

Abstract
The recent success in transformation of Chlamydia trachomatis represents a major advancement in Chlamydia research. Plasmid-free C. trachomatis serovar L2 organisms can be transformed with chlamydial plasmid-based shuttle vectors pGFP::SW2 and pBRCT. Deletion of plasmid genes coding for Pgp1 to Pgp8 in pBRCT led to the identification of Pgp1, -2, -6, and -8 as plasmid maintenance factors; Pgp4 as a transcriptional regulator of chlamydial virulence-associated gene expression; and Pgp3, -5, and -7 as being dispensable for chlamydial growth in vitro. Using the pGFP::SW2 vector system, we confirmed these findings in the current report. To further dissect the roles of pgp coding sequences and Pgp proteins in plasmid maintenance, we introduced premature stop codons into the pgp genes. Stable transformants were obtained with pGFP::SW2 derivatives carrying premature stop codons in pgp8 but not in pgp1, pgp2, and pgp6, suggesting that the pgp8 coding sequence but not the Pgp8 protein is required for maintaining the plasmid, while Pgp1, -2, and -6 proteins are necessary for plasmid maintenance. We also found that a minimum of 30 nucleotides in the pgp3 coding region was required for pgp4 expression. Finally, mCherry red fluorescent protein was successfully expressed when the mCherry gene was used to replace the pgp3, pgp4, or pgp5 coding region, indicating that these regions can be used to express nonchlamydial genes in chlamydial organisms. These novel observations have provided information for further use of chlamydial plasmid shuttle vectors as genetic tools to understand chlamydial biology and pathogenicity as well as to develop attenuated chlamydial vaccines.
INTRODUCTION
Chlamydia trachomatis is an obligate, intracellular, Gram-negative bacterium that is composed of various biovars. The ocular strains of trachoma biovar (consisting of serovars A to C) infect mainly ocular epithelial cells, which may lead to blindness (1). The genital strains of trachoma biovar (serovars D to K) cause sexually transmitted infections, which can result in complications such as ectopic pregnancy and tubal factor infertility (2). The lymphogranuloma venereum (LGV) biovar, consisting of serovars L1 to L3, invades mainly genital and rectal epithelial tissues and can cause disseminated infections (3). Although strains of these biovars share a high degree of genetic identity, it has been difficult to determine which of the limited genetic differences among these strains dictates biovar-specific pathogenicity, due to a lack of genetic tools. Nevertheless, it has been proposed that chlamydial intracellular infection-induced inflammatory responses may contribute significantly to pathogenesis (4–7). All C. trachomatis organisms have to replicate inside eukaryotic cells and share a highly conserved biphasic life cycle. C. trachomatis intracellular infection starts with an infectious elementary body (EB) entering an epithelial cell via endocytosis. The intravacuolar EB rapidly differentiates into a reticulate body (RB) that is no longer infectious but is metabolically active and can undergo replication. The progeny RBs eventually differentiate back into EBs for exiting infected cells and spreading to new cells. Although intracellular chlamydial organisms always restrict themselves within cytoplasmic vacuoles, also called inclusions, inflammatory responses are induced during both intracellular growth and cell-to-cell spreading (4, 7, 8), which may contribute significantly to Chlamydia-induced inflammatory pathology. However, it has been difficult to determine which chlamydial virulence factors are responsible and how these factors induce pathology-causing inflammation, due to a lack of convenient genetic tools.
Almost all C. trachomatis clinical isolates contain a highly conserved plasmid that has coevolved with the cognate chlamydial genome (9–11). The plasmid may play a significant role in C. trachomatis pathogenesis, since plasmid-free trachoma serovar A organisms no longer caused pathology in primate ocular tissues (12), which validated the previously reported finding that plasmid-free Chlamydia muridarum organisms were highly attenuated in mouse genital tract (13). The plasmid in C. trachomatis serovar L2 is known to regulate the expression of more than 20 genes at the transcription level (14). Plasmid-free C. trachomatis organisms displayed reduced expression levels of glycogen synthesis genes and lacked glycogen accumulation in inclusions (14). Recently, Wang et al. developed a chlamydial plasmid shuttle vector-based transformation system that allowed the authors to fully complement plasmid-free L2 organisms (15). This technology has offered an opportunity for chlamydiologists to genetically manipulate chlamydial organisms and define the molecular basis of plasmid-dependent pathogenesis (16). The shuttle vector used by Wang et al. was designated pGFP::SW2, which expresses a green florescence protein (GFP) and contains two origins of replication derived from bacterial plasmid pSP73 and a C. trachomatis serovar E plasmid, respectively. To make plasmid open reading frame (pORF) deletion mutants, Song et al. used the pBRCT shuttle vector, which is similar to pGFP::SW2 but without the GFP gene and being derived from L2 plasmid. pORF deletion analysis revealed that Pgp1, -2, -6, and -8 are critical for plasmid maintenance and that Pgp4 is a transcriptional regulator of chlamydial gene expression and glycogen synthesis, while Pgp3, -5, and -7 are dispensable for chlamydial growth in vitro (16). Some of these observations were confirmed by Wang et al. (17, 18).
This study was performed to further characterize chlamydial pORFs. By using a combination of pORF deletion and premature stop codon installation approaches, we found that the protein functions of Pgp1, -2, and -6 were necessary for plasmid maintenance, while the pgp8 DNA sequence but not the function of the Pgp8 protein was required for maintaining the plasmid. We also found that a sequence of 30 nucleotides or more in the pgp3 gene was required for optimal expression of pgp4. More importantly, we found that pgp3, pgp4, and pgp5 coding regions could be used to express foreign genes in chlamydial organisms. These observations have provided novel and important information for further use of the chlamydial plasmid shuttle vector systems to understand chlamydial pathogenic mechanisms and develop attenuated chlamydial vaccines.
MATERIALS AND METHODS
Cell culture and chlamydial infection.
HeLa cells (human cervical epithelial carcinoma cells) (ATCC CCL2), L929 cells (mouse fibroblast cells) (NCTC 929; ATCC CCL-1), as well as C. trachomatis L2 organisms (L2/LGV-434/Bu) were purchased from the ATCC, while L2R [L2(25667R)] organisms were kindly provided by Julius Schachter from the University of California at San Francisco (14, 19, 20). Chlamydial organisms were propagated, purified, aliquoted, and stored as described previously (21, 22). For infection, cells grown in 24-well plates with coverslips or 6-well plates containing Dulbecco's modified Eagle's medium (DMEM; Sigma, St. Louis, MO) with 10% fetal bovine serum (FBS; Gemini Bio-Products, West Sacramento, CA) at 37°C in an incubator supplied with 5% CO2 were inoculated with chlamydial organisms as described previously (23).
Construction of recombinant plasmids for transformation.
For making pORF deletion mutants, the primer pairs listed in Table S1 in the supplemental material were used to amplify two DNA fragments lacking the desired pORF by PCR using AccuPrime Pfx SuperMix (Life Technologies, Grand Island, NY) and pGFP::SW2 (kindly provided by Ian Clark) (15) as the template. For mCherry gene replacement, a third PCR product containing the mCherry gene was generated by using the corresponding primers listed in Table S2 in the supplemental material so that the mCherry gene replaced the coding region of pgp3, pgp4, or pgp5. The PCR conditions were as follows: an initial denaturation step at 95°C for 3 min followed by 35 cycles of amplification at 95°C for 15 s, 55°C for 30 s, and 68°C for 1 min per kb and a final extension step at 68°C for 10 min. PCR products were purified by using the GeneJET PCR purification kit (Fisher Scientific, Pittsburgh, PA) and digested with FastDigest DpnI (Fisher Scientific, Pittsburgh, PA) to remove template DNA. An In-Fusion HD cloning kit (Clontech Laboratories Inc., Mountain View, CA) was used to fuse the desired PCR products according to the manufacturer's instructions. The fusion products were transformed into Stellar competent cells (included in the Clontech In-Fusion HD cloning kit), and the transformants were selected for ampicillin resistance on an LB agar plate. Bacterial colonies were screened for positive green fluorescence by visualization under a fluorescence microscope and a for lack of the intended pORF and/or the presence of the mCherry gene in the correct position by PCR (the screening primers are listed in Table S3 in the supplemental material). Plasmids were extracted from the colonies with the desired screening results, and the purified plasmids were transformed into Escherichia coli K-12 ER2925 (Dam− Dcm− strain; New England BioLabs, Ipswich, MA) for amplification. The final plasmids were extracted, subjected to DNA sequencing analyses, and used for chlamydial transformation.
For constructing nonsense mutants of the pORF, one or two premature stop codons were introduced into each pORF in pGFP::SW2 according to instructions provided by the manufacturer of the QuikChange II site-directed mutagenesis kit (Agilent Technologies, Santa Clara, CA). Briefly, the pGFP::SW2 plasmid was amplified with the appropriate primers (see Table S4 in the supplemental material) that incorporate the desired nucleotide substitutions by using PCR with AccuPrime Pfx SuperMix. The PCR conditions were as follows: an initial denaturation step at 95°C for 30 s followed by 18 cycles of amplification at 95°C for 30 s, 55°C for 1 min, and 68°C for 1 min per kb and then a final extension step at 68°C for 10 min. PCR products were purified by using the GeneJET PCR purification kit and digested with FastDigest DpnI to remove the template DNA. The digested PCR products were purified, recombined, and transformed into E. coli XL-1 Blue competent cells. Plasmids were extracted from bacterial colonies positive for green florescence, and the extracted plasmids were partially sequenced for validation of mutations. The plasmids with the desired mutations were transformed into E. coli K-12 ER2925 for amplification. The amplified plasmids were fully sequenced and used for transforming chlamydial organisms. The final resultant plasmids were designated pGFPpgp1Y16stop (the 16th codon of pgp1 coding for the amino acid tyrosine was converted into a stop codon), pGFPpgp2C7stop, pGFPpgp3L71stop, pGFPpgp4K13stop, pGFPpgp5L9stop, pGFPpgp6K14stop, pGFPpgp7L11stop, pGFPpgp8E11stop, and pGFPpgp8K188stop.
Transformation of plasmid-free C. trachomatis L2R organisms.
Chlamydial transformation was performed by using the transformation protocol developed by Wang et al. (15), with modifications. Briefly, 10 μl L2R organisms (1 × 107 inclusion-forming units [IFU]) and 10 μl plasmid DNA (∼7 μg) were mixed in a total volume of 200 μl CaCl2 buffer for 45 min at room temperature. Freshly trypsinized L929 cells (6 × 106 cells) resuspended in 200 μl CaCl2 buffer were added to the EB-plasmid mixture and incubated for a further 20 min at room temperature, with occasional mixing. Each 70 μl of the final mixture was plated onto a single well of a six-well plate together with 1.5 ml of prewarmed DMEM plus 10% FBS. The cells were allowed to adhere to the culture plate by incubation at 37°C in 5% CO2 without cycloheximide or ampicillin for 24 h. The cultures were replenished with fresh DMEM–10% FBS containing cycloheximide (2 μg/ml) and ampicillin (5 μg/ml) and incubated for an additional 24 h. Inclusions positive for green fluorescence were identified under a fluorescence microscope and picked up for passage on a fresh monolayer of HeLa cells in the presence of ampicillin (20 μg/ml). The resultant inclusions remaining positive for green fluorescence were defined as generation 2 and were passed for an additional 3 to 4 generations to enrich fluorescence-positive organisms, which were finally plaque cloned, as described previously (24), for further experiments.
Live-cell-culture microscopy and indirect immunofluorescence assay.
An IX-81 inverted fluorescence microscope (Olympus, Center Valley, PA) was used to visualize live cells without or with infection by L2, L2R, or L2R transformants. Simple PCI imaging software (Olympus) was used to acquire both bright-field and green (GFP) or red (mCherry) fluorescence images, and Adobe Photoshop (Adobe, San Jose, CA) was used for postacquisition processing of the images.
For immunofluorescence images, HeLa cells with or without chlamydial infection were fixed at 30 h postinfection with 2% paraformaldehyde dissolved in phosphate-buffered saline (PBS) for 30 min at room temperature, followed by permeabilization with 2% saponin (Sigma, St. Louis, MO) for an additional 30 min. After blocking, the cell samples were subjected to antibody and chemical staining. Hoechst 33258 (blue; Sigma) was used to visualize DNA (blue). A rabbit antichlamydial organism antibody (R1L2 [25]) plus a goat anti-rabbit IgG secondary antibody conjugated with Alexa Fluor 488 (green; Jackson ImmunoResearch, West Grove, PA) were used to visualize chlamydial organisms. The mouse anti-GlgA (glycogen synthase A) antibody (raised with a glutathione S-transferase [GST]-GlgA fusion protein [our unpublished data]) plus a goat anti-mouse IgG conjugated with Cy3 (red; Jackson ImmunoResearch) were used to visualize GlgA. The immunofluorescence images were acquired by using an Olympus AX-70 fluorescence microscope equipped with multiple filter sets and Simple PCI imaging software, as described previously (26).
Iodine staining.
HeLa cells with or without chlamydial infection were fixed with ice-cold methanol for 10 min. The cell samples were stained with 5% iodine stain (5% potassium iodide and 5% iodine in 50% ethanol) for 40 min. The coverslips were mounted in 50% glycerol containing 5% potassium iodide and 5% iodine. The images were acquired by using an Olympus CH-30 microscope equipped with a Canon EOS Rebel T3i Digital SLR camera. The images were processed by using Adobe Photoshop.
Quantitative real-time RT-PCR.
HeLa cells grown in 6-well plates (1 × 106 cells/well) were infected with EBs at a multiplicity of infection (MOI) of 2. Thirty hours after infection, the cells were harvested by using TRIzol reagent (Life Technologies, Grand Island, NY), and the total RNA from each sample was extracted with a Direct-zol RNA Miniprep kit (Zymo Research, Irvine, CA). The RNA preparations were used for cDNA synthesis with random hexamer primers by using a ThermoScript reverse transcription-PCR (RT-PCR) system (Life Technologies, Grand Island, NY). TaqMan RT-PCR assays were then performed by using a CFX96 Touch Deep Well real-time PCR detection system (Bio-Rad, Hercules, CA) with iQ Supermix (Bio-Rad). The gene-specific primers used (see Table S5 in the supplemental material) included unlabeled primers and a double-quenched probe (5′FAM/ZEN/3′IBFQ; Integrated DNA Technologies, Coralville, IA). The PCR conditions were as follows: an initial denaturation step at 95°C for 3 min followed by 40 cycles of amplification at 95°C for 15 s and 60°C for 1 min. The transcript copy numbers for a given gene from triplicate samples were calculated based on a standard plasmid DNA preparation and further normalized to the copy numbers of chlamydial lpdA mRNA in the corresponding samples.
RESULTS
Targeted ORF deletion in pGFP::SW2 results in phenotypes similar to those observed for pBRCT ORF deletion mutants and reveals a requirement of 30 nucleotides in the pgp3 coding region for pgp4 expression.
The analysis of an individual plasmid ORF deletion mutation in the pBRCT shuttle vector led Song et al. to discover that Pgp1, -2, -6, and -8 are required for plasmid maintenance and that Pgp4 is a regulator of chlamydial gene expression and glycogen synthesis (16). We tested whether similar phenotypes can be obtained with the pGFP::SW2 shuttle vector, as pBRCT is derived from a serovar L2 strain (15, 16), while pGFP::pSW2 is derived from a natural mutant serovar E plasmid that possesses a distinct 377-bp deletion in pgp7 and a 44-bp duplication upstream of pgp1 (15). Deletion of each of the 8 full-length ORFs in the pGFP::SW2 plasmid was performed, and the resultant plasmids were designated pGFPΔPgp1 to pGFPΔPgp8 (Fig. 1A and B). These plasmids together with pGFP::SW2 and pBRCT were transformed into plasmid-free L2 organisms (L2R), as described previously (15, 16). The corresponding transformants were designated L2RpGFPΔPgp1 to L2RpGFPΔPgp8, L2RpGFP, and L2RpBRCT, respectively. Stable transformants were obtained only with pGFPΔPgp3, -4, -5, and -7 and not with pGFPΔPgp1, -2, -6, and -8 (Fig. 1C and data not shown), in complete agreement with previous observations made with the pBRCT vector system (16). Clearly, Pgp1, -2, -6, and -8 but not Pgp3, -4, -5, and -7 were required for plasmid maintenance. As shown in both Fig. 1C and 2, the impacts of the Pgp3, -4, -5, or -7 deletion on the expression of other chlamydial genes and glycogen synthesis were also similar to those observed previously (16). L2RpGFPΔPgp4 but not L2RpGFPΔPgp5 or L2RpGFPΔPgp7 displayed significantly reduced transcript levels of plasmid genes as well as the chromosomal genes CTL0397 and CTL0167 (glgA) but not CTL0814. Moreover, the transformant L2RpGFPΔPgp4 failed to produce GlgA protein or accumulate glycogen (Fig. 1C). Surprisingly, L2RpGFPΔPgp3 displayed a phenotype similar to that of L2RpGFPΔPgp4 (Fig. 1 and 2), which was quite different from what Song et al. observed (16). L2RpBRCTΔPgp3 produced 55 copies of normalized pgp4 transcripts, representing ∼46% of the pgp4 mRNA level in the control transformant L2RpBRCT (Fig. 3B). Apparently, the slight reduction in the pgp4 expression level did not affect the ability of L2RpBRCTΔPgp3 to express detectable levels of GlgA protein or to accumulate detectable levels of glycogen (Fig. 3B). However, L2RpGFPΔPgp3 expressed only ∼5 copies of the pgp4 transcript, representing only ∼6% of the pgp4 mRNA level in fully complemented L2R organisms (L2RpGFP). This severe reduction in Pgp4 expression resulted in a lack of detection of GlgA protein or glycogen. These observations suggested that deletion of the full-length pgp3 coding region in pGFP::SW2 negatively affected downstream Pgp4 expression.
Fig 1.

Effect of pORF deletion on GlgA protein expression and glycogen synthesis. (A) Each of the 8 individual ORFs from the shuttle vector pGFP::SW2 was deleted, with the deletion region indicated. (B) The deletion mutants, designated pGFPΔPgp1 to pGFPΔPgp8, together with the parent plasmid (pGFP::SW2), as listed on top of the gel image, were detected for the presence of each of the 8 pORFs (from top to bottom rows) by PCR. (C) The 9 plasmids were each transformed into plasmid-free L2 (L2R). However, stable L2R transformants were obtained with only 5 out of the 9 plasmids. The 5 stable transformants, designated L2RpGFP::SW2, L2RpGFPΔPgp3, L2RpGFPΔPgp4, L2RpGFPΔPgp5, and L2RpGFPΔPgp7, along with wild-type L2 (L2 Wt) and plasmid-free L2 (L2R) organisms were used to infect HeLa cells. The infected cells were monitored for live-cell bright-field appearance (a to g) and green fluorescence intensity (GFP) (h to n); triple immunofluorescence labeling for GlgA protein (red), chlamydial organisms (green), and DNA (blue) (o to u); and iodine staining of glycogen accumulation (v to bb). Inclusions positive for glycogen staining are marked with white arrows, while those with a lack of glycogen staining are marked with white arrowheads. Note that both GlgA expression and glycogen accumulation were restored in L2R organisms transformed with plasmid pGFP::SW2 (q and x) and plasmids with a deletion of pgp5 (t and aa) or pgp7 (u and bb) but not in plasmids with a deletion of pgp4 (s and z) or pgp3 (r and y).
Fig 2.

Effect of pORF deletion on expression of other genes. HeLa cell were infected with wild-type L2 (L2 Wt), plasmid-free L2 [L2R(25667)], or various stable L2R transformants, including L2RpGFP::SW2, L2RpGFPΔpgp3, L2RpGFPΔpgp4, L2RpGFPΔpgp5, and L2RpGFPΔpgp7, and the infected cultures were harvested at 30 h postinfection for total RNA preparation. The transcript copy number was determined from three replicates by using gene-specific primer/probe sets and TaqMan quantitative RT-PCR and was normalized to the lpdA mRNA copy number in the corresponding samples, since lpdA is constitutively expressed. Three groups of genes were detected, including plasmid-borne genes (pgp1 to pgp8), plasmid-regulated chromosomal genes (CTL0397, CTL0814, and CTL0167 [glgA]), and the non-plasmid-regulated chromosomal gene CTL0050 (ompA) (encoding the major outer membrane protein). Note that there is a significant reduction in the expression level of plasmid-regulated genes (except for CTL0814) by L2R, L2RpGFPΔPgp3, and L2RpGFPΔPgp4 but not by L2RpGFPΔPgp5 and L2RpGFPΔPgp7 organisms.
Fig 3.

Effect of pgp3 coding sequence deletion on pgp4 expression. (A) The pgp3 coding region in the pGFP::SW2 shuttle vector was completely deleted (L2RpGFPΔPgp3) or partially deleted by keeping the 3′ 72, 30, or 15 nucleotides (nt) of pgp3, resulting in L2RpGFPΔPgp3(72), L2RpGFPΔPgp3(30), and L2RpGFPΔPgp3(15), respectively. pBRCTΔPgp3 has the 3′ 72 nucleotides of the pgp3 sequence. (*Information on transcripts driven by promoter 3 [P3] and promoter 4 [P4] was reported in reference 9.) The relative amounts of transcripts are reflected by the thickness of the lines. (B) Both the pBRCT and pGFP::SW2 shuttle vectors and the derived constructs were used to transform L2R organisms. The corresponding transformants along with the plasmid-free L2R organisms, as listed on top of the images, were used to infect HeLa cells. The infected cultures were either harvested for quantitation of pgp4 and ompA transcript levels (top portion) by quantitative RT-PCR or processed for triple labeling of GlgA (red), chlamydial organisms (green), and DNA (blue) (a to h) or iodine staining (i to p). Inclusions positive for glycogen staining are marked with white arrows, while those with a lack of glycogen staining are marked with white arrowheads. Note that the pgp3 deletion mutants carrying 30 or more nucleotides in the 3′ pgp3 coding region express high levels of pgp4 transcripts and GlgA protein and accumulate glycogen.
Careful sequence comparison revealed that pBRCTΔPgp3 contained a 3′ region of 60 nucleotides of pgp3 (16) and 12 additional unrecognized nucleotides (data not shown), but pGFPΔPgp3 did not (Fig. 3A). To examine whether this 3′ pgp3 coding region was critical for pgp4 expression, we constructed a series of pgp3 deletion mutants in pGFP::SW2 by keeping 72, 30, or 15 nucleotides of the 3′ pgp3 coding sequence. The resultant L2R transformants, designated L2RpGFPΔPgp3(72), L2RpGFPΔPgp3(30), and L2RpGFPΔPgp3(15), respectively, were compared for their abilities to express pgp4 and synthesize GlgA protein and glycogen (Fig. 3B). We found that the longer the 3′ pgp3 region remained, the higher level of pgp4 was expressed. While 36 normalized copies of pgp4 transcripts were expressed in L2RpGFPΔPgp3(72) organisms, representing ∼46% of the normalized pgp4 transcripts in L2RpGFP organisms, there were 21 in L2RpGFPΔPgp3(30), representing 27%, and only 4 in L2RpGFPΔPgp3(15), representing only 5% of normalized pgp4 transcripts. Interestingly, both GlgA protein and glycogen were detected in cells infected with L2RpGFPΔPgp3(72) or L2RpGFPΔPgp3(30) but not in cells infected with L2RpGFPΔPgp3(15). We reasoned that a minimum of 30 nucleotides in the 3′ pgp3 region was sufficient for pgp4 to be expressed at a level high enough for GlgA protein and glycogen to be detectable. These observations demonstrated that a DNA sequence space between the pgp3 and pgp4 promoter regions is required for proper expression of pgp4.
Introduction of premature stop codons reveals the requirement for the pgp8 coding DNA sequence but not the pgp8-encoded protein for plasmid maintenance.
Although deletion of individual ORFs in their entirety can provide important information on the function of the deleted ORFs, it fails to distinguish the roles of the coding DNA sequences from those of the encoded protein products. Thus, we next undertook nonsense mutation by introducing a premature stop codon into each of the 8 plasmid-encoded ORFs (Fig. 4). The abilities of the resulting L2R transformants to synthesize GlgA protein and glycogen were examined. No stable transformants were obtained with plasmids that carry premature stop codons in Pgp1, -2, and -6, suggesting that these pgp DNA coding sequences are not sufficient but that the encoded proteins are required for plasmid maintenance. Surprisingly, stable transformants were obtained with plasmids carrying a stop codon at the 11th glutamic acid codon of Pgp8 (designated L2RpGFPPgp8E11stop) or a stop codon at the 188th lysine codon of Pgp8 (L2RpGFPPgp8K188stop). Please note that the mutant with a deletion of the entire pgp8 coding region (e.g., pGFPΔPgp8) failed to stably transform L2R (Fig. 1). These observations together suggested that it was the DNA sequence of pgp8 but not the Pgp8 protein that was required for maintaining plasmid viability in chlamydial organisms. The introduction of premature stop codons into Pgp3, -4, -5, and -7 coding regions resulted in the C. trachomatis transformants L2RpGFPPgp3L71stop, L2RpGFPPgp4K13stop, L2RpGFPPgp5L9stop, and L2RpGFPPgp7L11stop, respectively. All four transformant phenocopied the corresponding ORF deletion mutants (Fig. 4 versus Fig. 1). Neither ORF deletion nor a premature stop in pgp3, pgp5, and pgp7 affected plasmid replication or GlgA and glycogen syntheses, while both pgp4 deletion and the premature stop mutants significantly reduced GlgA levels and glycogen accumulation.
Fig 4.
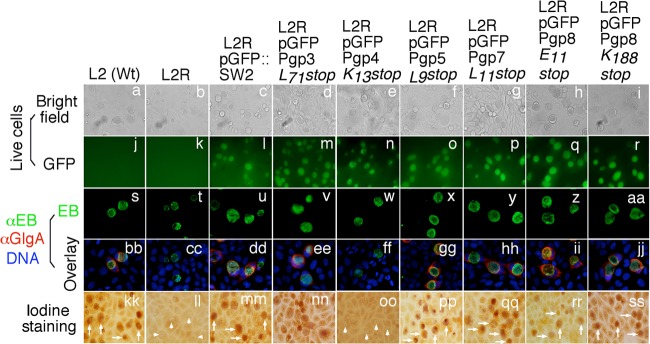
Effect of pORF premature stop on syntheses of GlgA protein and glycogen. A premature stop codon was introduced into each of the 8 pORFs. Stable transformants, designated L2RpGFPPgp3L71stop, L2RpGFPpgp4K13stop, L2RpGFPpgp5L9stop, L2RpGFPpgp7L11stop, L2RpGFPPgp8E11stop, and L2RpGFPPgp8K188stop, together with wild-type L2 [L2 (Wt)], plasmid-free L2 (L2R), and L2R transformed with plasmid L2RpGFP::SW2, as listed on top of the images, were used to infect HeLa cells. The infected cultures were monitored for live-cell bright-field appearance (a to i) and green fluorescence intensity (GFP) (j to r); triple labeled for GlgA protein (red) (bb to jj), chlamydial organisms (green) (s to aa), and DNA (blue) (bb to jj); and finally stained for glycogen accumulation with iodine (kk to ss). Inclusions positive for glycogen staining are marked with white arrows, while those with a lack of glycogen staining are marked with white arrowheads. Note that L2R transformed with pGFPPgp8E11stop or pGFPPgp8K188stop restored GlgA expression and glycogen accumulation (ii, jj, rr, and ss). L2RpGPFPgp3L71stop, L2RpGFPpgp4K13stop, L2RpGFPpgp5L9stop, and L2RpGFPpgp7L11stop phenocopied their corresponding ORF deletion mutants.
The mCherry red fluorescent protein gene can be used to replace the pgp3, pgp4, and pgp5 coding regions for expression in C. trachomatis.
To further explore the utility of the chlamydial plasmid shuttle vector pGFP::SW2, we used an mCherry red fluorescent protein gene to replace the coding region of pgp3, pgp4, or pgp5 so that the mCherry gene was controlled by the native chlamydial promoter(s) of pgp3, pgp4, or pgp5. The resultant L2R organisms transformed with the three mCherry replacement mutants were designated L2RpGFPmCherry(Pgp3), L2RpGFPmCherry(Pgp4), and L2RpGFPmCherry(Pgp5), respectively. These transformants were assessed for mCherry protein expression and glycogen synthesis (Fig. 5). We found that the mCherry protein was successfully expressed regardless of the pgp coding regions that were replaced. This observation indicates that the native chlamydial promoters and the ribosome-binding site upstream of the cloned mCherry gene are functional in the transformed chlamydial organisms. As expected, these C. trachomatis transformants largely phenocopied the corresponding ORF deletion mutants, including L2RpGFPDPgp3(72), L2RpGFPDPgp4, and L2RpGFPDPgp5. Both L2RpGFPmCherry(Pgp3) and L2RpGFPmCherry(Pgp5) displayed glycogen accumulation, whereas L2RpGFPmCherry(Pgp4) lacked glycogen accumulation, likely due to the reduced expression level of GlgA, which is consistent with the phenotype of the Pgp4 ORF deletion mutant (L2RpGFPDPgp4). Next, we evaluated the effect of mCherry expression on C. trachomatis gene expression by quantitative RT-PCR. Like L2RpGFPΔPgp4, L2RpGFPmCherry(Pgp4) organisms displayed significantly reduced levels of pgp3 but normal levels of ompA transcript, suggesting that Pgp4 positively regulates pgp3 expression, which is consistent with previously reported observations (16). Interestingly, L2RpGFPmCherry(Pgp3) but not L2RpGFPmCherry(Pgp5) organisms showed much higher expression levels of pgp4, glgA, and ompA transcripts than did L2RpGFP::SW2 organisms (Fig. 5B). In all, these mCherry replacement experiments have allowed us to obtain proof-of-concept evidence for using the chlamydial plasmid to express nonchlamydial genes.
Fig 5.

Expression of mCherry red fluorescent protein using chlamydial shuttle vector pGFP::SW2. The mCherry gene was used to replace the coding sequences of pgp3, pgp4, and pgp5. The replacement constructs were transformed into L2R, and stable transformants, designated L2RpGFPmCherry(pgp3), L2RpGFPmCherry(pgp4), and L2RpGFPmCherry(pgp5), respectively, were obtained. These transformants together with wild-type L2 [L2 (Wt)], plasmid-free L2 (L2R), and L2R transformed with a plasmid (L2RpGFP::SW2), as listed on top of the images, were used to infect HeLa cells. (A) The infected cultures were processed for monitoring red fluorescence intensity (mCherry; red) (a to f) and green fluorescence intensity (GFP; green) (g to l), labeling DNA (blue) (m to r), and, finally, staining for glycogen accumulation with iodine (s to x). Inclusions positive for glycogen staining are marked with white arrows, while those with a lack of glycogen staining are marked with white arrowheads. Note that red fluorescence was detected when the mCherry gene was used to replace pgp3 (d), pgp4 (e), or pgp5 (f). (B) The expression levels of pgp3, pgp4, and pgp5 as well as of the glgA and ompA genes were quantitated by quantitative RT-PCR. Note that the mCherry replacement mutants largely phenocopied the corresponding ORF deletion mutants, including L2RpGFPΔPgp3(72), L2RpGFPΔPgp4, and L2RpGFPΔPgp5.
DISCUSSION
Previous studies have shown that plasmid-free C. trachomatis serovar L2 organisms can be transformed with chlamydial plasmid-based shuttle vectors (15, 16) and that plasmid-encoded Pgp1, -2, -6, and -8 are required for plasmid maintenance, while Pgp4 is a transcriptional regulator of chlamydial gene expression and glycogen synthesis (16). In the current study, we have not only confirmed these previous findings using the pGFP::SW2 shuttle vector system but also, importantly, unraveled novel information on plasmid maintenance, gene expression, and the utility of this vector for expressing nonchlamydial genes. First, by using a combination of deletion and nonsense substitution mutation approaches, we were able to discern that the protein functions of Pgp1, -2, and -6 but not those of Pgp8 were required for plasmid maintenance. Second, we identified a minimum of 30 nucleotides in the coding region of pgp3 as necessary for optimal expression of pgp4. Finally, we found that nonchlamydial genes could be cloned to replace pgp3, pgp4, or pgp5 coding regions and expressed in chlamydial organisms. These novel observations are significant because they will promote the utility of the chlamydial plasmid shuttle vector systems for further understanding chlamydial pathogenic mechanisms and developing vaccines.
By simultaneously analyzing pORF deletion and nonsense substitution mutants, we have been able to differentiate the roles of the plasmid DNA coding sequences versus the encoded proteins in chlamydial plasmid maintenance. The observation that Pgp1 protein function is required for plasmid maintenance is consistent with the predicted function of Pgp1 as a DnaB-like helicase (10, 11, 27). Although Pgp2 and Pgp6 are Chlamydia-specific proteins of unknown function, both this study and the previous study by Song et al. (16) suggest that the Pgp2 and -6 proteins are required for plasmid maintenance. An unexpected finding is that while the pGFPΔPgp8 plasmid is nontransformable, both pGFPPgp8E11stop and pGFPPgp8K188stop stably transformed L2R. The dual mutants were created to address the concern of the potential alternative start codons in the 5′ region of the pgp8 gene. The fact that pGFPPgp8K188stop phenocopied pGFPPgp8E11stop suggests that Pgp8 protein initiated at any codons upstream of K188 is not required for plasmid maintenance. It is unlikely that Pgp8 protein fragments initiated downstream of K188 are functional. Thus, we can conclude that the DNA coding sequence but not the encoded Pgp8 protein is required for plasmid maintenance. The question is how the Pgp8 DNA coding sequence contributes to chlamydial plasmid maintenance. The chlamydial plasmid is known to encode various small noncoding RNAs (ncRNAs), including sRNA-2 (antisense to the 3′ end of pgp8) and sRNA-7 (antisense to the 3′ end of pgp5) (28–30). Despite the truncation or lack of Pgp8 protein in L2RpGFPPgp8E11stop organisms, the region encoding sRNA-2 remains intact and is likely active. Thus, sRNA-2 may play an important role in the viability of the plasmid in C. trachomatis. This hypothesis is consistent with previous findings that sRNA-2 was expressed in both EBs and RBs (29, 31) and was highly abundant (30). It remains unknown whether any other features of the pgp8 DNA coding sequence can contribute to plasmid maintenance. Nevertheless, we can conclude that sRNA-7 encoded in the pgp5 coding region may not be important for plasmid maintenance, since deletion of sRNA-7 did not affect the viability of the plasmid in L2RpGFPΔPgp5 organisms. Further studies to elucidate the precise roles of plasmid-encoded ncRNAs in chlamydial pathogenesis are ongoing.
Another surprising finding is the impact of pgp3 on pgp4 expression. The L2RpGFPΔPgp3 organisms with a deletion of the entire pgp3 gene displayed a 94% reduction in the pgp4 mRNA level compared to the parent L2RpGFP organisms. This severe reduction in pgp4 expression resulted in a lack of detection of GlgA protein or glycogen, which can be reversed by keeping 30 or more nucleotides of the 3′ pgp3 coding region, as in L2RpGFPDPgp3(30) organisms. It seems that the precise nucleotide composition of the DNA sequence is not important, since pgp4 was fully expressed when the entire pgp3 coding region was replaced with an mCherry gene (Fig. 5). An explanation for the disparity in pgp4 expression levels between L2RpGFPΔPgp3 and L2RpGFPΔPgp3(30) is that the shortened space between promoter 3 (P3) and promoter 4 (P4) in L2RpGFPΔPgp3 organisms might lead to interference with the functions of both P3 and P4. Thus, despite the fact that pgp4 expression can be initiated from both P4 and P3 (Fig. 3A) (9, 32), the close-proximity-induced interference between P3 and P4 still resulted in a significant reduction in pgp4 expression. Because pgp3 expression is regulated by Pgp4, as demonstrated in both the current (Fig. 2) and previous (16) studies, the finding of Pgp3 affecting pgp4 expression leaves open the possibility that there might be transcriptional and/or posttranscriptional feedback loops in regulating the expression of the pgp3 and pgp4 genes during C. trachomatis infection.
In the current study, we confirmed that Pgp3, -4, -5, and -7 are not required for plasmid maintenance, since mutant plasmids with deletions of these individual ORFs can be stably transformed in L2R organisms. This finding was validated by using the premature stop codon introduction approach. We then took advantage of this finding and used these sites to express foreign genes in chlamydial organisms. As a proof of principle, we used the mCherry red fluorescent protein gene to replace pgp3, pgp4, and pgp5 and found that mCherry was successfully expressed regardless of the site of replacement. Similarly, Wang et al. showed that β-galactosidase was expressed under the control of a chlamydial phage promoter when the β-galactosidase gene was used to replace pgp3 (18). Together, these observations have demonstrated the plasticity of the chlamydial plasmid as an expression vector. Florescence-labeled L2RpGFPmCherry(Pgp3), L2RpGFPmCherry(Pgp4), or L2RpGFPmCherry(Pgp5) organisms are being used to facilitate the identification of transcription elements and to understand how the transcription elements work during the chlamydial developmental cycle. We are also in the process of replacing pgp3, pgp4, and pgp5 with vaccine antigen and immunostimulatory molecule genes.
Supplementary Material
Footnotes
Published ahead of print 21 June 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.00511-13.
REFERENCES
- 1.Baneke A. 2012. Targeting trachoma: strategies to reduce the leading infectious cause of blindness. Travel Med. Infect. Dis. 10:92–96 [DOI] [PubMed] [Google Scholar]
- 2.Sherman KJ, Daling JR, Stergachis A, Weiss NS, Foy HM, Wang SP, Grayston JT. 1990. Sexually transmitted diseases and tubal pregnancy. Sex. Transm. Dis. 17:115–121 [DOI] [PubMed] [Google Scholar]
- 3.Vargas-Leguas H, Garcia de Olalla P, Arando M, Armengol P, Barbera M, Vall M, Vives A, Martin-Ezquerra G, Alsina M, Blanco J, Munoz C, Caballero E, Andreu A, Ros M, Gorrindo P, Dominguez A, Cayla J. 2012. Lymphogranuloma venereum: a hidden emerging problem, Barcelona, 2011. Euro Surveill. 17(2):pii=20057 http://www.eurosurveillance.org/ViewArticle.aspx?ArticleId=20057 [PubMed] [Google Scholar]
- 4.Stephens RS. 2003. The cellular paradigm of chlamydial pathogenesis. Trends Microbiol. 11:44–51 [DOI] [PubMed] [Google Scholar]
- 5.Rasmussen SJ, Eckmann L, Quayle AJ, Shen L, Zhang YX, Anderson DJ, Fierer J, Stephens RS, Kagnoff MF. 1997. Secretion of proinflammatory cytokines by epithelial cells in response to Chlamydia infection suggests a central role for epithelial cells in chlamydial pathogenesis. J. Clin. Invest. 99:77–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cheng W, Shivshankar P, Li Z, Chen L, Yeh IT, Zhong G. 2008. Caspase-1 contributes to Chlamydia trachomatis-induced upper urogenital tract inflammatory pathologies without affecting the course of infection. Infect. Immun. 76:515–522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cheng W, Shivshankar P, Zhong Y, Chen D, Li Z, Zhong G. 2008. Intracellular interleukin-1alpha mediates interleukin-8 production induced by Chlamydia trachomatis infection via a mechanism independent of type I interleukin-1 receptor. Infect. Immun. 76:942–951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Buchholz KR, Stephens RS. 2006. Activation of the host cell proinflammatory interleukin-8 response by Chlamydia trachomatis. Cell. Microbiol. 8:1768–1779 [DOI] [PubMed] [Google Scholar]
- 9.Ricci S, Ratti G, Scarlato V. 1995. Transcriptional regulation in the Chlamydia trachomatis pCT plasmid. Gene 154:93–98 [DOI] [PubMed] [Google Scholar]
- 10.Li Z, Chen D, Zhong Y, Wang S, Zhong G. 2008. The chlamydial plasmid-encoded protein pgp3 is secreted into the cytosol of Chlamydia-infected cells. Infect. Immun. 76:3415–3428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Seth-Smith HM, Harris SR, Persson K, Marsh P, Barron A, Bignell A, Bjartling C, Clark L, Cutcliffe LT, Lambden PR, Lennard N, Lockey SJ, Quail MA, Salim O, Skilton RJ, Wang Y, Holland MJ, Parkhill J, Thomson NR, Clarke IN. 2009. Co-evolution of genomes and plasmids within Chlamydia trachomatis and the emergence in Sweden of a new variant strain. BMC Genomics 10:239. 10.1186/1471-2164-10-239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kari L, Whitmire WM, Olivares-Zavaleta N, Goheen MM, Taylor LD, Carlson JH, Sturdevant GL, Lu C, Bakios LE, Randall LB, Parnell MJ, Zhong G, Caldwell HD. 2011. A live-attenuated chlamydial vaccine protects against trachoma in nonhuman primates. J. Exp. Med. 208:2217–2223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.O'Connell CM, Ingalls RR, Andrews CW, Jr, Scurlock AM, Darville T. 2007. Plasmid-deficient Chlamydia muridarum fail to induce immune pathology and protect against oviduct disease. J. Immunol. 179:4027–4034 [DOI] [PubMed] [Google Scholar]
- 14.Carlson JH, Whitmire WM, Crane DD, Wicke L, Virtaneva K, Sturdevant DE, Kupko JJ, III, Porcella SF, Martinez-Orengo N, Heinzen RA, Kari L, Caldwell HD. 2008. The Chlamydia trachomatis plasmid is a transcriptional regulator of chromosomal genes and a virulence factor. Infect. Immun. 76:2273–2283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang Y, Kahane S, Cutcliffe LT, Skilton RJ, Lambden PR, Clarke IN. 2011. Development of a transformation system for Chlamydia trachomatis: restoration of glycogen biosynthesis by acquisition of a plasmid shuttle vector. PLoS Pathog. 7:e1002258. 10.1371/journal.ppat.1002258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Song L, Carlson JH, Whitmire WM, Kari L, Virtaneva K, Sturdevant DE, Watkins H, Zhou B, Sturdevant GL, Porcella SF, McClarty G, Caldwell HD. 2013. Chlamydia trachomatis plasmid-encoded Pgp4 is a transcriptional regulator of virulence-associated genes. Infect. Immun. 81:636–644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang Y, Cutcliffe LT, Skilton RJ, Persson K, Bjartling C, Clarke IN. 2013. Transformation of a plasmid-free, genital tract isolate of Chlamydia trachomatis with a plasmid vector carrying a deletion in CDS6 revealed that this gene regulates inclusion phenotype. Pathog. Dis. 67:100–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang Y, Kahane S, Cutcliffe LT, Skilton RJ, Lambden PR, Persson K, Bjartling C, Clarke IN. 2013. Genetic transformation of a clinical (genital tract), plasmid-free isolate of Chlamydia trachomatis: engineering the plasmid as a cloning vector. PLoS One 8:e59195. 10.1371/journal.pone.0059195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ripa T, Nilsson PA. 2007. A Chlamydia trachomatis strain with a 377-bp deletion in the cryptic plasmid causing false-negative nucleic acid amplification tests. Sex. Transm. Dis. 34:255–256 [DOI] [PubMed] [Google Scholar]
- 20.Schachter J. 2007. The Chlamydia trachomatis plasmid deletion mutant—what does it mean to us? Sex. Transm. Dis. 34:257. [DOI] [PubMed] [Google Scholar]
- 21.Bose SK, Paul RG. 1982. Purification of Chlamydia trachomatis lymphogranuloma venereum elementary bodies and their interaction with HeLa cells. J. Gen. Microbiol. 128:1371–1379 [DOI] [PubMed] [Google Scholar]
- 22.Mukhopadhyay S, Clark AP, Sullivan ED, Miller RD, Summersgill JT. 2004. Detailed protocol for purification of Chlamydia pneumoniae elementary bodies. J. Clin. Microbiol. 42:3288–3290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhong G, Fan P, Ji H, Dong F, Huang Y. 2001. Identification of a chlamydial protease-like activity factor responsible for the degradation of host transcription factors. J. Exp. Med. 193:935–942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Matsumoto A, Izutsu H, Miyashita N, Ohuchi M. 1998. Plaque formation by and plaque cloning of Chlamydia trachomatis biovar trachoma. J. Clin. Microbiol. 36:3013–3019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gong S, Lei L, Chang X, Belland R, Zhong G. 2011. Chlamydia trachomatis secretion of hypothetical protein CT622 into host cell cytoplasm via a secretion pathway that can be inhibited by the type III secretion system inhibitor compound 1. Microbiology 157:1134–1144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fan T, Lu H, Hu H, Shi L, McClarty GA, Nance DM, Greenberg AH, Zhong G. 1998. Inhibition of apoptosis in chlamydia-infected cells: blockade of mitochondrial cytochrome c release and caspase activation. J. Exp. Med. 187:487–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Comanducci M, Ricci S, Cevenini R, Ratti G. 1990. Diversity of the Chlamydia trachomatis common plasmid in biovars with different pathogenicity. Plasmid 23:149–154 [DOI] [PubMed] [Google Scholar]
- 28.Ricci S, Cevenini R, Cosco E, Comanducci M, Ratti G, Scarlato V. 1993. Transcriptional analysis of the Chlamydia trachomatis plasmid pCT identifies temporally regulated transcripts, anti-sense RNA and sigma 70-selected promoters. Mol. Gen. Genet. 237:318–326 [DOI] [PubMed] [Google Scholar]
- 29.Albrecht M, Sharma CM, Reinhardt R, Vogel J, Rudel T. 2010. Deep sequencing-based discovery of the Chlamydia trachomatis transcriptome. Nucleic Acids Res. 38:868–877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ferreira R, Borges V, Nunes A, Borrego MJ, Gomes JP. 2013. Assessment of the load and transcriptional dynamics of Chlamydia trachomatis plasmid according to strains' tissue tropism. Microbiol. Res. 168(6):333–339 [DOI] [PubMed] [Google Scholar]
- 31.Fahr MJ, Sriprakash KS, Hatch TP. 1992. Convergent and overlapping transcripts of the Chlamydia trachomatis 7.5-kb plasmid. Plasmid 28:247–257 [DOI] [PubMed] [Google Scholar]
- 32.Pearce BJ, Fahr MJ, Hatch TP, Sriprakash KS. 1991. A chlamydial plasmid is differentially transcribed during the life cycle of Chlamydia trachomatis. Plasmid 26:116–122 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.