |
| SO3B9/pJB3TcGm::PP1637 | SO3B9/finR | Pvd− | 26.4 ± 3.4 |
aux, cysteine auxotroph; brad, cysteine bradytroph.
BD, below detection. Averages and standard deviations of at least three independent experiments are shown.
Fig 2.
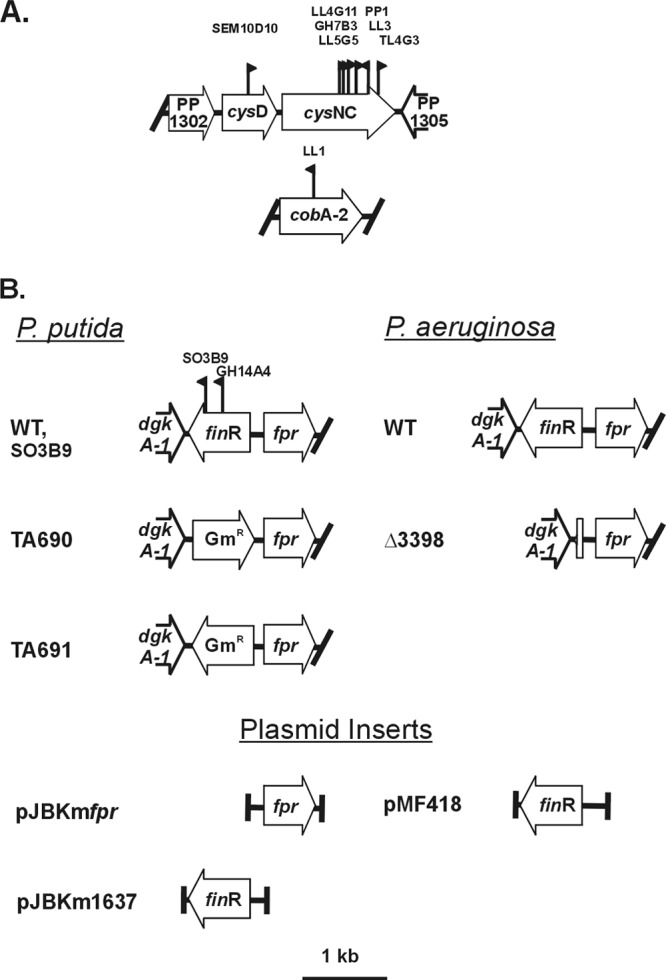
Genome segments and transposon insertions characterized. (A) Class I mutants; (B) class II mutants and P. putida and P. aeruginosa genomic segments analyzed involving the finR-fprA locus. Flag symbols indicate positions and orientations of mini-TnXylEKm insertions in respective mutants, with strain designations listed above. Half arrows above genes indicate the positions of PCR primers used to amplify genome segments for cloning/complementation.
Class I mutants.
Sulfate assimilation defects affect PDTC production by altering the mode of sulfur acquisition. With the single exception of the pdtI insertion described previously (9), all class I mutants obtained had defects in known sulfate assimilation genes. Both auxotrophs and prototrophs were found, defined by whether the strains could grow without preformed amino acid supplements.
Auxotrophic mutants.
The cobA2 gene, also denoted cysG in the literature regarding sulfate assimilation by enteric bacteria (25), is necessary for synthesis of the siroheme cofactor of sulfite reductase (CysI) and was identified as an auxotroph among the class I mutants. A hypermutator locus (mutL) was also retrieved in this screen. That strain (TL1) showed a Cys− phenotype, being unable to utilize sulfate or sulfite but able to utilize sulfide or cysteine as sources of sulfur (data not shown). The hypermutator effect of mutL defects makes it likely that a secondary mutation was responsible for the Cys phenotype, and consequently this mutant was not examined further.
Prototrophic mutants.
Insertions into the cysDNC operon, encoding subunits of adenosine phosphosulfate synthase, were the most frequently obtained class I mutations (Fig. 2). The cysDNC mutants were not capable of growth in minimal liquid medium with sulfate as sole sulfur source. Since the mutants grew on the screening medium lacking preformed amino acids, they were obviously capable of assimilating sulfur by some means. The only sulfur source present in CAS medium, other than sulfate (normally present in agar), was the pH buffer ingredient PIPES which contains sulfur in sulfonic acid groups. Pseudomonads are well known for their ability to utilize alkanesulfonates as sulfur sources (33, 34). The fact that strains limited to utilizing PIPES did not produce PDTC, whereas those capable of utilizing sulfate did indicated that the mode of sulfur assimilation can determine PDTC production. Utilization of alkanesulfonates as S sources requires induction of enzymes regulated as part of the sulfate starvation response; hence, their assignment as ssi (sulfate starvation-induced) genes (35). The wild-type and parental strains showed little or no detectable PDTC when provided with PIPES or methionine as sole sulfur sources (see Fig. S1 in the supplemental material and unpublished results), suggesting mutual exclusivity between the sulfate starvation response and PDTC production.
The reduced sulfur sources sulfite, sulfide, thiosulfate, or cysteine were also tested for their ability to support PDTC production by the wild type. These reduced sulfur sources were less effective than sulfate; however, the Pdt− phenotype of the cysNC and cobA2 mutants could be suppressed by those that supported growth (see Fig. S1 in the supplemental material). This eliminated APS (adenosine 5′-phosphosulfate), PAPS (2′-phosphoadenosine-5′-phosphosulfate), or sulfite as essential intermediates for PDTC biosynthesis since, for example, none of these compounds would be present in the cysNC or cobA2 mutants grown on sulfide or cysteine.
Class II mutants.
The FinR transcriptional regulator is required for wild-type PDTC production. The two class II mutants obtained contained disruptions of a conserved protein sharing homology with members of the LysR-type transcriptional regulator family FinR (PP1637). An ortholog of this gene was also found in a search for mutants defective in thioquinolobactin production by P. fluorescens ATCC 17400 (36). In P. putida FinR (fpr-inducing regulator) has been shown to regulate transcription of its upstream neighboring gene, fprA (ferredoxin:NADP+ oxidoreductase) in response to paraquat-induced oxidative stress (37). FinR-independent expression was also seen; FinR apparently being responsible for the bulk of the stress response but appreciable fprA expression was also observed in a finR truncation mutant (37). FprA is a redox mediator, shown to facilitate transfer of electrons between NADPH and ferredoxin (38). Mutants with defects in finR or fprA showed increased sensitivity to oxidative stress, and a growth defect in a minimal medium that could be suppressed by Casamino Acids (37). Sulfate assimilation is a reducing equivalent-intensive process. In enteric bacteria, sulfite reductase is a hetero-oligomeric enzyme, requiring the NADPH oxidoreductase activity of the CysJ flavoprotein, and the siroheme-containing CysI which directly reduces sulfite by six electrons to produce sulfide (25). The cysJIH genes form an operon on the E. coli chromosome. The 12 annotated Pseudomonas sp. genomes include cysI homologs that are not clustered with other cys genes (39). No cysJ homolog was identified in those genomes. Two genes encoding proteins with a high degree of sequence similarity to the E. coli CysJ (PA4513 and PA4130 [39]) were tested for roles in sulfate assimilation. Two mutants containing unique transposon insertions in each gene were found to be capable of growth on minimal media with sulfate as sole sulfur source (data not shown), indicating that neither PA4513 nor PA4130 are essential for sulfate assimilation by P. aeruginosa.
Plant CysI proteins have been shown to use reduced ferredoxin as a source of reducing power rather than a flavoprotein subunit such as CysJ (40). It is therefore likely that Pseudomonas Fpr proteins serve as components of an alternative CysI-reducing system. Based on the described properties of FinR, at least two alternative models can be invoked to explain how a defect in FinR may affect PDTC production; either indirectly due to a requirement for FprA to supply reducing equivalents for cysteine biosynthesis or directly via transcriptional regulation of pdt genes.
FprA is required for assimilatory sulfite reduction and PDTC biosynthesis.
We sought to resolve the effects due to fprA expression from other possible effects of finR mutation by constructing null mutants that varied in their potential for fprA expression (Fig. 2) and by complementation with fprA or with the E. coli cysJI genes. We were unsuccessful in constructing a complete deletion of fprA. Both ΔfinR alleles constructed in P. putida had identical chromosomal deletions encompassing the entire finR coding sequence plus upstream sequences that included the predicted fprA −35 promoter element (37) (Fig. 2). The resulting fprA gene lacked its native promoter, but was fused to an aacC1 gentamicin-resistance (Gmr) cassette which lacked a transcriptional terminator. In strain TA690, the Gmr cassette was oriented toward fprA (ΔfinR::Gmr→). In strain TA691 it was oriented oppositely (ΔfinR::Gmr←). Thus, the ΔfinR::Gmr→ strain was expected to show fprA expression under the control of the aacC1 promoter, whereas the ΔfinR::Gmr← strain was expected to have severely reduced fprA expression. Strains TA690 and TA691were compared to the originally isolated finR transposon insertion mutant, SO3B9 (finR::mini-Tn5), for fprA mRNA abundance and PDTC production. Since the finR::mini-Tn5 strain retained an intact fprA promoter, it should have retained FinR-independent fprA expression. Northern analysis of those strains confirmed robust expression of fpr-hybridizing RNA of a larger size than the wild-type transcript the ΔfinR::Gmr→ strain, and no detectable hybridization with RNA from the ΔfinR::Gmr← or finR::mini-Tn5 strains (Fig. 3). PDTC production was not impaired in the ΔfinR::Gmr→ strain, but no PDTC was detected from cultures of the ΔfinR::Gmr← strain (Table 4). Low but detectable levels of PDTC were seen with the finR::mini-Tn5 strain (Table 3). Those results recapitulate those which originally established FinR as a transcriptional regulator of fprA (37) and show that FprA is required for PDTC production, possibly through the assimilation of sulfate.
Fig 3.
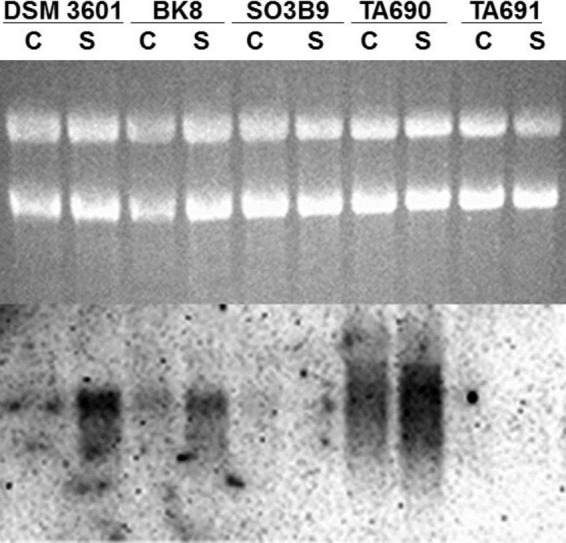
P. putida DSM 3601 fprA expression is dependent on sulfur source, as well as finR expression. Northern analysis of strains grown on M9 succinate medium with cystine (C) or sulfate (S) as sole sulfur sources. Strain genotypes are: DSM 3601, wild type; BK8, ΔpvdS (ΔpfrI) (pyoverdine-negative); SO3B9, finR::mini-Tn5; TA690, ΔfinR::Gmr (gentamicin resistance cassette oriented toward fprA); TA691, ΔfinR::Gmr (gentamicin resistance cassette oriented opposing fprA). Upper portion, ethidium-stained gel. Lower portion, Northern blot hybridized with fprA probe. 4.5 μg of RNA was loaded per lane.
Table 4.
PDTC production by finR-null mutants: complementation by cloned P. putida fprA and E. coli cysJI
| Strain | Genotype | Presence of arabinose | Avg PDTC production (μM/OD600) ± SDa |
|---|---|---|---|
| TA690 | pfrI ΔfinR::GmFW | − | 34.2 ± 8.5 |
| TA691 | pfrI ΔfinR::GmRV | − | BD |
| TA691/pJBKm1 | TA691/vector control | − | BD |
| TA691/pJBKm::finR | TA691/finRPp | − | BD |
| TA691/pJBKm::fprA | TA691/fprAPp | − | 28.1 ± 1.6 |
| SO3B9/pJN105 | pfrI finR/vector control | − | 5.9 ± 1.2 |
| SO3B9/pJN105 | pfrI finR/vector control | + | 10.0 ± 0.5 |
| SO3B9/pJN105::cysJI | SO3B9/arabinose-inducible cysJIEc | − | 31.0 ± 0.3 |
| SO3B9/pJN105::cysJI | SO3B9/arabinose-inducible cysJIEc | + | 41.5 ± 1.1 |
BD, below detection. Averages and standard deviations of triplicate assays are shown.
Comparisons of the growth of ΔfinR::Gmr→, ΔfinR::Gmr←, finR::mini-Tn5 and parental strains in minimal media are shown in Fig. 4. ΔfinR::Gmr→ showed growth that was indistinguishable from the parental strain, whereas ΔfinR::Gmr← and finR::miniTn5 strains showed decreased growth rates and an extended lag period on sulfate as sole sulfur source, with the ΔfinR::Gmr← strain showing the more severe impairment. The growth defects of the ΔfinR::Gmr← and finR::mini-Tn5 strains were suppressed by addition of cystine (Fig. 4) or thiosulfate, but not sulfite (data not shown).
Fig 4.
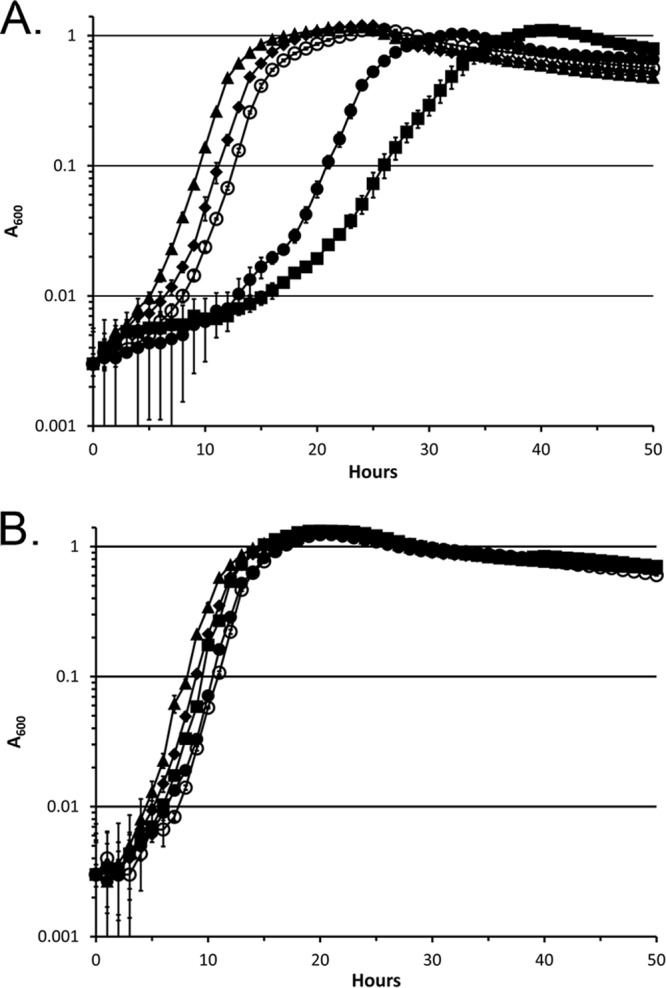
Growth of finR mutants with sulfate or cystine as sole sulfur sources. (A) M9 succinate medium with 1 mM sulfate as sulfur source; (B) M9 succinate medium with 0.5 mM cystine as sulfur source. Symbols: ○, DSM 3601 (WT); ◆, BK8 (Pvd− parental); ▲, TA690 (ΔfinR::Gmr-forward); ■, TA691 (ΔfinR::Gmr-reverse); ●, SO3B9 (finR::mini-Tn5KmxylE). The data are means of triplicate cultures. Error bars represent standard deviations.
The PDTC production defect and the cysteine bradytrophic phenotype of the ΔfinR::Gmr← strain could be complemented by fprA cloned in multicopy. However, finR cloned in the same vector could not complement either defect (Table 4 and data not shown). Those results are consistent with FinR acting as a transcriptional activator of fprA since the ΔfinR::Gmr← strain contains a truncation of the fprA promoter. The finR::mini-Tn5 strain was complemented for PDTC production by finR in trans (Table 3). The finR::mini-Tn5 strain was also complemented by either P. aeruginosa fprA or by E. coli cysJI cloned under the control of the araBAD promoter (Table 4; see also Fig. S2 in the supplemental material). Interestingly, while complementing the PDTC defect, cysJI only modestly improved the growth of the finR::mini-Tn5 strain and induction had a detrimental effect on growth by the mutant and wild-type strain with sulfate as sole sulfur source under the iron-limited conditions used (see Fig. S2 in the supplemental material). The data are consistent with a model in which FinR serves a role in PDTC production solely as a component of sulfite reduction/cysteine production. Transcriptional activation of fprA is critical but can be replaced by ectopic expression of E. coli cysJI. FinR is not necessary for pdt gene expression since the ΔfinR::Gmr→ strain showed significant PDTC production (Table 4).
Regulation of fpr in response to sulfur nutrition and oxidative stress.
The only previous description of the regulation of fprA expression in pseudomonads demonstrated finR-dependent induction in response to paraquat-induced oxidative stress (37). However, our observations indicated a role in sulfur metabolism and some effect of sulfur sources was seen in Northern analysis (Fig. 3). Those data indicated that fprA transcript abundance was higher in cultures grown with sulfate than with a reduced sulfur source (cystine; here we refer to sulfur at a formal oxidation state of 0 or less as ‘reduced'). We tested whether reduced sulfur sources had an effect on the robust, paraquat-induced stress response described by Lee et al. (37). This was done by exposing cells to paraquat in the presence of sulfate as sole sulfur source, or with added supplements of l-cysteine, l-cystine, or d-cystine. The results are shown in Fig. 5 in which a striking suppression of fprA induction was affected by l-cysteine or l-cystine supplementation, but not by d-cystine. The data indicated that intracellular reduced sulfur/cysteine levels can suppress paraquat-dependent fprA induction.
Fig 5.
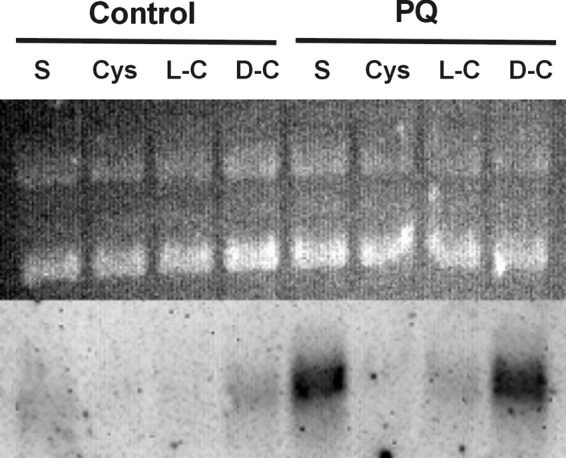
Reduced source of sulfur suppresses paraquat-induced fprA expression by P. putida DSM 3601. Northern analysis of control cultures grown on M9 succinate medium with sulfate as the sole sulfur source (S) or sulfate plus l-cysteine (Cys), l-cystine (L-C), or d-cystine (D-C). Paraquat-treated cultures (PQ) were exposed to 1 mM paraquat for 20 min with the respective sulfur supplements prior to RNA extraction. Upper portion, ethidium-stained gel. Lower portion, Northern blot hybridized with fprAPp probe. A total of 6 μg of RNA was loaded per lane.
To determine whether our observations of fprA transcription in P. putida were also relevant to other pseudomonads, we constructed a deletion of the P. aeruginosa PA01 finR gene (PA3398) and examined fprA expression in that species. P. aeruginosa also showed finR-dependent fprA expression that was repressed by reduced sulfur sources as shown by time course experiments with a chromosomally integrated reporter (fprA promoter fused to luxCDABE), and Northern analysis (Fig. 6). Comparison of a P. aeruginosa pyoverdine-defective mutant (i.e., the ΔpvdF mutant) and a ΔpvdF ΔfinR double mutant on CAS was used to assess affects of finR deletion on pyochelin production. Halo size was not significantly different between the two strains, i.e., finR mutants would not have been identified using the same methodology used to obtain the P. putida finR transposon mutants described in this work.
Fig 6.
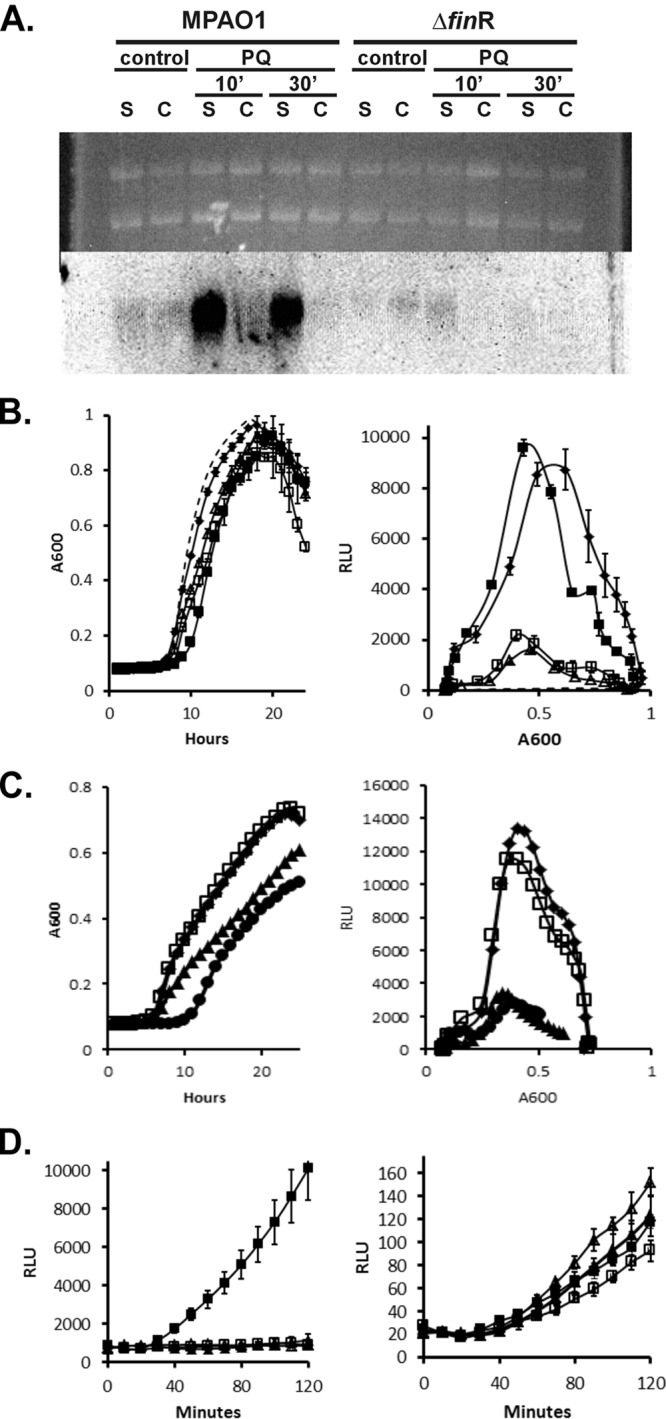
P. aeruginosa shows finR-dependent fprA expression which is responsive to reduced sulfur sources. (A) Northern analysis of cultures of wild type (MPA01) or finR deletion mutant (Δ3398) grown with sulfate as the sole sulfur source (S), or sulfate plus l-cystine (C). Paraquat-treated cultures (PQ) were exposed to 1 mM paraquat for 10 or 30 min with the respective sulfur supplements prior to RNA extraction. Upper portion, ethidium-stained gel; lower portion, Northern blot hybridized with an fprAPa probe. A total of 10 μg of RNA was loaded per lane. (B) Complementation analysis of the finR deletion strain Δ3398. Cultures were grown with sulfate as the sole sulfur source. The expression of a chromosomal luxCDABE operon fused to the PfprAPa promoter was monitored by luminescence. Left, growth; right, luminescence versus cell density (RLU, relative luminescence units). Symbols: ◆, MPA01; ▲, Δ3398; □, Δ3398/pMF54 (vector control); ■, 3398/pMF418 (plasmid-borne finRPa); dashed line, MPA01::Tn7lux control (no promoter). (C) Expression of PfprA-lux reporter by wild-type P. aeruginosa during growth with various sole sulfur sources. Symbols: ◆, sulfate; □, sulfite; ▲, cystine; ●, cysteine. (D) Expression of PfprA-lux reporter by wild type and ΔfinR mutant strains in response to paraquat and/or cysteine. Exponential-phase cells grown on minimal medium with sulfate as sulfur source were diluted into the same medium with or without additions of 1 mM paraquat or 0.5 mM cysteine. Left, wild type (MPA01); right, ΔfinR. Symbols: □, no addition; ■, 1 mM paraquat; △, cysteine; ▲, cysteine plus 1 mM paraquat.
DISCUSSION
Sulfur donors for PDTC biosynthesis.
The approach taken here and elsewhere (17), although aimed at further characterizing siderophore physiology, proved to be a rather effective means of identifying defects in sulfate assimilation since the majority of mutants isolated had Cys phenotypes. Although surprising, the screen for B. cepacia mutants defective in pyochelin production conducted by Farmer and Thomas (16) retrieved only a cysW (sulfate/thiosulfate transporter) mutant. That result and ours point out that transposon mutagenesis/screening protocols may not simply yield mutations based on target size alone; the pdt operon contains several genes that produce Pdt− phenotypes (41; T. Lewis, unpublished data) but only one insertion was retrieved.
Our collective data support a role for cysteine, rather than an intermediate of sulfate assimilation, as the sulfur donor for thiocarboxylate siderophore biosynthesis. The results obtained using mutants unable to produce intermediates of sulfate assimilation, or using media that circumvented their production by wild-type strains, eliminated all but sulfide as necessary substrates for PDTC production. It is not possible to resolve sulfide and cysteine for this role by the methods used since sulfide can be produced from cysteine through the activities of many enzymes in vivo. In fact, either may be chemically sufficient as sulfur donors for thiocarboxylate synthesis; however, the steady-state concentration of intracellular sulfide would be expected to be maintained at a relatively low level by the activities of the two known cysteine synthases (CysK and CysM) (25).
Our observations of PDTC synthesis are consistent with observations of B. cenocepacia (16) and P. aeruginosa (T. Lewis, unpublished data) in which cultures grown with various alternative sulfur sources such as methionine or organosulfonates did not produce detectable amounts of pyochelin. It is still unclear how this regulation is achieved; however, greater pyochelin biosynthetic gene transcription was seen by others comparing sulfate-grown and sulfamate-grown P. aeruginosa (42). The results are complicated by the fact that a pyochelin-proficient strain was used, making it possible that pyochelin had accumulated in the sulfate-amended cultures at the time of RNA extraction (i.e., autoinduction occurred due to pyochelin acting as a potent effector of pch gene induction) (43). Aside from transcriptional control, passive regulation of PDTC or pyochelin production could occur by virtue of PDTC biosynthetic enzymes having a higher Km for cysteine than cysteinyl tRNA synthase and/or other primary metabolic enzymes. The ssi sulfur sources likely do not allow the same rate of cysteine biosynthesis as sulfate, which would lead to decreased steady-state intracellular cysteine concentrations. It remains to be resolved whether active regulation or passive (kinetic) control are responsible for determining sulfur-containing siderophore production.
Implications for Pseudomonas sulfate assimilation.
Our findings have suggested a new scheme for sulfate assimilation by pseudomonads distinct from that of E. coli and enteric bacteria (Fig. 7). The identification of FprA as a component of bacterial sulfate assimilation supports the assertion that Pseudomonas CysI is a distinct type of sulfite reductase which partners with reduced ferredoxin, or FprA directly, rather than being CysJ dependent. No cysJ ortholog has been identified among 12 annotated Pseudomonas genomes (39), and transposon insertions in genes encoding proteins sharing the greatest identity with the E. coli CysJ in the P. aeruginosa genome do not confer Cys phenotypes. Ferredoxin-dependent sulfite reductases are characteristic of chloroplasts, some cyanobacteria, and other proteobacteria (44). fpr genes have been discovered as part of gene clusters that include the genes of sulfate assimilation (cysIHDN), as well as a putative ferredoxin-like protein (cysX) in Corynebacterium genomes (45). The fpr-2 gene of C. glutamicum was also shown to be coregulated with the sulfate assimilatory genes, and deletion of fpr-2 caused an increased lag time on minimal media. A ferredoxin-dependent sulfite reductase would not be the only aspect of Pseudomonas sulfate assimilation in common with chloroplasts: Pseudomonas sp. CysH are adenylylsulfate (APS) reductases (46) as opposed to phosphoadenylylsulfate (PAPS) reductase as is found in E. coli (25). However, it remains possible that the biochemistry of sulfite reduction in pseudomonads is unique. For example, it also remains possible that FprA transfers electrons directly to CysI. It has been shown that P. aeruginosa FprA efficiently transfers electrons from NADPH to heme oxygenase (47). Annotated conserved domains within bacterial and chloroplast CysI proteins include “ferredoxin-like” sequences (48).
Fig 7.
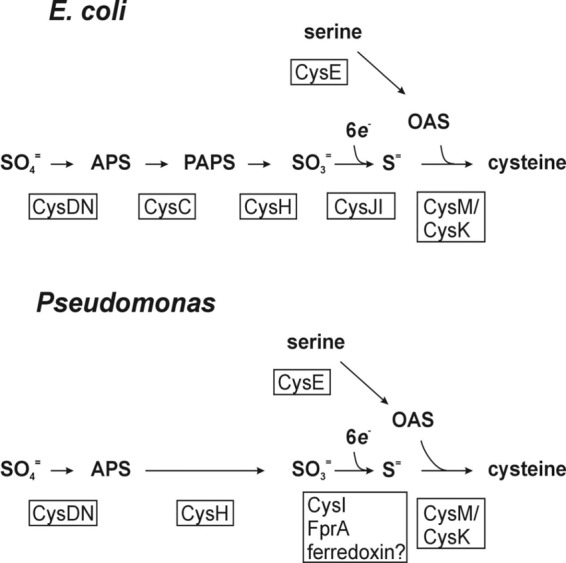
Pathway of sulfate assimilation/cysteine biosynthesis in enterics versus pseudomonads. Intermediates of cysteine biosynthesis are shown in bold. Gene products with relevant enzymatic activities (CysDN, CysC, etc.) are given below or beside the respective steps in boxes. Abbreviations: APS, adenosine phosphosulfate; PAPS, phosphoadenosine phosphosulfate; OAS, O-acetylserine. The negative allosteric regulation of CysE activity by intracellular cysteine, and the requirement for N-acetylserine (a product of spontaneous rearrangement of OAS) to induce sulfate assimilatory gene expression (via CysB), affords coordination between serine activation and sulfate activation/reduction activities in response to metabolic demand for cysteine (46).
Significance of FprA in pseudomonads.
The fact that growth on sulfate is still observed in strains showing severe fprA deficiency (e.g., P. putida TA691 or the P. putida KT2440 fprA truncation/merodiploid strain described by Lee et al. [37]) may be explained by the presence of less-effective, alternative means of transfer of electrons to CysI. These may include other flavoproteins or ferredoxins. Pseudomonads possess a second annotated NADP(H):ferredoxin reductase, FprB. FprA and FprB of P. putida have been examined for their relative abilities to reduce cellular redox mediators such as ferredoxins (both [2Fe:2S] and [4Fe:4S]) and flavodoxin (49). It was found that FprA served most efficiently as a flavodoxin reductase and that FprB was most effective at reducing the product of fdx-2 (referred to as FdA), and that FprB is also a ferric reductase (50). Sulfate assimilation by FprA− strains may also be due to remnant FprA activity. In order to resolve these possibilities, we attempted to construct complete deletions of fprA in the P. putida DSM 3601 and P. aeruginosa MPA01 backgrounds using the same approaches used to generate finR deletion strains but were unsuccessful. These results raise the possibility that some FprA activity must be present for viability. In addition, the alternatives to FprA may differ among pseudomonads. The P. aeruginosa finR mutant showed a strong defect in fprA expression but had a much milder growth phenotype (Fig. 7) and no significant siderophore phenotype (CAS halo size) compared to P. putida finR mutants.
Oxidative stress and sulfur assimilation.
In the present study we also observed a linkage between an oxidative stress response (paraquat-induced fpr expression) and sulfur metabolism. Oxidative stress and sulfur assimilation could be expected to be linked due to turnover of iron-sulfur clusters and glutathione resulting from intracellular oxidative damage. Purely chemical linkage is also expected due to general reactivity between oxidants and cysteine (51, 52). This linkage has been observed in studies of transcriptional and proteomic responses of pseudomonads to various sulfur sources (33, 42). It is not yet clear whether fprA expression is integrated into cellular stress responses or exclusively responds to cysteine biosynthetic demands. P. putida FinR has been characterized in order to define its ability to respond to paraquat-induced oxidative stress (38). Although extensive site-directed mutagenesis was carried out, a model involving reversible disulfide bond formation was not supported. We have shown that finR-dependent, paraquat-induced fprA expression was suppressed by reduced sulfur sources (Fig. 6 and 7). It would be interesting to know whether FinR promoter-binding affinity responds to an effector or effectors derived from specific intracellular sulfur metabolites. Potential effectors could include oxidized sulfur species such as sulfoxides or sulfinates. Considering its importance for sulfate assimilation, it is also of interest to know whether fprA is part of the CysB regulon in pseudomonads.
Supplementary Material
ACKNOWLEDGMENTS
This study was supported by the National Institutes of Health through Montana INBRE (2 P20 RR 016455-09) and the Montana State University Billings Research and Creative Endeavors program.
We also thank Kerry Williamson, Paula Austin, Hadley Hartwell, Grant Henderson, Lynne Leach, Sergio Morales, Stephanie Onyekaba, Parvez Pothiawala, and Chunxiao Yu for their technical assistance.
Footnotes
Published ahead of print 21 June 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.00528-13.
REFERENCES
- 1.Lee C-H, Lewis TA, Paszczynski A, Crawford RL. 1999. Identification of an extracellular catalyst of carbon tetrachloride dehalogenation from Pseudomonas stutzeri strain KC as pyridine-2,6-bis(thiocarboxylate). Biochem. Biophys. Res. Commun. 261:562–566 (Erratum, 265:770.) [DOI] [PubMed] [Google Scholar]
- 2.Ockels W, Römer Budzikiewicz AH. 1978. An Fe(III) complex of pyridine-2,6-di-(monothiocarboxylic acid): a novel bacterial metabolic product. Tetrahedron Lett. 36:3341–3342 [Google Scholar]
- 3.Cortese M, Paszczynski A, Lewis TA, Sebat J, Crawford RL. 2002. Metal chelating properties of pyridine-2,6-bis(thiocarboxylic acid) produced by Pseudomonas spp. and the biological activities of the formed complexes. Biometals 15:103–120 [DOI] [PubMed] [Google Scholar]
- 4.Leach L, Lewis TA. 2007. The role of the siderophore pyridine-2,6-bis (thiocarboxylic acid) (PDTC) in zinc utilization by Pseudomonas putida DSM 3601. Biometals 20:717–726 [DOI] [PubMed] [Google Scholar]
- 5.Lewis TA, Paszczynski A, Gordon-Wylie S, Jeedigunta S, Lee C-H, Crawford RL. 2001. Carbon tetrachloride dechlorination by the bacterial transition metal chelator pyridine-2,6-bis(thiocarboxylic acid). Environ. Sci. Technol. 35:552–559 [DOI] [PubMed] [Google Scholar]
- 6.Stolworthy JC, Paszczynski A, Korus RA, Crawford RL. 2001. Metal binding by pyridine-2,6-bis(monothiocarboxylic acid), a biochelator produced by Pseudomonas stutzeri and Pseudomonas putida. Biodegradation 12:411–418 [DOI] [PubMed] [Google Scholar]
- 7.Criddle CS, DeWitt JT, Grbic-Galic D, McCarty PL. 1990. Transformation of carbon tetrachloride by Pseudomonas sp. strain KC under denitrification conditions. Appl. Environ. Microbiol. 56:3240–3246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sepulveda-Torres LC, Huang A, Kim H, Criddle CS. 2002. Analysis of regulatory elements and genes required for carbon tetrachloride degradation in Pseudomonas stutzeri strain KC. J. Mol. Microbiol. Biotechnol. 4:151–161 [PubMed] [Google Scholar]
- 9.Leach L, Lewis TA. 2006. Identification and characterization of Pseudomonas membrane transporters necessary for utilization of the siderophore pyridine-2,6-bis(thiocarboxylic acid) (PDTC). Microbiology 152:3157–3166 [DOI] [PubMed] [Google Scholar]
- 10.Morales SE, Lewis TA. 2006. Transcriptional regulation of the pdt gene cluster of Pseudomonas stutzeri KC involves an AraC/XylS family transcriptional activator (PdtC) and the cognate siderophore pyridine-2,6-bis(thiocarboxylic acid) (PDTC). Appl. Environ. Microbiol. 72:6994–7002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Crosa JH, Walsh CT. 2002. Genetics and assembly line enzymology of siderophore biosynthesis in bacteria. Microbiol. Mol. Biol. Rev. 66:223–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cornelis P, Matthijs S. 2002. Diversity of siderophore-mediated iron uptake systems in fluorescent pseudomonads: not only pyoverdines. Environ. Microbiol. 4:787–798 [DOI] [PubMed] [Google Scholar]
- 13.Stintzi A, Evans K, Meyer J-M, Poole K. 1998. Quorum-sensing and siderophore biosynthesis in Pseudomonas aeruginosa: lasR/lasI mutants exhibit reduced pyoverdine biosynthesis. FEMS Microbiol. Lett. 166:341–345 [DOI] [PubMed] [Google Scholar]
- 14.Juhas M, Wiehlmann L, Huber B, Jordan D, Lauber J, Salunkhe P, Limpert AS, von Götz F, Steinmetz I, Eberl L, Tümmler B. 2004. Global regulation of quorum sensing and virulence by VqsR in Pseudomonas aeruginosa. Microbiology 150:831–841 [DOI] [PubMed] [Google Scholar]
- 15.Lewenza S, Sokol P. 2001. Regulation of ornibactin biosynthesis and N-acyl-l-homoserine lactone production by CepR in Burkholderia cepacia. J. Bacteriol. 183:2212–2218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Farmer KL, Thomas MS. 2004. Isolation and characterization of Burkholderia cenocepacia mutants deficient in pyochelin production: pyochelin biosynthesis is sensitive to sulfur availability. J. Bacteriol. 186:270–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Matthijs S, Tehrani KA, Laus G, Jackson RW, Cooper RM, Cornelis P. 2007. Thioquinolobactin, a Pseudomonas siderophore with antifungal and anti-pythium activity. Environ. Microbiol. 9:425–434 [DOI] [PubMed] [Google Scholar]
- 18.Lewis TA, Cortese M, Sebat J, Green T, Lee C-H, Crawford RL. 2000. A Pseudomonas stutzeri gene cluster encoding the biosynthesis of the CCl4-dechlorination agent pyridine-2,6-bis(thiocarboxylic acid). Environ. Microbiol. 2:407–416 [DOI] [PubMed] [Google Scholar]
- 19.Begley TP, Xi J, Kinsland C, Taylor SM, McLafferty FW. 1999. The enzymology of sulfur activation during thiamin and biotin biosynthesis. Curr. Opin. Chem. Biol. 3:623–629 [DOI] [PubMed] [Google Scholar]
- 20.Leimkühler S, Rajagopalan KV. 2001. A sulfurtransferase is required in the transfer of cysteine sulfur in the in vitro synthesis of molybdopterin from precursor Z in Escherichia coli. J. Biol. Chem. 276:22024–22031 [DOI] [PubMed] [Google Scholar]
- 21.Taylor SM, Kelleher NL, Kinsland C, Chiu H-J, Costello CA, Backstrom AD, McLafferty FW, Begley TP. 1998. Thiamin biosynthesis in Escherichia coli: identification of ThiS thiocarboxylate as the immediate sulfur donor in the thiazole formation. J. Biol. Chem. 273:16555–16560 [DOI] [PubMed] [Google Scholar]
- 22.Hildebrand U, Taraz K, Budzikiewicz H. 1986. 6-(Hydroxythio)carbonylpyridine-2-carboxylic acid and pyridine-2-carboxylic-6-monothiocarboxylic acid as intermediates in the biosynthesis of pyridine-2,6-di(monothiocarboxylic acid) from pyridine-2,6-dicarboxylic acid. Z. Naturforsch. 41c:691–694 [Google Scholar]
- 23.Budzikiewicz H. 2003. Heteroaromatic monothiocarboxylic acids from Pseudomonas spp. Biodegradation 14:65–72 [DOI] [PubMed] [Google Scholar]
- 24.Lewis TA, Leach L, Morales SE, Austin PR, Hartwell HJ, Kaplan B, Forker C, Meyer J-M. 2004. Physiological and molecular genetic evaluation of the dechlorination agent, pyridine-2,6-bis(monothiocarboxylic acid) (PDTC) as a secondary siderophore of Pseudomonas. Environ. Microbiol. 6:159–169 [DOI] [PubMed] [Google Scholar]
- 24a.Maniatis T, Sambrook J, Fritsch EF. 1982. Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 25.Kredich NM. 1987. Biosynthesis of cysteine, p 419–428 In Neidhardt NC, Curtiss R, III, Ingraham JL, Lin ECC, Low KB, Magasanik B, Reznikoff WS, Riley M, Schaechter M, Umbarger HE. Escherichia coli and Salmonella: cellular and molecular biology, 2nd ed American Society for Microbiology, Washington, DC [Google Scholar]
- 26.Nelson KE, Weinei C, Paulsen IT, Dodson RJ, Hilbert H, Martins dos Santos VAP, Fouts DE, Gill SR, Pop M, Holmes M, Brinkac L, Beanan M, DeBoy RT, Daugherty S, Kolonay J, Madupu R, Nelson W, White O, Peterson J, Khouri H, Hance I, Chris Lee P, Holtzapple E, Scanlan D, Tran K, Moazzez A, Utterback T, Rizzo M, Lee K, Kosack D, Moestl D, Wedler H, Lauber J, Stjepandic D, Hoheisel J, Straetz M, Heim S, Kiewitz C, Eisen J, Timmis K, Düsterhöft A, Tümmler B, Fraser CM. 2002. Complete genome sequence and comparative analysis of the metabolically versatile Pseudomonas putida KT2440. Environ. Microbiol. 4:799–808 [DOI] [PubMed] [Google Scholar]
- 27.Schwyn B, Neilands JB. 1987. Universal chemical assay for the detection and determination of siderophores. Anal. Biochem. 160:47–56 [DOI] [PubMed] [Google Scholar]
- 28.DeLorenzo V, Herrero M, Jakubzik U, Timmis K. 1990. Mini-Tn5 transposon derivatives for insertion mutagenesis, promoter probing, and chromosomal insertion of cloned DNA in Gram-negative eubacteria. J. Bacteriol. 172:6568–6572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.O'Toole GA, Pratt LA, Watnick PI, Newman DK, Weaver VB, Kolter R. 1999. Genetic approaches to study of biofilms. Methods Enzymol. 310:91–109 [DOI] [PubMed] [Google Scholar]
- 30.Bailey J, Manoil CJ. 2002. Genome-wide internal tagging of bacterial exported proteins. Nat. Biotechnol. 20:839–842 [DOI] [PubMed] [Google Scholar]
- 31.Choi K-H, Schweizer HP. 2006. Mini-Tn7 insertion in bacteria with single attTn7 sites: example Pseudomonas aeruginosa. Nat. Protoc. 1:153–161 [DOI] [PubMed] [Google Scholar]
- 32.Metcalf WW, Jiang W, Daniels LL, Kim SK, Haldimann A, Wanner BL. 1996. Conditionally replicative and conjugative plasmids carrying lacZ alpha for cloning, mutagenesis, and allele replacement in bacteria. Plasmid 35:1–13 [DOI] [PubMed] [Google Scholar]
- 33.Quadroni M, James P, Dainese-Hatt P, Kertesz MA. 1999. Proteome mapping, mass spectrometric sequencing and reverse transcription-PCR for characterization of the sulfate starvation-induced response in Pseudomonas aeruginosa PAO1. Eur. J. Biochem. 266:986–996 [DOI] [PubMed] [Google Scholar]
- 34.Endoh T, Habe H, Yoshida T, Nojiri H, Omori T. 2003. A CysB-regulated and sigma 54-dependent regulator, SfnR, is essential for dimethyl sulfone metabolism of Pseudomonas putida strain DS1. Microbiol. 149:991–1000 [DOI] [PubMed] [Google Scholar]
- 35.Hummerjohann J, Kuttel E, Quadroni M, Ragaller J, Leisinger T, Kertesz MA. 1998. Regulation of the sulfate starvation response in Pseudomonas aeruginosa: role of cysteine biosynthetic intermediates. Microbiol. 144:1375–1386 [DOI] [PubMed] [Google Scholar]
- 36.Matthijs S. 2002. The in-vitro antagonism of Pseudomonas fluorescens ATCC17400 against Pythium debaryanum: the role of trehalose in the interaction between both microorganims, and the identificaiton of the siderophores pyoverdine and quinolobactin as the antagonistic compounds. Ph.D. thesis Vrije Universiteit Brussels, Brussels, Belgium [Google Scholar]
- 37.Lee Y, Pena-Llopis S, Kang Y-S, Shin H-D, Demple B, Madsen EL, Jeon CO, Park W. 2006. Expression analysis of the fpr (ferredoxin-NADP+ reductase) gene in Pseudomonas putida KT2440. Biochem. Biophys. Res. Commun. 339:1246–1254 [DOI] [PubMed] [Google Scholar]
- 38.Yeom S, Yeom J, Park W. 2010. Molecular characterization of FinR, a novel redox-sensing transcriptional regulator in Pseudomonas putida KT2440. Microbiology 156:1487–1496 [DOI] [PubMed] [Google Scholar]
- 39.Winsor GL, Lam DK, Fleming L, Lo R, Whiteside MD, Yu NY, Hancock RE, Brinkman FS. 2011. Pseudomonas Genome Database: improved comparative analysis and population genomics capability for Pseudomonas genomes. Nucleic Acids Res. 39:596–600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Taiz L, Zeiger E. 2010. Plant physiology. Sinauer Associates, Sunderland, MA [Google Scholar]
- 41.Sepulveda-Torres LdC, Rajendran N, Dybas MJ, Criddle CS. 1999. Generation and initial characterization of Pseudomonas stutzeri KC mutants with impaired ability to degrade carbon tetrachloride. Arch. Microbiol. 171:424–429 [DOI] [PubMed] [Google Scholar]
- 42.Tralau T, Vuillemier S, Thibault C, Campbell BJ, Hart CA, Kertesz MA. 2007. Transcriptomic analysis of the sulfate starvation response of Pseudomonas aeruginosa. J. Bacteriol. 189:6743–6750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Michel L, Gonzales N, Jagdeep Nguyen-Ngoc ST, Reimmann C. 2005. PchR-box recognition by the AraC-type regulator PchR of Pseudomonas aeruginosa requires the siderophore pyochelin as an effector. Mol. Microbiol. 58:495–509 [DOI] [PubMed] [Google Scholar]
- 44.Neumann S, Wynen A, Truper HG, Dahl C. 2000. Characterization of the cys gene locus from Allochromatium vinosum indicates an unusual sulfate assimilation pathway. Mol. Biol. Rep. 27:27–33 [DOI] [PubMed] [Google Scholar]
- 45.Rückert C, Koch DJ, Rey DA, Albersmeier A, Mormann S, Pühler A, Kalinowski J. 2005. Functional genomics and expression analysis of the Corynebacterium glutamicum fpr2-cysIXHDNYZ gene cluster involved in assimilatory sulphate reduction. BMC Genomics 6:1–18. 10.1186/1471-2164-6-121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bick JA, Dennis JJ, Zylstra GJ, Nowack J, Leustek T. 2000. Identification of a new class of 5′-adenylylsulfate (APS) reductases from sulfate-assimilating bacteria. J. Bacteriol. 182:135–142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang A, Zeng Y, Han H, Weeratunga S, Morgan BN, Moënne-Loccoz P, Schönbrunn E, Rivera M. 2007. Biochemical and structural characterization of Pseudomonas aeruginosa Bfd and FPR: ferredoxin NADP+ reductase and not ferredoxin Is the redox partner of heme oxygenase under iron-starvation conditions. Biochem 46:12198–12211 [DOI] [PubMed] [Google Scholar]
- 48.Marchler-Bauer A, Lu S, Anderson JB, Chisaz F, Derbyshire MK, DeWeese-Scott C, Fong JH, Geer LY, Geer RC, Gonzales NR, Gwadz M, Jurwitz DI, Jackson JD, Ke Z, Lanczycki CJ, Lu F, Marchler GH, Mullokandov M, Omelchenko MV, Robertson CL, Song JS, Thanki N, Yamashita RA, Zhang N, Zheng C, Bryant SH. 2011. CDD: a Conserved Domain Database for the functional annotation of proteins. Nucleic Acids Res. 39:225–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yeom J, Jeon CO, Madsen EL, Park W. 2009. In vitro and in vivo interactions of ferredoxin-NADP+ reductases in Pseudomonas putida. J. Biochem. 145:481–491 [DOI] [PubMed] [Google Scholar]
- 50.Yeom J, Jeon CO, Madsen EL, Park W. 2009. Ferredoxin-NADP reductase from Pseudomonas putida functions as a ferric reductase. J. Bacteriol. 191:1472–1479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ohtsu I, Wiriyathanawudhiwong N, Nakatani T, Takagi H. 2010. The l-cysteine/l-cystine shuttle system provides reducing equivalents to the periplasm in Escherichia coli. J. Biol. Chem. 285:17479–17487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Park S, Imlay J. 2003. High levels of intracellular cysteine promote oxidative DNA damage by driving the Fenton reaction. J. Bacteriol. 185:1942–1950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Williams PA, Murray K. 1974. Metabolism of benzoate and the methylbenzoates by Pseudomonas putida (arvilla) mt-2: evidence for the existence of a TOL plasmid. J. Bacteriol. 120:416–423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jacobs MA, Alwood A, Thaipisuttikul I, spencer D, Haugen E, Ernst S, Will O, Kaul R, Raymond C, Levy R, Chun-Rong L, Guenther D, Bovee D, Olson MV, Manoil CJ. 2003. Comprehensive transposon mutant library of Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. U. S. A. 100:14339–14344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schweizer HP. 1993. Small broad-host-range gentamicin resistance gene cassettes for site-specific insertion and deletion mutagenesis. Biotechniques 15:831–833 [PubMed] [Google Scholar]
- 56.Blatny JM, Brautaset T, Winther-Larsen HC, Haugan K, Valla S. 1997. Construction and use of a versatile set of broad-host-range cloning and expression vectors based on the RK2 replicon. Appl. Environ. Microbiol. 63:370–379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Miller V, Mekalanos J. 1988. A novel suicide vector and its use in construction of insertion mutations: osmoregulation of outer membrane proteins and virulence determinants in Vibrio cholerae requires toxR. J. Bacteriol. 170:2575–2583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Newman JR, Fuqua C. 1999. Broad-host-range expression vectors that carry the l-arabinose-inducible Escherichia coli ara BAD promoter and the araC regulator. Gene 227:197–203 [DOI] [PubMed] [Google Scholar]
- 59.Hoang TT, Karkhoff-Schweizer R, Kutchma AJ, Schweizer HP. 1998. A broad-host-range Flp-FRT recombination system for site-specific excision of chromosomally located DNA sequences: application for isolation of unmarked Pseudomonas aeruginosa mutants. Gene 212:77–86 [DOI] [PubMed] [Google Scholar]
- 60.Franklin MF, Chitnis CE, Gacesa P, Sonesson A, White DC, Ohman DE. 1994. Pseudomonas aeruginosa AlgG is a polymer level alginate C5-mannuronan epimerase. J. Bacteriol. 176:1821–1830 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.