

Abstract
A stochastic, agent-based mathematical model of the coevolution of the archaeal and bacterial adaptive immunity system, CRISPR-Cas, and lytic viruses shows that CRISPR-Cas immunity can stabilize the virus-host coexistence rather than leading to the extinction of the virus. In the model, CRISPR-Cas immunity does not specifically promote viral diversity, presumably because the selection pressure on each single proto-spacer is too weak. However, the overall virus diversity in the presence of CRISPR-Cas grows due to the increase of the host and, accordingly, the virus population size. Above a threshold value of total viral diversity, which is proportional to the viral mutation rate and population size, the CRISPR-Cas system becomes ineffective and is lost due to the associated fitness cost. Our previous modeling study has suggested that the ubiquity of CRISPR-Cas in hyperthermophiles, which contrasts its comparative low prevalence in mesophiles, is due to lower rates of mutation fixation in thermal habitats. The present findings offer a complementary, simpler perspective on this contrast through the larger population sizes of mesophiles compared to hyperthermophiles, because of which CRISPR-Cas can become ineffective in mesophiles. The efficacy of CRISPR-Cas sharply increases with the number of proto-spacers per viral genome, potentially explaining the low information content of the proto-spacer-associated motif (PAM) that is required for spacer acquisition by CRISPR-Cas because a higher specificity would restrict the number of spacers available to CRISPR-Cas, thus hampering immunity. The very existence of the PAM might reflect the tradeoff between the requirement of diverse spacers for efficient immunity and avoidance of autoimmunity.
INTRODUCTION
The ubiquitous arms race between viruses and their hosts to a large extent shapes the evolution of both (1–3). All cellular life forms have evolved numerous, extremely diverse, and elaborate antiviral defense systems that occupy a substantial part of the genome, at least in free-living organisms (4–6). Although some widespread defense mechanisms of bacteria and archaea, in particular the restriction-modification systems, have been known for many years and thoroughly characterized, recent advances in comparative genomics and experimental study of virus-host interaction have revealed new antiviral defense mechanisms some of which function on novel, unexpected principles (6–10). Arguably, the foremost of these new advances is the discovery of the adaptive immunity system that became known as CRISPR (clustered regularly interspaced short palindromic repeats)-Cas (CRISPR-associated genes) (11–15).
The CRISPR-Cas system employs a unique defense mechanism that involves incorporation of virus DNA fragments into CRISPR repeat arrays and subsequent utilization of transcripts of these inserts (spacers) as guide RNAs to cleave the cognate virus genome (16–20). Thus, CRISPR-Cas represents bona fide adaptive immunity that until the discovery of this system has not been known to exist in prokaryotes (21). However, an important distinction between CRISPR-Cas and animal immune systems is that CRISPR-Cas modifies the host organism's genome in response to infection and hence provides heritable immunity. Thus, CRISPR-Cas is the most compelling known case of Lamarckian inheritance whereby an organism responds to an environmental cue by generating a heritable modification of the genome that provides an adaptive response to that specific cue (22). The role of CRISPR-Cas in antivirus defense in archaea and bacteria that initially was predicted on the basis of the detection of spacers identical to short sequence segments from virus and plasmid genomes and comparative analysis of Cas protein sequences (19) has been successfully demonstrated experimentally (23). In the few years that elapsed since this key breakthrough, the CRISPR research evolved into a highly dynamic field of microbiology with major potential for applications in epidemiology, biotechnology and genome engineering (24–26). The first applications of CRIPSR-Cas for genome manipulation and gene expression programming already have been developed (27–30).
The CRISPR-Cas system shows an enormous diversity of Cas protein repertoires and the architectures of the respective genomic loci. Comparative analysis of the sequences and structures of Cas proteins, combined with the analysis of genomic architectures, led to the classification of the CRISPR-Cas systems into three distinct types (I, II, and III) and several still unclassified minor variants (31, 32). For each type and subtype, a specific signature gene has been identified allowing straightforward classification of the highly variable CRISPR-Cas loci in the course of genome analysis (32).
The mechanism of CRISPR-Cas is usually divided into three stages: (i) adaptation, when new 30 to 84 bp long, unique spacers homologous to proto-spacer sequences in viral genomes or other alien DNA molecules are integrated into the CRISPR repeat cassettes; (ii) expression and processing of pre-crRNA into short guide crRNAs; and (iii) interference, when the alien DNA or RNA is targeted by a complex of Cas proteins containing a crRNA guide and cleaved within the unique target site (15, 32, 33).
Viruses can evade CRISPR-Cas through minimal mutational or recombinational changes in proto-spacer regions. In several experiments, single proto-spacer mutations have rendered CRISPR-Cas ineffectual (23, 34, 35), although other CRISPR-Cas systems showed less rigid specificity (36). Conversely, hosts can regain antiviral immunity through new spacer additions (34, 35, 37, 38), thus driving coevolutionary arms races between the mutating virus and the spacer-incorporating host. This arms race apparently can go multiple rounds and takes unexpected turns as demonstrated by the recent finding that certain bacteriophages encode their own CRISPR-Cas system which targets host innate immunity loci, thus turning a defense mechanism into an assault weapon (39).
The CRISPR-Cas systems show a remarkably nonuniform distribution among prokaryotes, with nearly all sequenced hyperthermophiles (mostly archaea) but <50% of the mesophiles (largely bacteria) encompassing CRISPR-Cas loci (19, 32, 40). In bacteria, the CRISPR-Cas loci demonstrate notable evolutionary volatility, with many cases reported when some of several closely related bacterial strains possessed CRISPR-Cas, but the others lacked it (41, 42). Numerous cases of apparent horizontal gene transfer (HGT) of CRISPR-Cas loci also have been reported (43, 44). Furthermore, the CRISPR-Cas loci have been shown to abrogate acquisition of foreign DNA via HGT (25, 45) and consequently are rapidly lost under selective pressure for HGT as demonstrated by the propagation of antibiotic-resistant CRISPR− strains of Enterococcus faecalis derived from a CRISPR+ progenitor in a hospital environment (46). Rapid acquisition and loss of CRISPR spacers leading to intrapopulation heterogeneity also has been observed in experiments on both archaeal (47) and bacterial (48) models. Findings like these introduce the more general subject of the fitness cost incurred by the maintenance of the CRISPR-Cas loci (40, 49) that, in addition to the curtailment of HGT, is likely to involve the strong deleterious effect of autoimmunity caused by an occasional incorporation of proto-spacers from the self-DNA (50, 51).
The arms race between the immune system and viruses, the common events of loss and horizontal transfer of CRISPR-Cas loci and the fitness cost apparently incurred by CRISPR-Cas combine to yield complex evolutionary dynamics. These types of dynamics provide fertile ground for mathematical modeling with a potential to elucidate the interactions between different evolutionary processes and possibly discover unexpected evolutionary regimes. Thus, recently, several mathematical models of CRISPR-Cas-virus coevolution have been developed and studied, using different assumptions and approaches. Essentially, these modeling efforts focused on explaining the striking features of the CRISPR-Cas systems that became apparent through comparative genomic analyses (52), namely, their fast evolution, enormous diversity and old end uniformity.
Kupczok and Bollback used maximum-likelihood estimates to analyze purely mechanistic models of CRISPR evolution in which spacers are added and removed stochastically (53). The fits of the model to the observed CRISPR-Cas loci content in collections of closely related bacteria yielded estimates of the spacer addition and deletion rates and indicated that single spacer deletions were more likely than deletions of groups of spacers.
He and Deem were the first to introduce a stochastic population model with explicit CRISPR dynamics (54) to analyze the dependence of the spacer diversity on the relative position in the CRISPR array. Their approach has substantial limitations in that the CRISPR cassettes in the model have an unrealistically short fixed CRISPR length (as few as two CRISPR units in the mean field approach) and the immunity is decoupled from the virus growth rate. In a follow-up study, the model was extended to include the effect of viral recombination (52).
A similar approach, but with an explicit coupling between immunity and virus growth rate, also resulted in the observation that leading spacers were more likely to confer immunity and that a small probability of CRISPR failure was irrelevant to the dynamics (55).
Haerter et al. investigated the conditions under which the diversity of CRISPR loci can be maintained in a spatially inhomogeneous, agent-based model with a small finite number of viral strains (56, 57). These studies have concluded that spatial structure was required to explain the observed diversity of the CRISPR loci.
Levin explored the conditions for the maintenance of a costly CRISPR-Cas locus using a parameter-rich mean field model in which CRISPR immunity was parameterized rather than derived from an explicit coevolution dynamics of the spacer and proto-spacer populations (58). This study led to the conclusion that there were narrow parameter regimes under which CRISPR-Cas provided bacteria with an advantage over CRISPR-lacking counterparts with a higher Malthusian fitness due to antivirus immunity and that selection for maintaining CRISPR-Cas was weak, suggesting that antivirus defense might not be the principal function of CRISPR-Cas. When the model was compared to experiments that measured the phage/host population dynamics, several apparent disagreements prompted the authors to conclude that the basic assumptions of the coevolutionary arms race models of CRISPR had to be reevaluated (59).
Weinberger et al. aimed to explain the old end uniformity of the CRISPR loci by examining the dynamics of the diversities of the host and viral populations while keeping the total population size fixed in a model that derived immunity directly from the CRISPR locus dynamics (60). A variant of this model with the additional dynamics of acquisition and loss of the entire CRISPR-Cas locus yielded the prediction of a viral diversity threshold above which CRISPR-Cas became ineffective and was therefore lost due to the fitness cost associated with its maintenance (40). This study further tested the hypothesis that CRISPR-Cas is nearly ubiquitous in hyperthermophiles but much less common in mesophiles due to the decreased rate of mutation fixation in viruses infecting hyperthermophiles (40). Simulations that included competition between CRISPR+ and CRISPR− hosts, as well as loss and HGT of CRISPR-Cas loci, showed that the immunological benefits provided by CRISPR-Cas outweigh the costs under moderate virus diversity that appears to be characteristic of hyperthermophilic environments. These results offered a possible explanation for the higher prevalence of CRISPR-Cas in hyperthermophiles compared to mesophiles and more generally identified the conditions for the evolutionary stability of sensor-type defense mechanisms.
To summarize, efforts using CRISPR modeling appear to be in the early stages of development when the ingredients and mechanisms required for an accurate description of the empirical observations are still being hashed out. Because modeling efforts to date have all made radical simplifications for the sake of tractability, interpretation of the results can be problematic, and direct comparisons to empirical data are not particularly informative. In the present study, we sought to avoid the tradeoffs present in the previous models of CRISPR such as the lack of explicit population dynamics, decoupling of immunity from viral dynamics, and the finite, limited CRISPR length and/or viral diversity. The model presented here is capable of representing the arms race, in which viruses mutate stochastically to escape immunity, in conjunction with the effect of the CRISPR array content on the fraction of immune interactions and therefore on the dynamics of the host and virus populations. The virus and host diversities are the outcomes of the model dynamics and not prescribed a priori.
The explicit population dynamics, as well as the possibility of virus and host extinction, distinguishes this work from our earlier model (40) that formally assumed constant population sizes. The inclusion of explicit virus and host population dynamics into the model allowed us to exploit the thoroughly characterized stochastic agent based predator-prey framework, which reduces to the Lotka-Volterra (LV) formalism in the limit of the infinite population size when fluctuations can be neglected (61–63), for understanding virus-host coevolution in the presence of CRISPR-Cas immunity. The results of the model analysis indicate that CRISPR-Cas stabilizes the stochastic LV system in the intermediate range of viral mutation rate, i.e., leads to extended coexistence of viruses and their microbial hosts. The model further reveals the dependence of CRISPR-Cas efficacy on the population size, spacer incorporation efficiency, number of proto-spacers per virus, and viral mutation rate. When the fitness cost of maintaining CRISPR-Cas is factored into the model, the efficacy of the immunity system determines whether it is maintained in a population under the deletion bias conditions.
MATERIALS AND METHODS
Model.
The model developed here aims to reproduce the coevolutionary process shaped by immune interactions between viruses and bacteria or archaea carrying the CRISPR-Cas system, with an underlying ecological dynamics that controls the population of hosts and viruses. The model takes into account the fitness cost of the CRISPR-Cas loci, as well as the possibility of their loss and gain via horizontal transfer. Thus, the model is suitable to study the evolutionary and ecological conditions that determine the efficacy of CRISPR-Cas and its long-term fate in the host population.
The analyzed system consists of variable numbers of CAS-positive hosts (Nb+), CAS-negative hosts (Nb−), and viruses (Nv). Hosts (bacteria or archaea) are abstracted as variable size sets of (possibly nonunique) spacers. Viruses are represented as sets of Ns distinct proto-spacers. Importantly, unlike in several previous models, neither the size nor the content of CRISPR locus are fixed a priori in our model but are the outcomes of the dynamics, which is described next. The host-virus system evolves according to a stochastic dynamics that is simulated using the Gillespie algorithm (64). We consider the following events: (i) growth of a CAS− host population with rate Nb− (this sets the scale of time); (ii) growth of a CAS+ host population with rate Nb+/(1 + c), where c is the fitness cost of the CRISPR system; (iii) encounter of viruses and hosts with rates bNb+Nv and bNb−Nv for CAS+ and CAS−, respectively; (iv) viral degradation with rate dNv; and (v) CRISPR-Cas locus horizontal transfer from CAS+ to CAS− hosts with rate σNb+Nb-/(Nb+ + Nb-). This implementation of HGT as frequency independent assumes that the DNA exchange mechanisms are saturated. The effect of a nonsaturated scenario is briefly described below in the context of a mean field model.
An encounter between a virus and a host may be immune or productive. An immune encounter occurs if a CAS+ host contains at least one spacer that matches any of the viral proto-spacers. Alternatively, both CAS+ and CAS− hosts can experience “innate” immune encounters (whereby the immunity is provided by defense systems other than CRISPR-Cas that do not depend on spacer acquisition), with a small probability s. Otherwise, the encounter is productive and results in the death of the host and a viral burst of size M.
The model further incorporates genome-level dynamics of the host spacers and viral proto-spacers. Every time a CAS+ host divides, the daughter cell may lose its CRISPR-Cas locus and all of the spacers with probability λ. Moreover, single spacers are deleted with probability ℓ (per spacer). New spacers can be incorporated every time an immune encounter takes place: each proto-spacer of the infecting virus is added to the spacer list of the host with probability a. Finally, the new viruses produced at every viral burst mutate their proto-spacers with probability μ (per proto-spacer). We assume an infinite allele scenario where mutations always give rise to novel proto-spacers.
Parameter setting.
Since we are dealing with a stochastic, agent-based model, the choice of parameter values is limited by the computational cost. We set the model parameters in such a way that simulation times become affordable, while the key properties of the population remain as realistic as possible. We varied the viral burst size M between 2 and 90, while the degradation rate was fixed at d = 0.5. This parameter choice provides an equilibrium composition, with viruses being 10- to 100-fold more abundant than hosts, which is close to the actual virus/cell ratios observed in various habitats (65–67). The mutation rate per proto-spacer μ was varied between 0 and 0.1; the encounter rate b (whose inverse controls the population size) was varied between 10−3 and 10−4. The CRISPR-related parameters were set as follows: virus size Ns = 10 to 50 proto-spacers per virus, spacer loss probability ℓ = 0.05 or 0.1, and the incorporation probability a between 0 and 0.1. The probability of innate immunity was fixed to s = 0.1. These parameters translate into spacer deletion bias so that in the absence of adaptive immunity, the host loses its spacers. In the region of the parameter space that was chosen to explore, the parameter values combine to yield a realistic range (10 to 100) of the steady-state size of the CRISPR cassette (12).
Simulations start with viral and host population sizes equal to their Lotka-Volterra (LV) equilibrium values (see Results). Viruses were allowed to mutate once prior to the beginning of the simulation, whereas hosts start as CAS+ with no spacers. Simulation results were averaged over 100 independent realizations.
RESULTS
Effect of CRISPR-Cas on the host-virus system dynamics.
We first studied the effect of the CRISPR-induced immunity on the dynamics of the host-virus system. As a starting point, we simplified the model by assuming that the CRISPR-Cas loci are constitutively maintained in the host population, i.e., there is neither loss nor HGT of CRISPR-Cas loci, and the fitness cost incurred by the CRISPR-Cas system is negligible. In terms of the model parameters, these assumptions translate into λ = σ = c = 0.
To evaluate the effect of CRISPR-induced immunity, it was first necessary to characterize the behavior of the virus-host system in the absence of CRISPR-Cas. When the population size is large and fluctuations can be neglected, the hosts and the viruses comprise a Lotka-Volterra (LV) system that oscillates around an equilibrium state with Nb* hosts and Nv* viruses. As shown in the supplemental material, the equilibrium sizes of the host and virus populations are Nb* = d/[b(M − Ms − s)] and Nv* = 1/[b(1 − s)], respectively, and the period of the LV oscillations is 2π/√d. Because the host and virus populations are finite, either viruses or hosts can become extinct. Simulations of the model in the absence of adaptive immunity (a = 0) with b = 10−3 show that the mean survival time for the hosts under the chosen parameters setting is ∼102 generations, whereas survival probability at T = 103 generations is negligible. Thus, stochastic extinction (68) of the hosts within a time span of T = 103 generations is the expected outcome whenever the CRISPR system is unable to provide antiviral immunity.
We assessed the effect of CRISPR-induced immunity on the host-virus system at various values of the viral mutation rate μ and the spacer incorporation rate a, after a simulation time of T = 103 generations (Fig. 1). According to the final fate of the system, three regimes become apparent: (i) viral extinction at low μ and moderate a, (ii) long-term coexistence at an intermediate range of the parameters, and (iii) stochastic extinction of hosts at greater μ values. Viral extinction is fast and occurs if the hosts achieve an average fraction of immune encounters greater than 1 − M−1, which makes the mean viral yield drop below one per encounter (this regime corresponds to the black, upper left regions in Fig. 1A and B). In contrast, host extinction is the result of stochastic fluctuations in the discrete LV model and requires much longer times to occur (main, right region in Fig. 1A and 1B). Within a rather narrow range of both parameters lies the regime of stable virus-host coexistence (the colored areas in Fig. 1).
Fig 1.
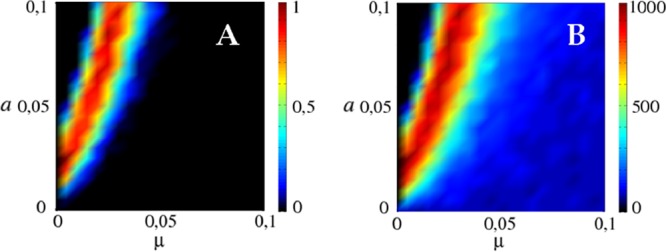
(A) Fraction of simulations that survive extinction for at least 103 generations (color bar) for various values of the viral mutation rate μ and the spacer incorporation probability a. The small black region in the upper left corner corresponds to viral extinction driven by adaptive immunity, whereas the main black region corresponds to the stochastic extinction of hosts. (B) Mean survival time of the population (color bar). The maximum survival time, T = 103, corresponds to populations with coexistence at the end of the simulation. Note that the survival time has a peak as a function of μ or a when all other parameters are fixed.
An important by-product of the CRISPR-induced immunity is that it increases the host and the viral population sizes (Fig. 2). This effect might appear paradoxical at the first glance, but it is a direct consequence of the degree of CRISPR-mediated adaptive immunity pc achieved by the hosts and can be captured by using pc instead of the innate immunity s in the above expressions for Nb* and Nv* (solid lines in Fig. 2). A higher level of immunity leads to increased survival of the hosts, and the larger the host population, the more viruses it can sustain. There exists an optimal virus mutation rate that maximizes the mean lifetime of the system before extinction and therefore the total amount of viral particles produced during the infection (Fig. 3B). This optimal mutation rate is associated with a relatively high level of CRISPR-induced immunity but not as high as to quickly extinguish the virus. Immunity allows for large populations and long-term coexistence, which translates into sustained production of viral particles. Conversely, virus mutation rates that render CRISPR-Cas ineffective result in a decreased size of the host populations and consequently lead to a low total production of viral particles. In a qualitatively similar manner, both the mean lifetime of the system and the probability of virus extinction show sharp dependencies on the spacer acquisition rate a, with the longest lifetime at intermediate a values and a steep drop in the lifetime associated with the deterministic virus extinction at higher a values (Fig. 3C).
Fig 2.
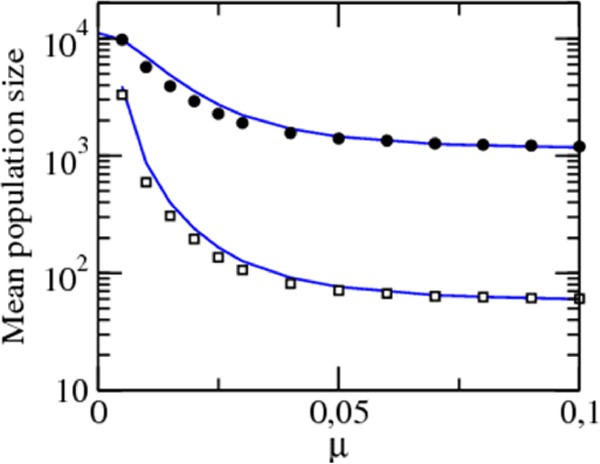
Average number of hosts (□) and viruses (●) in the population at different values of μ. The incorporation probability is fixed at a = 0.05. The blue curves correspond to the analytical estimates based on the average degree of immunity in each simulation.
Fig 3.

(A) Exponential growth of the mean lifetime before extinction indicates that CRISPR stabilizes the stochastic virus-host system when hosts have the upper hand. (B) The constant a transect shows that stabilization is most effective in the intermediate viral mutation regime when viruses and hosts coexist. (C) A transect of Fig. 1B for a constant μ also shows that virus/host coexistence and LV stabilization are most pronounced in the intermediate range of spacer acquisition rates (a).
Furthermore, the exponential increase of the mean lifetime before extinction with the population size (Fig. 3A) indicates that effective CRISPR-Cas stabilizes the stochastic LV virus-host system (a linear increase in the lifetime is expected without the stabilization effect). When viruses coexist with the hosts (see Fig. 1A and B), the populations are in a quasi-steady state in which the length L of the CRISPR array, the number Nt of distinct proto-spacers in the viral population, and the probability pc that CRISPR provides immunity fluctuate around their time-average values. We show in the supplemental material that the probability pc of a match between a spacer and a proto-spacer in an encounter between a random virus and a random host can be expressed as a function of the ratio L/Nt and the magnitude of the correlation between the relative abundances of proto-spacers and the matching spacers. Figure 4 illustrates that the steady-state value of the CRISPR-associated immunity pc is well approximated by:
| (1) |
where α is a constant that depends on the strength of the correlation between the relative abundances of matching spacers and proto-spacers. The spacer–proto-spacer correlation reflected by α increases with the burst size M but does not seem to depend on virus size Ns. However, because the L/Nt ratio does not seem to depend on Ns, and Ns appears in the exponent, adaptive immunity pc grows rapidly with Ns.
Fig 4.

(A) CRISPR immunity pc is a function of only the ratio of the length L of the CRISPR array and the total number Nt of distinct viral proto-spacers. The graph is obtained by varying the spacer incorporation probability a and the viral mutation rate μ across the range of virus-host coexistence while keeping the rest of the parameters fixed at b = 10−4, d = 0.5, ℓ = 0.05, and s = 0.1. Solid lines are the predictions of equation 1 with the parameter α = 4, which reflects the strength of correlation between spacers and proto-spacers. This correlation seems to be independent of the number Ns of protospacers per virus. (B) Varying the viral burst size M at a fixed Ns shows that the correlation between the spacers and proto-spacers reflected by the parameter α grows roughly linearly with the burst size M.
To get a handle on the dependence of the CRISPR immunity pc on the model parameters, we examined L and Nt in equation 1 separately. In steady state, the decay of the CRISPR array due to spacer loss is balanced by the growth due to immune encounters with viruses
| (2) |
where p = s + pc(1 − s) is the total immunity, and we used the expression Nb = 1/[b(1 − p)] for the average viral population size. Equation 2 is obtained under the assumption that fluctuations in L and p are small and uncorrelated with each other across a particular population. The empirically computed steady-state value of L is consistently above the prediction, indicating that, not surprisingly, L and p are positively correlated, leading to a larger average L for the same p (Fig. 5).
Fig 5.

The steady-state length L of the CRISPR array computed for a range of parameters (spacer incorporation probability a is varied in each group of like-colored points) is above the prediction (solid curve) of equation 2.
Effect of CRISPR-Cas on viral diversity.
The selective pressure exerted by the CRISPR-Cas system on frequent viral proto-spacers suggests that CRISPR-Cas might directly promote viral diversity. On the other hand, new viruses that manage to escape adaptive immunity tend to rapidly proliferate, thereby reducing viral diversity. We found that neither mechanism operates and that the steady-state number of viral proto-spacers Nt is closely approximated by the diversity in a viral population of the same size evolving in the absence of CRISPR-mediated immunity (a = 0) (see Fig. 6). Thus, the proto-spacer diversity increases in the presence of CRISPR-Cas only inasmuch as the virus population grows. This counterintuitive finding is likely the result of the high number of proto-spacers per viral genome, which means that the beneficial effect of a mutation in a single proto-spacer is small and, accordingly, positive selection driving the evolution of new proto-spacers is weak if not negligible.
Fig 6.

The number of distinct viral proto-spacers per virus Nt/Nv (symbols, y axis) computed for a wide range of a, μ, and b = 10−4 is slightly below what it would be in a freely evolving viral population of the same size (solid curves).
The diversity of a freely evolving virus population (Fig. 6) is described by a remarkably simple expression. In the limit of the large burst size M we obtain
| (3) |
Perhaps counterintuitively, Nt is a declining function of the burst size M for a fixed viral population size Nv (Fig. 7). This behavior can be explained by noting that when μ is small and the total number of proto-spacers in a viral population is fixed, each burst of a virus carrying a particular set of proto-spacers produces a relatively greater fraction of these spacers in the whole population and thus results in the reduction of the number of distinct proto-spacer types.
Fig 7.

The number of distinct proto-spacers Nt in a freely evolving viral population divided by the product of the viral population size Nv, and the virus size Ns is proportional to μ (solid line has unit slope) in the limit of the large burst size M.
Conditions for the maintenance of the CRISPR system.
In the previous sections, we elucidated the behavior of the model in a situation where CRISPR-Cas cannot be completely lost. In this section, motivated by the patchy distribution of the CRISPR-Cas system in prokaryotic genomes (6, 18, 40), we allow for deletion of the entire CRISPR-Cas system and explore the conditions that govern its maintenance in the host population. Obviously, when carrying a CRISPR-Cas locus incurs a fitness cost, its maintenance depends on the fitness benefit it provides. Below, we elucidate the conditions under which CRISPR is effective enough to be retained in the population. Not surprisingly, and in accord with previous findings (40), CRISPR is most effective and is therefore maintained in the intermediate regime when the viruses evolve fast enough to challenge CRISPR but not fast enough to evade it.
We first sought to determine how the virus mutation rate affects the efficacy of CRISPR-Cas. Equation 1 can be used to derive a characteristic viral mutation rate μc at which CRISPR-associated immunity pc is equal to innate immunity s. When the viral mutation rate is much smaller than μc, CRISPR immunity dominates over the innate immunity and vice versa. When s ≪ 1, we obtain:
| (4) |
where we used equation 2 to obtain the second expression. Equation 4 predicts that the threshold mutation rate of the virus, below which CRISPR-Cas is effective, is proportional to the virus size Ns and the spacer acquisition probability a and inversely proportional to the viral population size Nv. If viruses present a larger target for CRISPR or if hosts are more efficient at incorporating viral genetic material, the viruses have to mutate faster to escape immunity. Conversely, as the viral population grows, the concomitant growth of the proto-spacer diversity renders CRISPR ineffective. In other words, if the viral mutation rate is fixed, there exists a critical viral population size below which CRISPR provides immunity and above which it is useless.
To further investigate the evolutionary dynamics of CRISPR-Cas, we explore the “three-species system” that consists of CRISPR+ and CRISPR− hosts and viruses. Here, we drop the simplifying assumptions of the preceding sections and assume that the CRISPR-Cas system entails some fitness cost c and that the CRISPR-Cas loci can be lost or horizontally transferred at rates λ and σ, respectively. As a first approach to the problem, let us introduce the mean field approximation that is valid when fluctuations can be ignored and the fraction of immune encounters in CAS+ hosts is assumed to be a constant parameter p. The population of CAS+ hosts (Nb+), CAS− hosts (Nb−) and virus (Nv) follows the equations:
| (5) |
The analysis of the system of equations (i.e., equations 5) shows that a minimum degree of CRISPR-induced immunity is required if CAS+ hosts are to survive. That efficacy threshold, denoted as pmin, is equal to:
| (6) |
If the degree of immunity provided by CRISPR-Cas is smaller than pmin, the CRISPR-Cas loci are lost. Horizontal transfer and deletion rates are involved in the expression for pmin, together with the fitness cost. Thus, deletion bias plays a role equivalent to that of the fitness cost with respect to the maintenance of cas genes. Conversely, an enhanced rate of horizontal transfer might compensate for fitness cost. Here, we analyze a scenario with equal rates of deletion and horizontal transfer, λ = σ = 0.1, and focus on the consequences of the fitness cost. In modeling horizontal transfer, a scenario with saturated DNA exchange was chosen. It is easy to generalize the model to include nonsaturated scenarios where the horizontal transfer rate is proportional to the number of hosts (see the supplemental material). In such a case, the value of pmin that allows for CAS maintenance depends on the population size, with greater pmin required in smaller populations.
The results of simulations addressing the maintenance of a costly CRISPR system are plotted in Fig. 8. With the fitness cost set to c = 1, the fate of cas genes in the host population has been studied for various values of the mutation and spacer incorporation rates (Fig. 8A). Three regimes can be distinguished: (i) viral extinction, (ii) coexistence of virus and host, with CRISPR-Cas maintained, and (iii) CRISPR-Cas loss. Not surprisingly, these regimes roughly correspond to those obtained in Fig. 1A for the case without cost. When CRISPR-Cas is ineffective, it is rapidly lost (Fig. 8B), whereas the stochastic extinction of hosts takes much longer, especially in large populations. When CRISPR-Cas drives viruses to extinction, the absence of new infections renders CRISPR-Cas useless and eventually causes its loss. That would not be the case if new viruses were introduced stochastically before CRISPR-Cas loss occurred.
Fig 8.

The maintenance of a costly CRISPR-Cas system depends on its effectiveness. (A) Fraction of simulations where host and virus coexist and cas genes are maintained after 103 generations (color bar), as a function of a and μ. The small black region at the upper left corner corresponds to viral extinction. The main black region corresponds to the loss of cas genes. (B) The average duration of CAS maintenance in the population. In the white region, where the virus gets extinct, CRISPR becomes useless and cas genes are lost unless periodic reinfections occur. (C) Fraction of encounters between virus and CAS+ hosts with an immune outcome. The dashed line at the bottom corresponds to the innate immunity s = 0.1. The dashed line at the top corresponds to the 90% probability of an immune encounter at which the viral productivity is <1 for the burst size M = 10, causing viral extinction.
The fate of the cas genes is determined by the effectiveness of the CRISPR-Cas immune system. With the parameters used in Fig. 8, equation 6 predicts that a minimum fraction of immune encounters pmin = 0.505 is required in order to retain CRISPR-Cas. Such a degree of immune encounters is achieved if the viral mutation rate is μ < 0.03 (at a = 0.05) or μ < 0.04 (at a = 0.1) (Fig. 8C). The coincidence between these values and the boundary of the CRISPR-Cas maintenance region (Fig. 8A) supports the idea of an efficacy threshold given by equation 2.
Population size, fitness cost, burst size, and the number of proto-spacers.
To study the effect of the population size on the efficacy and evolutionary fate of CRISPR-Cas, we focused on the encounter parameter b that is inversely proportional to the time average population size in the model. By varying parameter b across 1 order of magnitude, we found that CRISPR-Cas fails to provide immunity in large populations, and, as a result, large populations lose CRISPR-Cas loci (Fig. 9). This is a direct consequence of the increase in the viral diversity reflected in equation 3 and the resulting decrease in CRISPR-induced adaptive immunity predicted by equation 1.
Fig 9.

Large population size leads to CAS loss. The upper panel shows a fraction of simulations where cas genes are retained (red) and lost (blue) as a function of the population size parameter (inverse encounter rate b−1). The lower panels show typical evolution of single realizations in small (b−1 = 102, left) and large (b−1 = 103, right) populations. (A) Numbers of viruses (red), CAS+ hosts (blue), and CAS− hosts (green). (B) Fraction of immune encounters. (C) Fraction of hosts that conserve CAS.
A closer examination of the explicit dynamics of the model yields further insight into the mechanism of CRISPR-Cas loss (Fig. 9). There is an initial phase where the degree of immunity is high in both small and large populations. However, the increasing viral diversity reached in large populations leads to a gradual decrease in the efficacy of CRISPR, resulting in the eventual loss of CRISPR-Cas. Again, it is the increasing diversity of viruses, reflected by the total number Nt of proto-spacers in the viral population, which gradually makes the immunological memory ineffectual.
The effect of other biological parameters on the maintenance of CAS is summarized in Fig. 10. The evolutionary outcome does not depend on the value of the CRISPR-Cas fitness cost as long as it is moderately high. Even for a small fitness cost, an ineffective CRISPR system becomes an almost neutral trait that may be lost through neutral drift and population bottlenecks. This feature seems to explain the loss of CRISPR-Cas at small fitness costs and moderate mutation rates (Fig. 10A) even when equation 6 predicts its retention. To summarize, the magnitude of the fitness cost for CRISPR-Cas does not qualitatively affect the outcome of virus-host interaction. The viral burst size does not seem to perceptibly affect the results either (Fig. 10B). In contrast, changes in the number Ns of proto-spacers per virus dramatically change the evolutionary fate of CRISPR-Cas. It is easy to see that the greater the number of proto-spacers per virus, the more difficult it is for the virus to escape the immune memory. This dependence translates into a sharp transition in the long-term maintenance of CRISPR when immunity becomes greater than the threshold value pmin as the number of proto-spacers per virus increases slightly.
Fig 10.
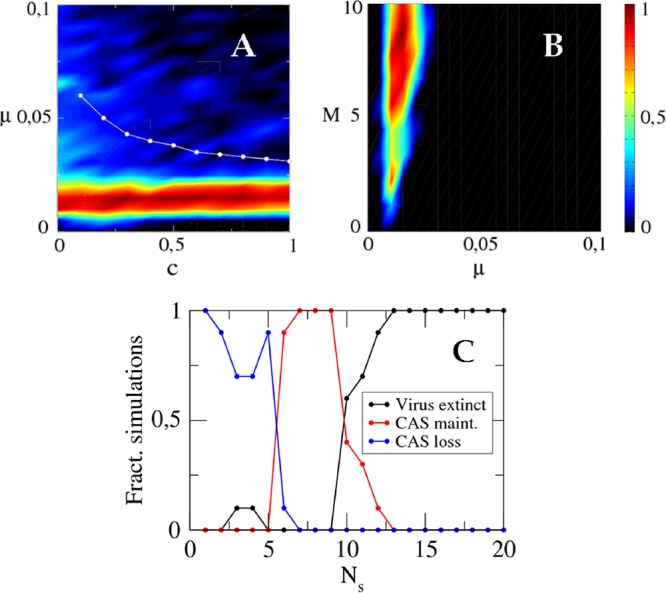
Effect of the fitness cost (A), burst size (B), and number of proto-spacers per virus (C) on the evolutionary fate of CRISPR-Cas. (A) Fraction of simulations where CRISPR-Cas is retained after 103 generations, as a function of the mutation rate μ and fitness cost c. Cases of stochastic host extinction before CRISPR-Cas is lost are also included. The white dots correspond to the threshold values predicted by equation 6. (B) Fraction of simulations with CRISPR-Cas conservation at different values of the mutation rate and the burst size (M). The black band to the left is due to viral extinction. The main black area corresponds to CRISPR-Cas loss. (C) Fraction of simulations with viral extinction (black), CRISPR-Cas conservation (red), and CRISPR-Cas loss (blue) at increasing values of the number of proto-spacers per viral genome. Parameter values: a = 0.05 (μ = 0.01 in panel C).
DISCUSSION
The model described here seems to be more realistic than the previous models of CRISPR-Cas evolution because it makes fewer oversimplifying assumptions by including changing population sizes of both the hosts and the viruses coupled to the explicit dynamics of the CRISPR array. In addition, the size and content of the CRISPR arrays are not fixed a priori, as was the case in several previous models, but rather are determined by the evolution of the system. Accordingly, this model enables an explicit analysis of the population dynamics within a stochastic agent-based predator-prey framework that reduces to an LV system in the large population limit. This general modeling framework provides for an explicit analysis of the virus and host population dynamics and the evolution of the CRISPR-Cas loci and leads to several nontrivial results.
Although it might seem counterintuitive, CRISPR-Cas immunity stabilizes the virus-host system for intermediate values of the viral mutation rate, i.e., promotes the long-term virus-host coexistence, rather than leading to the extinction of the virus. Alternatively, if the mutation rate is fixed, this stabilization only occurs for intermediate values of the host and virus population size. In large populations CRISPR-Cas is lost, due to the increase in viral diversity, whereas when populations are small, stochastic extinction reduces the mean lifetime of the system. This observation links the present results to those of our earlier modeling study of CRISPR-Cas in which we have shown that the immunological memory provided by CRISPR-Cas can be effective only at moderate virus diversities (40). This result has been suggested to explain the ubiquity of CRISPR-Cas in hyperthermophiles as opposed to the substantially lower prevalence in mesophiles given that the rates of mutation fixation are much lower in hyperthermophiles (both hosts and viruses) than in mesophiles (69, 70). The present findings offer a complementary and simpler perspective on the difference in the prevalence of CRISPR-Cas between hyperthermophiles and mesophiles: extremely large populations in which CRISPR-Cas becomes useless according to the model results are known only for mesophiles, whereas hyperthermophiles typically exist as smaller populations (71–74) in which CRISPR-Cas immunity is predicted to be efficient. More specifically, in deep-sea marine hydrothermal sediments, the cell densities of hyperthermophilic prokaryotes (primarily, Archaea) appear to vary approximately from 105 to 108 cells per g (72, 75–79). In contrast, substantially higher prokaryotic cell densities, on the order of 109 cells per g and even greater have been reported for shallow water, mesophilic sediments (80–86).
Because the mean lifetime of the system (LV stabilization) has a sharp peak at a certain intermediate value of the viral mutation rate, the maximum total virus yield before either viruses or hosts become extinct is reached at the same intermediate value of the viral mutation rate. This value is neither so high that CRISPR-Cas would become ineffective, nor so low that CRISPR-Cas would extinguish the virus.
Counterintuitively, in the present model, CRISPR-Cas immunity does not specifically promote viral diversity in the sense of driving positive selection for emergence of new spacers, presumably because the selection pressure on any single spacer is too weak. In fact, the model results indicate that, when the viral mutation rate is sufficiently low, the clonal bloom dynamics results in slightly reduced viral diversity compared to a freely evolving (without pressure to escape adaptive immunity) population. Virus diversity is directly proportional to the mutation rate and the population size and, accordingly, CRISPR-Cas promotes virus diversity only inasmuch as the immunity leads to an increase in the population size.
The model results indicate that the efficacy of CRISPR increases with the number Ns of proto-spacers per viral genome. Because maintenance of CRISPR-Cas is a threshold phenomenon, a small decrease in Ns can lead to CRISPR-Cas loss. This finding might explain, at least in part, why the proto-spacer-associated motif (PAM), the presence of which in a viral genome is essential for a proto-spacer acquisition, has a low information content (i.e., consists of only two or three nucleotides) (87, 88): it is critical for the host to be able to use multiple proto-spacers. The fact that the specificity in the selection of proto-spacers exists at all might reflect the tradeoff between the benefits of the utilization of multiple proto-spacers for efficient immunity and the avoidance of autoimmunity. Clearly, the mechanisms of self/non-self discrimination in CRISPR-Cas requires further, detailed exploration.
This model makes explicit, detailed predictions of the dependencies between the diversities of viruses and hosts, as well as the dynamics of virus and host populations. At present, the available data on virus and host diversity in the same habitat is insufficient for direct testing of these predictions, However, the rapid advances of metagenomics are expected to yield such data in the near future. Moreover, this model can provide the framework for setting up and exploring experimental evolution system for the study of the arms races between viruses and CRISPR-Cas-carrying hosts.
Supplementary Material
ACKNOWLEDGMENTS
J.I. is supported by Comunidad de Madrid through a research grant and project MODELICO S2009/ESP-1691 and Spanish MINECO through project FIS2011-27569.
A.E.L., Y.I.W., and E.V.K. are supported by intramural funds from the U.S. Department of Health and Human Services (to the National Library of Medicine).
Footnotes
Published ahead of print 21 June 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.00412-13.
REFERENCES
- 1.Stern A, Sorek R. 2011. The phage-host arms race: shaping the evolution of microbes. Bioessays 33:43–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Koonin EV, Wolf YI. 2012. Evolution of microbes and viruses: a paradigm shift in evolutionary biology? Front. Cell Infect. Microbiol. 2:119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Forterre P, Prangishvili D. 2009. The great billion-year war between ribosome- and capsid-encoding organisms (cells and viruses) as the major source of evolutionary novelties. Ann. N. Y. Acad. Sci. 1178:65–77 [DOI] [PubMed] [Google Scholar]
- 4.Haaber J, Samson JE, Labrie SJ, Campanacci V, Cambillau C, Moineau S, Hammer K. 2010. Lactococcal abortive infection protein AbiV interacts directly with the phage protein SaV and prevents translation of phage proteins. Appl. Environ. Microbiol. 76:7085–7092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Makarova KS, Wolf YI, Snir S, Koonin EV. 2011. Defense islands in bacterial and archaeal genomes and prediction of novel defense systems. J. Bacteriol. 193:6039–6056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Makarova KS, Wolf YI, Koonin EV. 2013. Comparative genomics of defense systems in archaea and bacteria. Nucleic Acids Res. 41:4360–4377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Blower TR, Salmond GP, Luisi BF. 2011. Balancing at survival's edge: the structure and adaptive benefits of prokaryotic toxin-antitoxin partners. Curr. Opin. Struct. Biol. 21:109–118 [DOI] [PubMed] [Google Scholar]
- 8.Blower TR, Evans TJ, Przybilski R, Fineran PC, Salmond GP. 2012. Viral evasion of a bacterial suicide system by RNA-based molecular mimicry enables infectious altruism. PLoS Genet. 8:e1003023. 10.1371/journal.pgen.1003023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Leplae R, Geeraerts D, Hallez R, Guglielmini J, Dreze P, Van Melderen L. 2011. Diversity of bacterial type II toxin-antitoxin systems: a comprehensive search and functional analysis of novel families. Nucleic Acids Res. 39:5513–5525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vasu K, Nagaraja V. 2013. Diverse functions of restriction-modification systems in addition to cellular defense. Microbiol. Mol. Biol. Rev. 77:53–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Koonin EV, Makarova KS. 2013. CRISPR-Cas: evolution of an RNA-based adaptive immunity system in prokaryotes. RNA Biol. 10:679–686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bhaya D, Davison M, Barrangou R. 2011. CRISPR-Cas systems in bacteria and archaea: versatile small RNAs for adaptive defense and regulation. Annu. Rev. Genet. 45:273–297 [DOI] [PubMed] [Google Scholar]
- 13.Deveau H, Garneau JE, Moineau S. 2010. CRISPR/Cas system and its role in phage-bacteria interactions. Annu. Rev. Microbiol. 64:475–493 [DOI] [PubMed] [Google Scholar]
- 14.Marraffini LA, Sontheimer EJ. 2010. CRISPR interference: RNA-directed adaptive immunity in bacteria and archaea. Nat. Rev. Genet. 11:181–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wiedenheft B, Sternberg SH, Doudna JA. 2012. RNA-guided genetic silencing systems in bacteria and archaea. Nature 482:331–338 [DOI] [PubMed] [Google Scholar]
- 16.Haft DH, Selengut J, Mongodin EF, Nelson KE. 2005. A guild of 45 CRISPR-associated (Cas) protein families and multiple CRISPR/Cas subtypes exist in prokaryotic genomes. PLoS Comput. Biol. 1:e60. 10.1371/journal.pcbi.0010060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jansen R, Embden JD, Gaastra W, Schouls LM. 2002. Identification of genes that are associated with DNA repeats in prokaryotes. Mol. Microbiol. 43:1565–1575 [DOI] [PubMed] [Google Scholar]
- 18.Koonin EV, Makarova KS. 2009. CRISPR-Cas: an adaptive immunity system in prokaryotes. F1000 Biol. Rep. 1:95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Makarova KS, Grishin NV, Shabalina SA, Wolf YI, Koonin EV. 2006. A putative RNA-interference-based immune system in prokaryotes: computational analysis of the predicted enzymatic machinery, functional analogies with eukaryotic RNAi, and hypothetical mechanisms of action. Biol. Direct 1:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mojica FJ, Diez-Villasenor C, Garcia-Martinez J, Soria E. 2005. Intervening sequences of regularly spaced prokaryotic repeats derive from foreign genetic elements. J. Mol. Evol. 60:174–182 [DOI] [PubMed] [Google Scholar]
- 21.Goren M, Yosef I, Edgar R, Qimron U. 2012. The bacterial CRISPR/Cas system as analog of the mammalian adaptive immune system. RNA Biol. 9:549–554 [DOI] [PubMed] [Google Scholar]
- 22.Koonin EV, Wolf YI. 2009. Is evolution Darwinian and/or Lamarckian? Biol. Direct 4:42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Barrangou R, Fremaux C, Deveau H, Richards M, Boyaval P, Moineau S, Romero DA, Horvath P. 2007. CRISPR provides acquired resistance against viruses in prokaryotes. Science 315:1709–1712 [DOI] [PubMed] [Google Scholar]
- 24.Fabre L, Zhang J, Guigon G, Le Hello S, Guibert V, Accou-Demartin M, de Romans S, Lim C, Roux C, Passet V, Diancourt L, Guibourdenche M, Issenhuth-Jeanjean S, Achtman M, Brisse S, Sola C, Weill FX. 2012. CRISPR typing and subtyping for improved laboratory surveillance of Salmonella infections. PLoS One 7:e36995. 10.1371/journal.pone.0036995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bikard D, Hatoum-Aslan A, Mucida D, Marraffini LA. 2012. CRISPR interference can prevent natural transformation and virulence acquisition during in vivo bacterial infection. Cell Host Microbe 12:177–186 [DOI] [PubMed] [Google Scholar]
- 26.Carroll D. 2012. A CRISPR approach to gene targeting. Mol. Ther. 20:1658–1660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Qi L, Haurwitz RE, Shao W, Doudna JA, Arkin AP. 2012. RNA processing enables predictable programming of gene expression. Nat. Biotechnol. 30:1002–1006 [DOI] [PubMed] [Google Scholar]
- 28.Mali P, Yang L, Esvelt KM, Aach J, Guell M, Dicarlo JE, Norville JE, Church GM. 2013. RNA-guided human genome engineering via Cas9. Science 339:823–826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, Zhang F. 2013. Multiplex genome engineering Using CRISPR/Cas systems. Science 339:819–823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cain AK, Boinett CJ. 2013. A CRISPR view of genome sequences. Nat. Rev. Microbiol. 11:226. [DOI] [PubMed] [Google Scholar]
- 31.Makarova KS, Aravind L, Wolf YI, Koonin EV. 2011. Unification of Cas protein families and a simple scenario for the origin and evolution of CRISPR-Cas systems. Biol. Direct 6:38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Makarova KS, Haft DH, Barrangou R, Brouns SJ, Charpentier E, Horvath P, Moineau S, Mojica FJ, Wolf YI, Yakunin AF, van der Oost J, Koonin EV. 2011. Evolution and classification of the CRISPR-Cas systems. Nat. Rev. Microbiol. 9:467–477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.van der Oost J, Jore MM, Westra ER, Lundgren M, Brouns SJ. 2009. CRISPR-based adaptive and heritable immunity in prokaryotes. Trends Biochem. Sci. 34:401–407 [DOI] [PubMed] [Google Scholar]
- 34.Deveau H, Barrangou R, Garneau JE, Labonte J, Fremaux C, Boyaval P, Romero DA, Horvath P, Moineau S. 2008. Phage response to CRISPR-encoded resistance in Streptococcus thermophilus. J. Bacteriol. 190:1390–1400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Andersson AF, Banfield JF. 2008. Virus population dynamics and acquired virus resistance in natural microbial communities. Science 320:1047–1050 [DOI] [PubMed] [Google Scholar]
- 36.Semenova E, Jore MM, Datsenko KA, Semenova A, Westra ER, Wanner B, van der Oost J, Brouns SJ, Severinov K. 2011. Interference by clustered regularly interspaced short palindromic repeat (CRISPR) RNA is governed by a seed sequence. Proc. Natl. Acad. Sci. U. S. A. 108:10098–10103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Heidelberg JF, Nelson WC, Schoenfeld T, Bhaya D. 2009. Germ warfare in a microbial mat community: CRISPRs provide insights into the co-evolution of host and viral genomes. PLoS One 4:e4169. 10.1371/journal.pone.0004169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Horvath P, Romero DA, Coute-Monvoisin AC, Richards M, Deveau H, Moineau S, Boyaval P, Fremaux C, Barrangou R. 2008. Diversity, activity, and evolution of CRISPR loci in Streptococcus thermophilus. J. Bacteriol. 190:1401–1412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Seed KD, Lazinski DW, Calderwood SB, Camilli A. 2013. A bacteriophage encodes its own CRISPR/Cas adaptive response to evade host innate immunity. Nature 494:489–491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Weinberger AD, Wolf YI, Lobkovsky AE, Gilmore MS, Koonin EV. 2012. Viral diversity threshold for adaptive immunity in prokaryotes. mBio 3:e00456–12. 10.1128/mBio.00456-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jorth P, Whiteley M. 2012. An evolutionary link between natural transformation and CRISPR adaptive immunity. mBio 3:e00309. 10.1128/mBio.00309-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pleckaityte M, Zilnyte M, Zvirbliene A. 2012. Insights into the CRISPR/Cas system of Gardnerella vaginalis. BMC Microbiol. 12:301. 10.1128/mBio.00309-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Horvath P, Coute-Monvoisin AC, Romero DA, Boyaval P, Fremaux C, Barrangou R. 2009. Comparative analysis of CRISPR loci in lactic acid bacteria genomes. Int. J. Food Microbiol. 131:62–70 [DOI] [PubMed] [Google Scholar]
- 44.Chakraborty S, Snijders AP, Chakravorty R, Ahmed M, Tarek AM, Hossain MA. 2010. Comparative network clustering of direct repeats (DRs) and cas genes confirms the possibility of the horizontal transfer of CRISPR locus among bacteria. Mol. Phylogenet. Evol. 56:878–887 [DOI] [PubMed] [Google Scholar]
- 45.Weinberger AD, Gilmore MS. 2012. CRISPR-Cas: to take up DNA or not-that is the question. Cell Host Microbe 12:125–126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Palmer KL, Gilmore MS. 2010. Multidrug-resistant enterococci lack CRISPR-Cas. mBio 1:e00227. 10.1128/mBio.00227-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Erdmann S, Garrett RA. 2012. Selective and hyperactive uptake of foreign DNA by adaptive immune systems of an archaeon via two distinct mechanisms. Mol. Microbiol. 85:1044–1056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lopez-Sanchez MJ, Sauvage E, Da Cunha V, Clermont D, Ratsima Hariniaina E, Gonzalez-Zorn B, Poyart C, Rosinski-Chupin I, Glaser P. 2012. The highly dynamic CRISPR1 system of Streptococcus agalactiae controls the diversity of its mobilome. Mol. Microbiol. 85:1057–1071 [DOI] [PubMed] [Google Scholar]
- 49.Westra ER, Swarts DC, Staals RH, Jore MM, Brouns SJ, van der Oost J. 2012. The CRISPRs, they are a-changin': how prokaryotes generate adaptive immunity. Annu. Rev. Genet. 46:311–339 [DOI] [PubMed] [Google Scholar]
- 50.Stern A, Keren L, Wurtzel O, Amitai G, Sorek R. 2010. Self-targeting by CRISPR: gene regulation or autoimmunity? Trends Genet. 26:335–340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Paez-Espino D, Morovic W, Sun CL, Thomas BC, Ueda K, Stahl B, Barrangou R, Banfield JF. 2013. Strong bias in the bacterial CRISPR elements that confer immunity to phage. Nat. Commun. 4:14–30 [DOI] [PubMed] [Google Scholar]
- 52.Han P, Niestemski LR, Barrick JE, Deem MW. 2013. Physical model of the immune response of bacteria against bacteriophage through the adaptive CRISPR-Cas immune system. Phys Biol. 10:025004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kupczok A, Bollback JP. 2013. Probabilistic models for CRISPR spacer content evolution. BMC Evol. Biol. 13:54. 10.1186/1471-2148-13-54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.He J, Deem MW. 2010. Heterogeneous diversity of spacers within CRISPR (clustered regularly interspaced short palindromic repeats). Phys. Rev. Lett. 105:128102. [DOI] [PubMed] [Google Scholar]
- 55.Childs LM, Held NL, Young MJ, Whitaker RJ, Weitz JS. 2012. Multiscale model of CRISPR-induced coevolutionary dynamics: diversification at the interface of Lamarck and Darwin. Evolution 66:2015–2029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Haerter JO, Sneppen K. 2012. Spatial structure and Lamarckian adaptation explain extreme genetic diversity at CRISPR locus. mBio 3:e00126–12. 10.1128/mBio.00126-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Haerter JO, Trusina A, Sneppen K. 2011. Targeted bacterial immunity buffers phage diversity. J. Virol. 85:10554–10560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Levin BR. 2010. Nasty viruses, costly plasmids, population dynamics, and the conditions for establishing and maintaining CRISPR-mediated adaptive immunity in bacteria. PLoS Genet. 6:e1001171. 10.1371/journal.pgen.1001171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Levin BR, Moineau S, Bushman M, Barrangou R. 2013. The population and evolutionary dynamics of phage and bacteria with CRISPR-mediated immunity. PLoS Genet. 9:e1003312. 10.1371/journal.pgen.1003312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Weinberger AD, Sun CL, Pluciński MM, Denef VJ, Thomas BC, Horvath P, Barrangou R, Gilmore MS, Getz WM, Banfield JF. 2012. Persisting low-abundance viral sequences shape microbial CRISPR-based immunity. PLoS Comput. Biol. 8:e1002475. 10.1371/journal.pcbi.1002475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hofbauer J, Sigmund K. 1998. Evolutionary games and population dynamics. Cambridge University Press, Cambridge, United Kingdom [Google Scholar]
- 62.Grimm V, Railsback SF. 2005. Individual-based modeling and ecology. Princeton University Press, Princeton, NJ [Google Scholar]
- 63.May RM. 2001. Stability and complexity in model ecosystems. Princeton University Press, Princeton, NJ [Google Scholar]
- 64.Gillespie DT. 1977. Exact stochastic simulation of coupled chemical reactions. J. Phys. Chem. 81:2340–2361 [Google Scholar]
- 65.Breitbart M, Rohwer F. 2005. Here a virus, there a virus, everywhere the same virus? Trends Microbiol. 13:278–284 [DOI] [PubMed] [Google Scholar]
- 66.Suttle CA. 2007. Marine viruses: major players in the global ecosystem. Nat. Rev. Microbiol. 5:801–812 [DOI] [PubMed] [Google Scholar]
- 67.Rohwer F, Truber RV. 2009. Viruses manipulate the marine environment. Nature 459:207–212 [DOI] [PubMed] [Google Scholar]
- 68.Donalson DD, Nisbet RM. 1999. Population dynamics and spatial scale: effects of system size on population persistence. Ecology 80:2492–2507 [Google Scholar]
- 69.Zeldovich KB, Berezovsky IN, Shakhnovich EI. 2007. Protein and DNA sequence determinants of thermophilic adaptation. PLoS Comput. Biol. 3:e5. 10.1371/journal.pcbi.0030005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Drake JW. 2009. Avoiding dangerous missense: thermophiles display especially low mutation rates. PLoS Genet. 5:e1000520. 10.1371/journal.pgen.1000520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Fuhrman JA. 2002. Community structure and function in prokaryotic marine plankton. Antonie Van Leeuwenhoek 81:521–527 [DOI] [PubMed] [Google Scholar]
- 72.Schrenk MO, Kelley DS, Delaney JR, Baross JA. 2003. Incidence and diversity of microorganisms within the walls of an active deep-sea sulfide chimney. Appl. Environ. Microbiol. 69:3580–3592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Whitaker RJ, Banfield JF. 2006. Population genomics in natural microbial communities. Trends Ecol. Evol. 21:508–516 [DOI] [PubMed] [Google Scholar]
- 74.Wilmes P, Simmons SL, Denef VJ, Banfield JF. 2009. The dynamic genetic repertoire of microbial communities. FEMS Microbiol. Rev. 33:109–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hoaki T, Nishijima M, Miyashita H, Maruyama T. 1995. Dense community of hyperthermophilic sulfur-dependent heterotrophs in a geothermally heated shallow submarine biotope near Kodakara-Jima Island, Kagoshima, Japan. Appl. Environ. Microbiol. 61:1931–1937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Takai K, Komatsu T, Inagaki F, Horikoshi K. 2001. Distribution of archaea in a black smoker chimney structure. Appl. Environ. Microbiol. 67:3618–3629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Nakagawa S, Takai K, Inagaki F, Chiba H, Ishibashi J, Kataoka S, Hirayama H, Nunoura T, Horikoshi K, Sako Y. 2005. Variability in microbial community and venting chemistry in a sediment-hosted backarc hydrothermal system: impacts of subseafloor phase-separation. FEMS Microbiol. Ecol. 54:141–155 [DOI] [PubMed] [Google Scholar]
- 78.Zhou H, Li J, Peng X, Meng J, Wang F, Ai Y. 2009. Microbial diversity of a sulfide black smoker in main endeavour hydrothermal vent field, Juan de Fuca Ridge. J. Microbiol. 47:235–247 [DOI] [PubMed] [Google Scholar]
- 79.Flores GE, Wagner ID, Liu Y, Reysenbach AL. 2012. Distribution, abundance, and diversity patterns of the thermoacidophilic “deep-sea hydrothermal vent euryarchaeota 2”. Front. Microbiol. 3:47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Llobet-Brossa E, Rossello-Mora R, Amann R. 1998. Microbial community composition of Wadden Sea sediments as revealed by fluorescence in situ hybridization. Appl. Environ. Microbiol. 64:2691–2696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sahm K, MacGregor BJ, Jorgensen BB, Stahl DA. 1999. Sulphate reduction and vertical distribution of sulphate-reducing bacteria quantified by rRNA slot blot hybridization in a coastal marine sediment. Environ. Microbiol. 1:65–74 [DOI] [PubMed] [Google Scholar]
- 82.Wieringa EB, Overmann J, Cypionka H. 2000. Detection of abundant sulfate-reducing bacteria in marine oxic sediment layers by a combined cultivation and molecular approach. Environ. Microbiol. 2:417–427 [DOI] [PubMed] [Google Scholar]
- 83.Rusch A, Huettel M, Reimers CE, Taghon GL, Fuller CM. 2003. Activity and distribution of bacterial populations in Middle Atlantic Bight shelf sands. FEMS Microbiol. Ecol. 44:89–100 [DOI] [PubMed] [Google Scholar]
- 84.Mussmann M, Ishii K, Rabus R, Amann R. 2005. Diversity and vertical distribution of cultured and uncultured deltaproteobacteria in an intertidal mud flat of the Wadden Sea. Environ. Microbiol. 7:405–418 [DOI] [PubMed] [Google Scholar]
- 85.Musat N, Werner U, Knittel K, Kolb S, Dodenhof T, van Beusekom JE, de Beer D, Dubilier N, Amann R. 2006. Microbial community structure of sandy intertidal sediments in the North Sea, Sylt-Romo Basin, Wadden Sea. Syst. Appl. Microbiol. 29:333–348 [DOI] [PubMed] [Google Scholar]
- 86.Carr SA, Vogel SW, Dunbar RB, Brandes J, Spear JR, Levy R, Naish TR, Powell RD, Wakeham SG, Mandernack KW. 2013. Bacterial abundance and composition in marine sediments beneath the Ross Ice Shelf, Antarctica. Geobiology 11:377–395 [DOI] [PubMed] [Google Scholar]
- 87.Mojica FJ, Diez-Villasenor C, Garcia-Martinez J, Almendros C. 2009. Short motif sequences determine the targets of the prokaryotic CRISPR defense system. Microbiology 155:733–740 [DOI] [PubMed] [Google Scholar]
- 88.Fischer S, Maier LK, Stoll B, Brendel J, Fischer E, Pfeiffer F, Dyall-Smith M, Marchfelder A. 2012. An archaeal immune system can detect multiple protospacer adjacent motifs (PAMs) to target invader DNA. J. Biol. Chem. 287:33351–33363 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.