

Abstract
Enzymes of the isoprenoid biosynthetic pathway in halophilic archaea remain poorly characterized, and parts of the pathway remain cryptic. This situation may be explained, in part, by the difficulty of expressing active, functional recombinant forms of these enzymes. The use of newly available expression plasmids and hosts has allowed the expression and isolation of catalytically active Haloferax volcanii 3-hydroxy-3-methylglutaryl coenzyme A (CoA) synthase (EC 2.3.310). This accomplishment has permitted studies that represent, to the best of our knowledge, the first characterization of an archaeal hydroxymethylglutaryl CoA synthase. Kinetic characterization indicates that, under optimal assay conditions, which include 4 M KCl, the enzyme exhibits catalytic efficiency and substrate saturation at metabolite levels comparable to those reported for the enzyme from nonhalophilic organisms. This enzyme is unique in that it is the first hydroxymethylglutaryl CoA synthase that is insensitive to feedback substrate inhibition by acetoacetyl-CoA. The enzyme supports reaction catalysis in the presence of various organic solvents. Haloferax 3-hydroxy-3-methylglutaryl CoA synthase is sensitive to inactivation by hymeglusin, a specific inhibitor known to affect prokaryotic and eukaryotic forms of the enzyme, with experimentally determined Ki and kinact values of 570 ± 120 nM and 17 ± 3 min−1, respectively. In in vivo experiments, hymeglusin blocks the propagation of H. volcanii cells, indicating the critical role that the mevalonate pathway plays in isoprenoid biosynthesis by these archaea
INTRODUCTION
Archaea represent an evolutionarily distinct class of organisms that include many extremophiles that function under conditions of high temperature, acidity, or salinity (1). These properties prompt interest in the potential use of their enzymes for biotechnology applications (2). While archaea exhibit eukaryotic traits related to DNA replication, transcription, and protein translation, they are similar to bacteria in the absence of cellular organelles (3). Archaea are characterized by cell membrane lipids that contain, as a major component, polyisoprenoids in ether linkages to glycerol rather than the ester-linked fatty acids that characterize membranes of other organisms (4). Metabolite labeling experiments have suggested that archaeal polyisoprenoids derive from isopentenyl 5-diphosphate produced by enzymes of the mevalonate (MVA) pathway (5). However, some uncertainty regarding possible diversity in the terminal enzymatic reactions leading to isopentenyl 5-diphosphate has been noted (6). Annotations and functional demonstrations of some of these putative enzymes remain cryptic.
Characterization of MVA pathway enzymes from halophilic archaea has been hindered by the fact that these enzymes function at high salt levels (>2.5 M). When the open reading frames for such proteins are expressed in bacteria such as Escherichia coli, the target protein is typically recovered in insoluble form. Denaturation and renaturation processes are commonly attempted with the uncertain goal of recovering the functional enzyme from the inactive protein precipitate. Such a process has met with some success in the case of E. coli expression of Haloferax volcanii 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase (7), but widespread success of this approach for the production of active enzymes critical to halophilic metabolism remains to be documented.
Recently, technology has been developed and reported for the expression of halophilic target proteins in H. volcanii (8). Such an approach appeared to offer a productive way to isolate active forms of MVA pathway enzymes and ultimately to explore cryptic reactions in the MVA pathway. In order to provide an initial test of this hypothesis, expression of an open reading frame proposed to encode H. volcanii HMG-CoA synthase (HvHMGCS), which catalyzes the condensation of acetyl-CoA with acetoacetyl-CoA in the first irreversible reaction (Fig. 1) in the MVA pathway, was attempted. Progress in the expression and purification of the native enzyme and the characterization of HvHMGCS are presented in this report.
Fig 1.
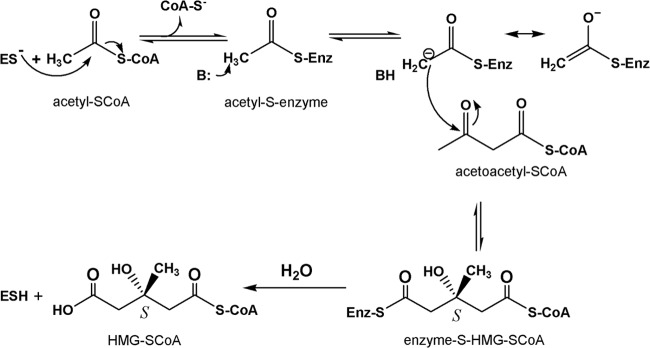
Details of the HMGCS reaction. ES, enzyme-Cys-S−; B, active-site base; BH, protonated active-site base; SCoA, free (unacylated) coenzyme A; Enz, enzyme.
MATERIALS AND METHODS
Materials.
Acetyl-CoA and acetoacetyl-CoA were synthesized by the method of Simon and Shemin (9) by using CoA with acetic anhydride and diketene, respectively. Hymeglusin was provided by M. D. Greenspan (Merck Research Laboratories). H. volcanii H1209 (ΔpyrE2 ΔhdrB Δmrr nph-pitA) and pTA963 were generously made available by Thorsten Allers and Julie Maupin-Furlow. All of the other biological or chemical products used were purchased from Sigma-Aldrich or Fisher Scientific.
Cloning, overexpression, and purification of HvHMGCS.
An H. volcanii open reading frame annotated as a putative HvHMGCS that successfully aligned (BLASTP) with bacterial and animal HMGCSs was amplified from H. volcanii H1209 genomic DNA by PCR adding 5′ EcoRI and 3′ BamHI restriction sites. The PCR product and pTA963 were digested with the EcoRI and BamHI endonucleases and ligated with T4 DNA ligase. This vector directs the tryptophan-inducible expression of an N-terminally six-His-tagged HvHMGCS protein. Ligations were transformed into H. volcanii H1209 cells, and positive transformants were selected for by growth on Hv-Ca (8) plates. Hv-Ca plates are deficient in uracil and thymidine, and rescue copies of pyrE2 (uracil) and hdrB (thymidine) on pTA963 allow the selection of transformants of H. volcanii H1209 (ΔpyrE2 ΔhdrB Δmrr nph-pitA) (8). Five milliliters of Hv-YPC medium (H. volcanii medium containing yeast extract, peptone, Casamino Acids, and 18% saltwater) (8) was inoculated with individual colonies, colonies were grown for 2 days at 45°C with shaking at 200 rpm, and then glycerol was added to a final concentration of 20% and the mixture was stored at −80°C. Five milliliters of Hv-YPC medium in sterile culture tubes was then inoculated with frozen transformed cells, incubated at 45°C and 200 rpm for 2 days, transferred to 50 ml of Hv-YPC medium, and incubated at 45°C and 200 rpm overnight. For expression of the target protein, 50 ml of cell culture was transferred to three 4-liter baffled flasks with 1 liter of Hv-YPC medium and cells were incubated with 1.3 mM tryptophan at 42°C with shaking at 200 rpm. After 16 h, additional tryptophan was added to a final concentration of 3 mM and incubation was continued for 2 h at 42°C at 200 rpm. Cells were then pelleted at 6,000 × g for 10 min at 4°C; the supernatant was removed, and cells were resuspended in 100 mM Tris-Cl (pH 8)–2 M KCl–1 mM dithiothreitol. Cells were homogenized with a Microfluidizer, and the lysate was centrifuged at 11,000 × g at 4°C. His-tagged protein was isolated by gravity Ni column chromatography with a gradient maker (40-ml total volume) with 20 to 200 mM imidazole. Fractions free of contaminants as judged by SDS-PAGE were pooled, and the imidazole was removed by passing the eluate over a Sephadex G-50 size exclusion column. Fractions with activity were pooled and concentrated with a 20-ml, 10-kDa Millipore concentrator at 7,500 × g for 15 min.
Molecular mass estimation.
The molecular mass of HvHMGCS was determined by matrix-assisted laser desorption ionization–time of flight mass spectrometry (MALDI-TOF MS). HvHMGCS was desalted with a reversed-phase matrix (C18 ZipTip; Millipore) and eluted directly onto the MALDI plate with 50% acetonitrile containing 10 mg/ml sinapinic acid matrix (3,5-dimethoxy-4-hydroxycinnamic acid). MALDI-TOF (Perseptive Biosystems Voyager DE Pro) was performed in positive, linear mode, acquiring 100 scans per spectrum, with the estimate of 51,975 ± 26 Da representing the average of five spectra.
Enzyme assay and characterization of salt, pH, temperature dependence, and organic solvent compatibility.
Spectrophotometric activity assays were performed on a PerkinElmer λ19 equipped with a water bath for temperature control or a PerkinElmer λ35 equipped with a Peltier temperature controller. The dependence of enzyme activity on the salt concentration was measured by monitoring the disappearance of acetoacetyl-CoA at 300 nm (εmM = 3.6) in Tris-Cl (100 mM unless lower concentrations are specified, pH 8, 30°C) containing acetyl-CoA (250 μM); acetoacetyl-CoA (10 μM); and 0, 1, 2, 3, or 4 M NaCl or KCl. Observation of protein concentration-dependent catalytic activity confirmed that the open reading frame does, in fact, encode HvHMGCS. The pH dependency of HvHMGCS enzyme activity was measured by the same assay by increasing the pH in 0.5-unit increments from 7.5 to 10 with 100 mM Tris-Cl (7.5 to 9) or 100 mM potassium N-cyclohexyl-2-aminoethanesulfonate (CHES-K) (9.5 to 10) in 4 M KCl at 30°C. Extinction coefficients (300 nm) appropriate for each pH value were used to calculate enzyme rates. Temperature dependence was determined by increasing the assay temperature in 5°C increments from 25 to 60°C in 100 mM Tris-Cl (pH 8)–4 M KCl; activity was monitored with the same 300-nm assay.
Kinetic characterization of HMGCS from H. volcanii.
To determine the maximum velocity (Vmax) and Km of acetyl-CoA, HvHMGCS was incubated in 100 mM Tris-Cl (pH 8)–4 M KCl at 30°C with acetoacetyl-CoA (10 μM). The reaction was started by the addition of various concentrations (6.25 to 400 μM) of acetyl-CoA, and activity was determined by monitoring the disappearance of acetoacetyl-CoA at 300 nm. The Vmax and Km of acetoacetyl-CoA were determined at 30°C by incubating HvHMGCS in 100 mM Tris-Cl (pH 8)–4 M KCl–250 μM acetyl-CoA with the HMGCS 5,5′-dithiobis-(2-nitrobenzoic acid) (DTNB) assay, which estimates reaction velocities equivalent to those observed with the acetoacetyl-CoA-based 300-nm assay (10). The reaction, performed with 250 μM DTNB, measures the rate of CoA production and is started by the addition of various concentrations (0.25 to 100 μM) of acetoacetyl-CoA. Similarly, the purification and kinetic characterization of Enterococcus faecalis HMGCS were performed by using methods previously documented by Skaff et al. (11). The data obtained were analyzed by nonlinear regression analysis with GraphPad 5.0 or SigmaPlot 10.
Inhibition of purified HvHMGCS by hymeglusin.
Enzyme activity was measured at 412 nm by the HMGCS DTNB assay. Purified HvHMGCS (49 nM) was incubated with hymeglusin (100 to 400 nM) in 100 mM Tris-Cl (pH 8)–4 M KCl at 18°C. This lower temperature diminishes the reaction rate and allows inhibition to be studied at inhibitor concentrations approximating those appropriate for Michaelis-Menten kinetic analysis. Saturating levels of acetyl-CoA (250 μM) were added at specified time points to acetylate any free enzyme and block hymeglusin from forming additional inactivated enzyme. Acetoacetyl-CoA (10 μM) was added to start the reaction, which was measured at 412 nm with 250 μM DTNB. The data were fitted to semilog plots of residual activity versus time with a linear model. Nonlinear regression analysis (GraphPad Prism 5.0) was used to determine Ki and kinact.
Inhibition of H. volcanii H1209 culture growth by hymeglusin.
Three samples (10 ml) of Hv-YPC supplemented with thymidine (60 μg/ml) were inoculated with an overnight culture of H. volcanii H1209 to an A600 of 0.050 and grown in the presence or absence of hymeglusin (25 μM) for 4 h at 45°C and 200 rpm.
Cells were pelleted at 6,000 × g for 10 min and resuspended in fresh Hv-YPC medium (10 ml) supplemented with thymidine (60 μg/ml). Samples (1.5 ml) were plated in 24-well plates and incubated at 45°C and 200 rpm in the presence or absence hymeglusin (25 μM) with A600 measured at the indicated times with a BioTek H1 Synergy plate reader. Data represent averages of three replicates.
Phylogenetic analyses.
Twenty open reading frames from each of the eukaryotic, prokaryotic, and archaeal domains (60 total) retrieved from GenBank were selected after successful alignment with known bacterial and animal HMGCSs with BLASTP. The protein sequences were then aligned with Mega 5.1 (12). The Muscle package with gap open, extend, and hydrophobicity penalty of −2.9, 0, and 1.2, respectively, was used. A maximum-likelihood tree was constructed with the previously aligned sequences by the Mega 5.1 (12) maximum-likelihood package with 100 bootstrap replications and the partial deletions missing data treatment site coverage cutoff at 95%. As discussed in the supplemental material, to create maximum-parsimony trees of the conserved catalytic residues, the 60 potential HMGCS protein sequences were aligned with Mega 5.1 Muscle package (12) with gap open, extend, and hydrophobicity penalty of −2.9, 0, and 1.2, respectively. For the maximum-parsimony tree of the catalytic cysteine, all flanking amino acids were removed from the sequence alignment except a two-amino-acid overhang surrounding the catalytic conserved cysteine residue and conserved upstream alanine (++AC++). The retained sequences were used to create a maximum-parsimony tree with the Mega 5.1 (12) maximum-parsimony package with 100 bootstrap replications and the partial deletions missing data treatment site coverage cutoff at 95%. The maximum-parsimony tree of the catalytic glutamate was created as previously described (++E++). The maximum-parsimony tree of the catalytic histidine was created as previously described, except with a two-amino-acid overhang outside the conserved histidine and proline (++H+P++).
RESULTS
Sequence comparisons of a putative HvHMGCS with eukaryotic and prokaryotic HMGCS enzymes.
The H. volcanii open reading frame that encodes a putative HMGCS corresponds to a 445-residue protein. In comparison, human cytosolic HMGCS is a 522-residue protein and Staphylococcus aureus HMGCS (MvaS) is a 388-residue protein. BLAST comparison of HvHMGCS and human HMGCS indicates only 30% identity, comparable to the 31% identity indicated by a BLAST of HvHMGCS versus the S. aureus enzyme. Despite these low levels of identity, sequence alignment of the archaeal, animal, and bacterial proteins (Fig. 2) makes it apparent that several residues critical to catalytic function are conserved in the putative HvHMGCS. For example, C120 contributes the active-site thiol (13) involved in the formation of the acetyl-S-enzyme and CoA-HMG-S-enzyme reaction intermediates (14). Also, H244 contains the imidazole moiety shown to be important in the binding of substrate acetoacetyl-CoA (15). This binding contribution is explained by its interaction with both of the oxygen substituents on C1 and C3 of the substrate's acyl group (16). While these HvHMGCS residues are undoubtedly functionally important, there are homologous cysteine and histidine residues (17) in other members of the family of initial condensing enzymes (e.g., β-ketothiolase, as well as the β-ketoacyl synthase of fatty acid biosynthesis). Any ambiguity concerning the functional assignment is, however, removed by the observation of E83, which is homologous to animal HMGCS residue E95, which has been shown to be the catalytic base critical to the condensation reaction (18). For other members of the initial condensing enzyme family, this catalytic base does not exist and is not required. The observation of E83 convinced us that the HMGCS annotation was correct and stimulated our serious attempts to accomplish the expression, isolation, and characterization of HMGCS from this halophile.
Fig 2.

HMGCS multiple-sequence alignment. The sequence of HvHMGCS was aligned with previously characterized HMGCSs from E. faecalis (EfMVAS) and Homo sapiens (HsHMGCS). Conserved residues are in black, and similar residues are in gray. Catalytically important residues are indicated by asterisks. Residues flanking catalytically conserved residues used to create maximum-parsimony trees are indicated by plus signs.
Phylogenetic analysis of HMGCS, which produces a precursor of the isopentenyl 5-diphosphate that accounts for thousands of diverse polyisoprenoids.
The evolutionary history and origin of archaea have been objects of constant research over the past 30 years, with various opinions and findings driving everything from minor revisions of clades to entire revisions of the basic domain structure of living organisms. From our perspective, there are two issues; the first is whether the gene that encodes HMGCS supports the general three-domain phylogeny of archaea, bacteria, and eukaryotes (19). Perhaps unsurprisingly, a maximum-likelihood phylogeny generated from the primary sequence of HMGCS proteins does, indeed, support the general structure of the currently accepted domains (Fig. 3). However, there are some interesting notes that we can add regarding the evolution of these genes in particular. First, we see a distinct split in the archaea clade that suggests that this enzyme has experienced either some divergence event (possibly driven by selection) or perhaps convergent evolution that makes it look more like eukaryotes in certain species of archaea (notably, H. volcanii).
Fig 3.

Maximum-likelihood tree of HMGCSs from the domains Eukaryota, Bacteria, and Archaea. HMGCS protein sequences were aligned by Muscle. The tree was constructed by using the subsequent alignment with Mega 5.1 (12).
Perspective on a second issue, which involves functional aspects of Hv HMGCS, can be gathered from looking at the evolution of specific regions of the protein, considering them as proxies for the different functionalities of the protein. We broke the protein down into three functional units (indicated in Fig. 2 by residues marked with * and + symbols) and built a parsimony tree for the residues and their near neighbors involved in these functions. This perspective yielded some interesting insight into the functional evolution of these proteins; however, the trees had low bootstrap support because of the small number of residues examined. Looking at the conserved residues (asterisks in the Fig. 2 alignment), we see that the catalytic regions examined do not necessarily fit the full specific species tree. The catalytic cysteine, which is involved in the covalent binding of the reaction intermediates (Fig. 1), is most compatible with the canonically accepted species tree (see Fig. S1A in the supplemental material). The catalytic histidine (stabilization) shows the archaea being the most derived species group (see Fig. S1B). The catalytic glutamate (general base responsible for extraction of an acetyl methyl proton) shows bacteria and archaea generally grouping together within separate clades (see Fig. S1C) and a derivative eukaryote clade (that is not monophyletic) with some eukaryotes in fact grouping together with both bacteria and archaea. An examination of these conserved regions suggests that the functional aspects of this protein have been under selection for differing traits across these three domains, and the overall evolutionary pattern of the gene is therefore a chimera of the different forces driving this enzyme over evolutionary time. It also shows that the catalytic glutamate region is the most variable of the three regions we have examined, with the least relationship to the overall tree (Fig. 3). Overall, this suggests a divergence between the general evolution of the protein and the specific, selected functions of this protein over evolutionary time. The protein has therefore likely been shaped by both convergent evolution and selection for particular functions necessary for functionality in each phylum.
H. volcanii expression of HMGCS.
Initial attempts to express in E. coli the open reading frame annotated as HvHMGCS met, not surprisingly, with limited success. The expressed protein target was recovered as a largely insoluble, inactive enzyme. Little HMGCS activity was measured in the supernatant fraction recovered from the lysate of induced bacteria. The methodology of Allers et al. (8) seemed promising, and with the expression host strain and vector that were documented, an expression plasmid encoding HvHMGCS was prepared and expressed in H. volcanii. A His-tagged target protein was recovered, mainly in the soluble fraction of cell lysates. Ni-resin chromatography was used to isolate a highly purified form of this protein (1.5 mg from 1 liter of culture), as indicated by SDS-PAGE (Fig. 4). The protein exhibits substantial HMGCS activity; the specific activity of the purified enzyme is certainly comparable to values reported for other HMGCS enzymes (Table 1). The protein's mobility on SDS-PAGE suggests a subunit molecular mass of ∼65 kDa, a value much higher than the calculated mass of the expressed protein (51,990 Da) and the MALDI estimate of 51,975 ± 26 Da. Anomalies in the SDS-PAGE mobility of halophilic proteins have previously been documented (20) and are attributed to their typically high content of acidic residues. In accord with this prediction, HvHMGCS is characterized by a high acidic residue content (42 Asp and 46 Glu residues, 19.8% of the total); the calculated pI of HvHMGCS (4.5) may explain its SDS-PAGE mobility properties. In comparison with the Haloferax enzyme, human HMGCS contains 37 Asp and 28 Glu residues (12.5% of the total; calculated pI = 5.2) while S. aureus HMGCS/MvaS contains 29 Asp and 27 Glu residues (14.5% of the total; calculated pI = 5.0).
Fig 4.
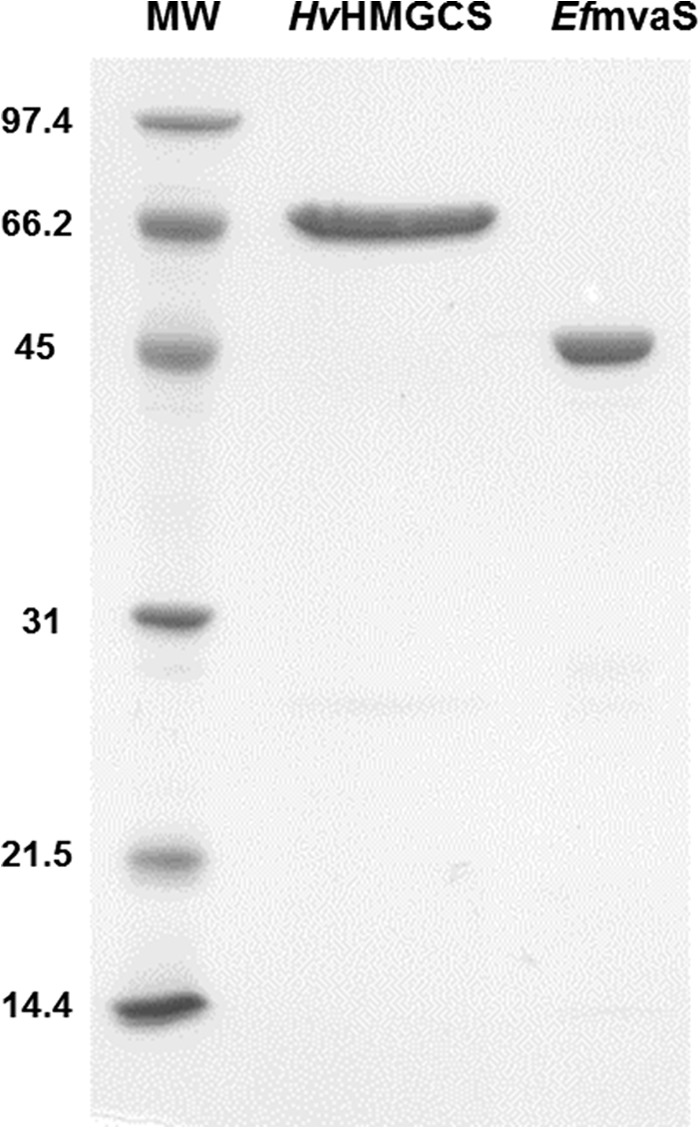
SDS-PAGE of HvHMGCS. HvHMGCS was purified by gradient Ni column chromatography. Lanes: MW, molecular mass standards; HvHMGCS, 2 μg of affinity-purified HvHMGCS (molecular mass calculated on the basis of amino acid composition, 52.0 kDa); EfmvaS, 2 μg of affinity-purified E. faecalis MvaS (molecular mass calculated on the basis of amino acid composition, 42.2 kDa). The values to the left are molecular masses in kilodaltons.
Table 1.
Kinetic parameters of H. volcanii, E. faecalis, and H. sapiens HMGCS enzymes
| Enzyme |
Km (μM) |
Vmax (μmol/min/mg) | kcat (s−1) | kcat/Km (s−1 mM−1) | ki, hymeglusin (nM) | kinact (min−1) | |
|---|---|---|---|---|---|---|---|
| Acetyl-CoA | Acetoacetyl-CoA | ||||||
| HvHMGCS | 50 ± 6a | 1.4 ± 0.1 | 5.3 ± 0.2 | 4.6 ± 0.2 | 92 ± 11 | 570 ± 120 | 17 ± 3 |
| E. faecalis MvaSb | 400 ± 60 | 0.5 ± 0.1 | 1.6 ± 0.3 | 1.1 ± 0.2 | 2.8 ± 0.2 | 700 ± 18 | 3.5 ± 0.6 |
| H. sapiens HMGCSc | 29 ± 7 | NDd | 0.7 ± 0.1 | 0.7 | 23 | 53.7 | 1.06 |
Values are means and standard error of the means.
Data for E. faecalis HMGCS were previously reported by Skaff et al. (11).
Data for H. sapiens HMGCS were previously reported by Rokosz et al. (23). The kcat and kcat/Km values were not explicitly reported but were calculated by using the Vmax and molecular weight.
ND, not determined.
Protein characterization.
As expected for a halophilic organism, the enzyme activity of HvHMGCS increases upon the inclusion of either NaCl or KCl in the assay mixture (see Fig. S2A in the supplemental material). There is some advantage with KCl rather than NaCl at a 4 M concentration. Chloride from Tris-Cl buffer or KCl stimulates catalytic activity by ∼15-fold over the values measured without halide salt. The pH rate profile of HvHMGCS indicates an optimum at ∼8.5 (see Fig. S2B and Table S1 in the supplemental material). This value is comparable to reports for the enzyme from eukaryotic (avian, pH 9.4) and prokaryotic (E. faecalis, pH 9.8) sources (21, 22) and could reflect either the ionization state of key amino acids (e.g., at lower pH) or the observed lability of the thioester substrates (e.g., acetoacetyl-CoA) during the assay at alkaline pH. The optimum temperature for catalytic activity (see Fig. S2C and Table S1) is 45°C, in accord with the temperature used to optimize the growth of H. volcanii (8). For comparison with prokaryotic HMGCS, the enzyme from E. faecalis (22) has a reported optimum reaction temperature of 37°C. The temperature stability of HvHMGCS over extended time periods has been measured (see Table S1 in the supplemental material). In the presence of an excess of acetyl-CoA, the half-life of the active enzyme is 27 h at 45°C. Assay of catalytic activity in the presence of 5 to 10% (vol/vol) of various organic solvents (dimethyl sulfoxide [DMSO], acetonitrile, and methanol; see Table S1) demonstrated that substantial activity persists in the presence of DMSO and acetonitrile but is somewhat diminished in methanol. Experiments at higher solvent levels were precluded by the formation of precipitates in the 4 M salt-containing assay mixtures.
Kinetic characterization.
HvHMGCS exhibits a Vmax of 5.3 U/mg upon a hyperbolic curve fit of observed rates versus variable acetyl-CoA concentrations; a Vmax of 4.0 U/mg is estimated from a similar fit of rates to variable acetoacetyl-CoA concentrations. These values fall into the range of Vmax values reported for eukaryotic and prokaryotic HMGCS enzymes (Table 1). The calculated Km for acetyl-CoA is 50 μM, which is lower than most other Km values in the literature (Table 1); it approaches the 10−5 M range reported (14) for the Km of acetyl-CoA in the hydrolysis partial reaction, which occurs slowly when acetyl-CoA is incubated with enzyme in the absence of the second substrate, acetoacetyl-CoA. The discrepancy between Michaelis constants for acetyl-CoA in the overall versus partial reactions has been attributed to enzyme inhibition of the overall reaction in the presence of the second substrate, acetoacetyl-CoA, at typical assay levels. New data available (see below) for HvHMGCS support this hypothesis.
The dependence of the overall rate of HMG-CoA synthesis upon the acetoacetyl-CoA concentration is depicted in Fig. 5 for both HvHMGCS and E. faecalis HMGCS. While data for the latter demonstrate the onset of strong substrate inhibition at an [acetoacetyl-CoA] of >2 μM, the data for HvHMGCS fit a hyperbolic saturation curve well, providing no indication of comparable substrate inhibition. This kinetic characteristic distinguishes the archaeal enzyme from those eukaryotic (23) and bacterial (22) HMGCS enzymes that have been extensively characterized. An acetoacetyl-CoA Km of 1.4 μM (Table 1) is estimated for HvHMGCS; we estimated a Km value of 0.5 μM for the productive binding of acetoacetyl-CoA to the E. faecalis enzyme. For nonproductive acetoacetyl-CoA binding to the latter enzyme as a substrate inhibitor, the data fit indicates a Ki of 1.9 μM. Thus, for this prokaryotic enzyme, productive and nonproductive (inhibitory) second-substrate binding occurs in concentration ranges that are not markedly different.
Fig 5.

Dependence of HvHMGCS and E. faecalis MvaS reaction rates on [acetoacetyl-CoA]. The specific activities of both HvHMGCS and E. faecalis MvaS for acetoacetyl-CoA were determined by monitoring the production of free CoA in the presence of DTNB at 412 nm in 100 mM Tris-Cl, pH 8. (A) HvHMGCS (■) assay containing 4 M KCl and saturating acetyl-CoA (250 μM). (B) Double-reciprocal plot of HvHMGCS (■) [acetoacetyl-CoA] dependence. (C) E. faecalis MvaS (●) assay containing saturating acetyl-CoA (500 μM). (D) Double-reciprocal plot of E. faecalis MvaS (●) [acetoacetyl-CoA] dependence. Data points represent specific activities at the indicated concentrations of acetoacetyl-CoA and were analyzed with GraphPad 5.0 or SigmaPlot 10.
In vitro and in vivo inhibition of HvHMGCS by the specific inhibitor hymeglusin.
Hymeglusin (depicted in Fig. 6; also designated in the literature as 1233A, F244, and L-659-699) is a fungal metabolite that exhibits high specificity for inhibition of HMGCS (24). Inhibition occurs upon reaction of its β-lactone functional group with the active-site cysteine of HMGCS. This results in a time-dependent loss of activity as inhibitor forms a thioester linkage to the cysteine (11). In order to test whether C120 of HvHMGCS may react similarly to the active-site cysteines of eukaryotic and prokaryotic HMGCS, the enzyme was incubated with a range of hymeglusin concentrations and activity monitored over a 2-min incubation period (Fig. 7). A time-dependent loss of activity was observed; inactivation occurred with first-order kinetics, as expected for covalent modification. Computational fits of these data indicate an affinity for hymeglusin (Ki = 570 ± 120 nM; Table 1) that is over an order of magnitude weaker than the value (53.7 nM) reported for human HMGCS (23). However, the rate of inactivation (kinact = 17 ± 3 min−1) is an order of magnitude greater than that observed for the human enzyme (1.06 min−1). Intrinsic reactivity of C120 may be influenced by thioester access to solvent or by active-site residues that interact with this critical amino acid's sulfhydryl to account for these contrasts in the rate of inactivation.
Fig 6.
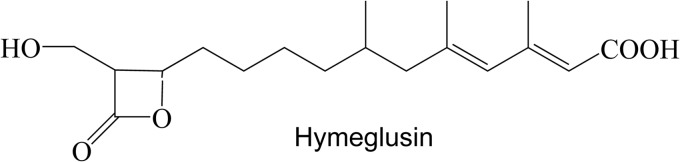
Chemical structure of hymeglusin (also designated 1233A, F244, and L-659-699).
Fig 7.

Time-dependent inactivation of HvHMGCS. Hymeglusin was incubated with purified HvHMGCS protein, and the inactivation process was stopped by the addition of acetyl-CoA at the time points indicated. The assay of residual activity was initiated by the addition of acetoacetyl-CoA, and the reaction was monitored by measuring A412 with the DTNB assay described in Materials and Methods. The hymeglusin concentrations used were 100 (▲), 150 (■), 200 (●), 300 (☐), and 400 (△) nM. Progress curves were fitted to a linear model with GraphPad Prism 4 with R2 values of >0.97 for all time courses. The inset is a replotting of the reciprocal of the apparent kobserved values (derived from the half-life values of the data sets depicted in the main graph) versus the reciprocal of the hymeglusin concentration. From this double-reciprocal plot, the x and y intercepts were used to determine a Ki of 570 ± 120 nM and a kinact of 17 ± 3 min−1, respectively.
After inactivation experiments demonstrated the sensitivity of HvHMGCS to hymeglusin, it seemed reasonable to determine whether this compound could be useful in an in vivo test of the importance of HvHMGCS and, by inference, the MVA pathway to essential metabolism in H. volcanii. In this experiment, H. volcanii H1209 cells were grown for 4 h in the presence or absence of 25 μM hymeglusin. Cells were separated from the medium by centrifugation. At t = 0 h, the untreated cells were resuspended in fresh medium (without hymeglusin) while the cells previously treated with hymeglusin were resuspended in medium that contained either 0 or 25 μM hymeglusin. The growth of cells (incubated at 45°C) in these three samples was monitored (A600) for 36 h. Cells never treated with hymeglusin exhibited a sigmoidal growth curve that exhibited an inflection point at 11.2 h (Fig. 8). In contrast, cells previously treated with hymeglusin prior to resuspension in medium without this inhibitor exhibited a lag before growth resumed. The inflection point in the growth curve of this sample was observed at 20 h. In contrast, pretreated cells that remained in hymeglusin-containing medium never demonstrated any recovery of growth over the 36-h experiment. The ability of inhibited cells to resume growth upon the removal of hymeglusin from their medium is likely to reflect the slow endogenous hydrolase activity of HMGCS. In the absence of a condensation reaction partner for the thioester-bound moiety (in this case, ring-opened hymeglusin), solvent water reacts with the thioesterified moiety to release it as a hydrolysis product and the active-site cysteine is liberated for a productive reaction. This slow process presumably accounts for the >6-h lag time before growth resumes after cells are pretreated with inhibitor, as reflected in the delay in reaching an inflection point in the growth curve of this sample. Thus, the submicromolar affinity of hymeglusin for purified HvHMGCS (Fig. 7) and the efficacy and selectivity of this inhibitor for this key enzyme of isoprenoid biosynthesis are useful in implicating both the enzyme and the pathway as essential elements in the propagation of H. volcanii.
Fig 8.

Hymeglusin inhibition and recovery of H. volcanii cellular growth. H. volcanii H1209 was grown in the presence or absence of hymeglusin in Hv-YPC medium at 45°C for 4 h and pelleted by centrifugation. Cells were resuspended in Hv-YPC medium in the presence or absence of hymeglusin and incubated at 45°C and 200 rpm in the wells of a 24-well plate. Growth was monitored by measuring A600. Plots depict H. volcanii H1209 never grown in the presence of hymeglusin (●), grown in the presence of hymeglusin only before resuspension (▲), and grown in the presence of hymeglusin before and after resuspension (▼). Curves represent nonlinear regression fits of the data.
DISCUSSION
Allers and colleagues (8) have developed and shared the experimental tools that offer the possibility of expressing His-tagged native Haloferax proteins under high-salt conditions in an H. volcanii host. The level of expression of any particular target protein may vary, but since the proteins are His tagged, isolation of highly purified native protein yields several-milligram amounts that are satisfactory for characterization, as well as a variety of mechanistic or biosynthetic experiments. In contrast, the use of an E. coli expression system for halophilic proteins commonly resulted in the production of inactive protein or insoluble protein in inclusion bodies. Denaturation, renaturation, and/or reactivation of those proteins has been required, and while examples of success with such regimens have been described (7, 25), not all protein targets are expected to be recovered as samples with characteristics that closely approximate those of the physiologically relevant proteins. The rationale outlined above prompted us to explore Haloferax expression methodology in our exploration of the enzymes of Haloferax isoprenoid biosynthesis.
The successful recovery of HvHMGCS in a native state as a result of Haloferax expression allows us to more convincingly contrast its properties with those of other eukaryotic and prokaryotic HMGCS proteins. Noteworthy contrasts include the lack of observed substrate inhibition by acetoacetyl-CoA, which distinguishes HvHMGCS from both prokaryotic and eukaryotic forms of the enzymes. Such substrate inhibition is most clearly demonstrated by results for E. faecalis HMGCS/MvaS (Fig. 5). The very small difference between the Km and Ki values for acetoacetyl-CoA (0.5 and 1.9 μM, respectively; Table 1) leads to a sharp departure of the rate-versus-concentration curve from the hyperbolic shape observed for HvHMGCS where there is no strong substrate inhibition. For eukaryotic HMGCS (e.g., the yeast [26] and human [23] enzymes), substrate inhibition by acetoacetyl-CoA has also been reported but more gradual rate decreases occur at elevated substrate concentrations. Estimates of their Ki values for acetoacetyl-CoA are lower in affinity (8 and 12 μM, respectively). Any interpretation of the contrast between negligible substrate inhibition for one archaeal (H. volcanii) HMGCS and the significant substrate inhibition observed for one bacterial (E. faecalis) enzyme is, of course, speculative. However, it seems plausible that, since archaeal HMGCS commits biosynthetic acetate to a quantitatively major isoprenoid product, phytanic acid, any constraint on flux through HMGCS (or the MVA pathway in general) is undesirable. In contrast, while bacterial isoprenoids contribute to important biosynthetic products (e.g., dolichol, undecaprenyl phosphate, etc.), these do not represent as quantitatively large a pool of materials as does the phytanic acid component of archaeal membranes. On this basis, regulation of the partition of bacterial acetate between biosynthesis and trichloroacetic acid cycle energy production may represent an important factor in metabolism.
There are also clear contrasts between HMGCS enzymes in the context of hymeglusin inhibition or inactivation. The in vitro experiment demonstrates a kinact for HvHMGCS (17.3 min−1) that is much greater than the corresponding values for the human (1.06 min−1) and E. faecalis (3.5 min−1) HMGCS enzymes. In the in vivo experiment, contrasts were also observed. Recovery of cells treated with hymeglusin involves a substantial lag time for Haloferax (growth curve inflection point at 20 h versus 11 h for the untreated control) and also a notable lag for E. faecalis (inflection point at 3.1 h versus 0.7 h for the untreated control). For animal HepG2 cells, 50% recovery of HMGCS from hymeglusin inactivation occurs more rapidly (∼1 h) (27).
The partial annotation of Haloferax open reading frames does not yet permit an unambiguous prediction of the detailed biosynthetic route to isopentenyl 5-diphosphate. The results of this study, as well as results of E. coli expression work in Rodwell's lab (7), indicate that HMGCS and reductase function as MVA pathway enzymes, but the latter steps of this pathway are cryptic. It is unclear whether reactions catalyzed by phospho-MVA kinase and MVA 5-diphosphate decarboxylase are critical to isopentenyl 5-diphosphate production or whether this product is produced by a shunt involving a putative MVA 5-phosphate decarboxylase, as well as an isopentenyl 5-phosphate kinase of the type described in other archaea (28, 29). Available annotations of Haloferax open reading frames provide no resolution of this ambiguity, since proteins for both metabolic routes are proposed. For the proposed shunt proteins, there is no extensive collection of reports from which to draw if it becomes necessary to compare the functional properties of new preparations of native enzymes with those of the denatured/renatured proteins that probably would have to be produced after heterologous expression (e.g., in E. coli). On this basis, expression and isolation of the Haloferax enzymes in their native state seem to represent the most reliable approach to pursue.
There has been significant effort directed at engineering the MVA pathway in order to support biotechnology projects aimed at elevated production of either key precursors or final target compounds that represent high-value polyisoprenoids. For example, yeast strains have been engineered to produce the immediate precursor of the antimalarial drug artemisinin in a process that includes optimization of MVA pathway reactions (30). Likewise, enhanced production of bioisoprene has been accomplished by engineering into E. coli the MVA pathway enzymes from E. faecalis (31). Yields were elevated when a mutated, high-specific-activity HMGCS enzyme replaced the wild-type enzyme, suggesting the possible significance of HMGCS as a rate-limiting step in the process. Halophilic enzymes may make future contributions to bioengineered metabolism. Their abilities to function at high salt levels, to tolerate moderate process temperatures, and to exhibit catalytic activity in the presence of mixed aqueous and organic solvents should prompt inquiries into their potential utility in biotechnology applications. In such a context, the ability to characterize the native forms of halophilic metabolic enzymes will certainly expedite the evaluation of their potential.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported, in part, by endowment funds (to H.M.M.) from the Marion-Merrell-Dow foundation.
A. Keightley (SBS MS Core lab) provided helpful assistance in MALDI experiments on protein molecular mass.
Footnotes
Published ahead of print 21 June 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.00485-13.
REFERENCES
- 1.Timpson LM, Liliensiek AK, Alsafadi D, Cassidy J, Sharkey MA, Liddell S, Allers T, Paradisi F. 2013. A comparison of two novel alcohol dehydrogenase enzymes (ADH1 and ADH2) from the extreme halophile Haloferax volcanii. Appl. Microbiol. Biotechnol. 97:195–203 [DOI] [PubMed] [Google Scholar]
- 2.Margesin R, Schinner F. 2001. Potential of halotolerant and halophilic microorganisms for biotechnology. Extremophiles 5:73–83 [DOI] [PubMed] [Google Scholar]
- 3.Dennis PP. 1986. Molecular biology of archaebacteria. J. Bacteriol. 168:471–478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Koga Y, Mori H. 2005. Recent advances in structural research on ether lipids from archaea including comparative and physiological aspects. Biosci. Biotechnol. Biochem. 69:2019–2034 [DOI] [PubMed] [Google Scholar]
- 5.De Rosa M, Gambacorta A, Gliozzi A. 1986. Structure, biosynthesis, and physicochemical properties of archaebacterial lipids. Microbiol. Rev. 50:70–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Smit A, Mushegian A. 2000. Biosynthesis of isoprenoids via mevalonate in Archaea: the lost pathway. Genome Res. 10:1468–1484 [DOI] [PubMed] [Google Scholar]
- 7.Bischoff KM, Rodwell VW. 1996. 3-Hydroxy-3-methylglutaryl-coenzyme A reductase from Haloferax volcanii: purification, characterization, and expression in Escherichia coli. J. Bacteriol. 178:19–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Allers T, Barak S, Liddell S, Wardell K, Mevarech M. 2010. Improved strains and plasmid vectors for conditional overexpression of His-tagged proteins in Haloferax volcanii. Appl. Environ. Microbiol. 76:1759–1769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Simon EJ, Shemin D. 1953. The preparation of S-succinyl coenzyme A. J. Am. Chem. Soc. 75:2520 [Google Scholar]
- 10.Skaff DA, Miziorko HM. 2010. A visible wavelength spectrophotometric assay suitable for high-throughput screening of 3-hydroxy-3-methylglutaryl-CoA synthase. Anal. Biochem. 396:96–102 [DOI] [PubMed] [Google Scholar]
- 11.Skaff DA, Ramyar KX, McWhorter WJ, Barta ML, Geisbrecht BV, Miziorko HM. 2012. Biochemical and structural basis for inhibition of Enterococcus faecalis hydroxymethylglutaryl-CoA synthase, mvaS, by hymeglusin. Biochemistry 51:4713–4722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. 2011. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol. Evol. 28:2731–2739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Miziorko HM, Lane MD. 1977. 3-Hydroxy-3-methylgutaryl-CoA synthase. Participation of acetyl-S-enzyme and enzyme-S-hydroxymethylgutaryl-SCoA intermediates in the reaction. J. Biol. Chem. 252:1414–1420 [PubMed] [Google Scholar]
- 14.Misra I, Narasimhan C, Miziorko HM. 1993. Avian 3-hydroxy-3-methylglutaryl-CoA synthase. Characterization of a recombinant cholesterogenic isozyme and demonstration of the requirement for a sulfhydryl functionality in formation of the acetyl-enzyme reaction intermediate. J. Biol. Chem. 268:12129–12135 [PubMed] [Google Scholar]
- 15.Misra I, Miziorko HM. 1996. Evidence for the interaction of avian 3-hydroxy-3-methylglutaryl-CoA synthase histidine 264 with acetoacetyl-CoA. Biochemistry 35:9610–9616 [DOI] [PubMed] [Google Scholar]
- 16.Theisen MJ, Misra I, Saadat D, Campobasso N, Miziorko HM, Harrison DH. 2004. 3-Hydroxy-3-methylglutaryl-CoA synthase intermediate complex observed in “real-time.” Proc. Natl. Acad. Sci. U. S. A. 101:16442–16447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Miziorko HM. 2011. Enzymes of the mevalonate pathway of isoprenoid biosynthesis. Arch. Biochem. Biophys. 505:131–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chun KY, Vinarov DA, Zajicek J, Miziorko HM. 2000. 3-Hydroxy-3-methylglutaryl-CoA synthase. A role for glutamate 95 in general acid/base catalysis of C-C bond formation. J. Biol. Chem. 275:17946–17953 [DOI] [PubMed] [Google Scholar]
- 19.Woese CR, Kandler O, Wheelis ML. 1990. Towards a natural system of organisms: proposal for the domains Archaea, Bacteria, and Eucarya. Proc. Natl. Acad. Sci. U. S. A. 87:4576–4579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Madern D, Ebel C, Zaccai G. 2000. Halophilic adaptation of enzymes. Extremophiles. 4:91–98 [DOI] [PubMed] [Google Scholar]
- 21.Clinkenbeard KD, Sugiyama T, Lane MD. 1975. Cytosolic 3-hydroxy-3-methylglutaryl-CoA synthase from chicken liver. Methods Enzymol. 35:160–167 [PubMed] [Google Scholar]
- 22.Sutherlin A, Hedl M, Sanchez-Neri B, Burgner JW, II, Stauffacher CV, Rodwell VW. 2002. Enterococcus faecalis 3-hydroxy-3-methylglutaryl coenzyme A synthase, an enzyme of isopentenyl diphosphate biosynthesis. J. Bacteriol. 184:4065–4070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rokosz LL, Boulton DA, Butkiewicz EA, Sanyal G, Cueto MA, Lachance PA, Hermes JD. 1994. Human cytoplasmic 3-hydroxy-3-methylglutaryl coenzyme A synthase: expression, purification, and characterization of recombinant wild-type and Cys129 mutant enzymes. Arch. Biochem. Biophys. 312:1–13 [DOI] [PubMed] [Google Scholar]
- 24.Tomoda H, Kumagai H, Takahashi Y, Tanaka Y, Iwai Y, Omura S. 1988. F-244 (1233A), a specific inhibitor of 3-hydroxy-3-methylglutaryl coenzyme A synthase: taxonomy of producing strain, fermentation, isolation and biological properties. J. Antibiot. (Tokyo) 41:247–249 [DOI] [PubMed] [Google Scholar]
- 25.Connaris H, Chaudhuri JB, Danson MJ, Hough DW. 1999. Expression, reactivation, and purification of enzymes from Haloferax volcanii in Escherichia coli. Biotechnol. Bioeng. 64:38–45 [PubMed] [Google Scholar]
- 26.Middleton B. 1972. The kinetic mechanism of 3-hydroxy-3-methylglutaryl-coenzyme A synthase from baker's yeast. Biochem. J. 126:35–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Greenspan MD, Bull HG, Yudkovitz JB, Hanf DP, Alberts AW. 1993. Inhibition of 3-hydroxy-3-methylglutaryl-CoA synthase and cholesterol biosynthesis by beta-lactone inhibitors and binding of these inhibitors to the enzyme. Biochem. J. 289:889–895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Grochowski LL, Xu H, White RH. 2006. Methanocaldococcus jannaschii uses a modified mevalonate pathway for biosynthesis of isopentenyl diphosphate. J. Bacteriol. 188:3192–3198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen M, Poulter CD. 2010. Characterization of thermophilic archaeal isopentenyl phosphate kinases. Biochemistry 49:207–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ro DK, Paradise EM, Ouellet M, Fisher KJ, Newman KL, Ndungu JM, Ho KA, Eachus RA, Ham TS, Kirby J, Chang MC, Withers ST, Shiba Y, Sarpong R, Keasling JD. 2006. Production of the antimalarial drug precursor artemisinic acid in engineered yeast. Nature 440:940–943 [DOI] [PubMed] [Google Scholar]
- 31.Yang J, Xian M, Su S, Zhao G, Nie Q, Jiang X, Zheng Y, Liu W. 2012. Enhancing production of bio-isoprene using hybrid MVA pathway and isoprene synthase in E. coli. PLoS One 7:e33509. 10.1371/journal.pone.0033509 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.