

Abstract
Infectious diarrhea can be classified based on its clinical presentation as noninflammatory or inflammatory disease. In developing countries, among inflammatory diarrhea cases, Shigella is the most common cause, followed by Campylobacter and Salmonella. Because the time frame in which treatment choices must be made is short and conventional stool cultures lack good sensitivity, there is a need for a rapid, sensitive, and inexpensive detection technique. The purpose of our study was to develop a multiplex real-time PCR procedure to simultaneously identify Campylobacter spp., Salmonella spp., and Shigella spp. Primers were designed to amplify the invA, ipaH, and 16S rRNA genes simultaneously in a single reaction to detect Salmonella, Shigella, and Campylobacter, respectively. Using this approach, we correctly identified 102 of 103 strains of the targeted enteropathogens and 34 of 34 other pathogens. The melting temperatures were 82.96 ± 0.05°C for invA, 85.56 ± 0.28°C for ipaH, and 89.21 ± 0.24°C for 16S rRNA. The limit of accurate quantification for the assay in stool samples was 104 CFU g−1; however, the limit of detection was 103 CFU g−1. This assay is a simple, rapid, inexpensive, and reliable system for the practical detection of these three enteropathogens in clinical specimens.
INTRODUCTION
Infectious diarrhea is a global health problem that is still responsible for thousands of deaths worldwide, especially in children (1). It can be classified based on its clinical presentation as one of two syndromes—noninflammatory or inflammatory diarrhea (2). Among cases of inflammatory diarrhea, Shigella is the most common cause, followed by Campylobacter and Salmonella (3). These invasive organisms primarily target the lower bowel; they invade the intestinal mucosa to induce an acute inflammatory reaction and activate cytokines and inflammatory mediators (4).
Because the time frame in which treatment choices must be made is short and the conventional stool cultures lack good sensitivity, there is a need for a rapid, sensitive, and inexpensive detection technique. We searched for DNA sequences that were highly conserved between the different species of each genera, and we selected the following genes as targets, invA (invasion A gene) for Salmonella spp., ipaH (invasion plasmid antigen H) for Shigella spp., and 16S rRNA for Campylobacter spp., to develop a fluorescence-based real-time PCR procedure to simultaneously identify these enteropathogens. In this method the post-PCR products are identified based on melting-point curve analysis. We have also standardized the technique to quantify these bacteria directly from stool samples.
MATERIALS AND METHODS
Bacterial strains.
A total of 147 enteropathogenic strains (Table 1) were analyzed, including clinical isolates representative of Salmonella spp., Shigella spp., and Campylobacter spp., as well as other enteropathogens. These clinical strains had previously been identified based on serology, biochemical assays, and real-time PCR for the diarrheagenic Escherichia coli (5). In addition, we used Salmonella enteritidis strain ATCC 13076, Shigella flexneri strain ATCC 12022, and Campylobacter jejuni subsp. jejuni strain ATCC 33560 as positive controls.
Table 1.
Clinical enteropathogenic strains analyzed in the real-time PCR assay system
| Strain/species | n |
|---|---|
| Salmonella | 26 |
| S. enteritidis strain ATCC 13076 | 1 |
| S. enteritidis | 6 |
| S. infantis | 2 |
| Other Salmonella spp. | 17 |
| Shigella | 49 |
| S. flexneri strain ATCC 12022 | 1 |
| S. boydii | 6 |
| S. dysenteriae | 3 |
| S. flexneri | 5 |
| S. sonnei | 6 |
| Other Shigella spp. | 28 |
| Campylobacter | 41 |
| C. jejuni subspp. jejuni strain ATCC 33560 | 1 |
| C. coli | 10 |
| C. jejuni | 10 |
| Other Campylobacter spp. | 20 |
| Other enteropathogens (negative controls) | 34 |
| Diffusely adherent E. coli | 6 |
| Enteroaggregative E. coli | 6 |
| Enteroinvasive E. coli | 1 |
| Enteropathogenic E. coli | 6 |
| Enterotoxigenic E. coli | 6 |
| Shiga toxin-producing E. coli | 5 |
| E. coli K-12 | 1 |
| Pseudomonas aeruginosa | 1 |
| Klebsiella pneumoniae | 1 |
| Proteus mirabilis | 1 |
| Total | 147a |
Total does not include the 3 ATCC strains, which were positive controls.
DNA isolation from pure culture.
Strains were subcultured from frozen or peptone stocks onto MacConkey agar (Merck, Darmstadt, Germany) for Salmonella and Shigella and chocolate agar (Trypticase soy agar [TSA] [Oxoid; Basingstoke, Hampshire, United Kingdom] with 5% sheep blood) for Campylobacter using quadrant streaking methods and were incubated at 37°C to produce isolated colonies. After overnight incubation (48 to 72 h for Campylobacter cultures), a bacterial suspension was carefully prepared (0.5 McFarland scale), avoiding agar contamination, an important cause of erratic amplification. Crude lysates were prepared and used directly as the templates for the PCR. DNA was extracted by boiling the bacterial suspension in 500 μl of PCR mixture or molecular grade water for 5 min, followed by room temperature incubation for 10 min, and centrifugation at 12,000 rpm for 10 min. Two microliters of this crude lysate was used as the template, with 18 μl PCR master mix to make a 20-μl total reaction mixture volume.
Primer design.
The primers were designed to detect three different virulence genes simultaneously in a single reaction (Table 2). Primers were designed so that amplicons would be produced having melting temperatures (Tm) ranging from 77°C to 95°C, with >1°C between peaks. We targeted the amplicon Tm as the first parameter, seeking appropriate primer sequences to amplify unique regions in a given virulence gene that would result in an amplicon of the desired Tm. Sequences of each gene were examined for features such as areas of high or low GC content, size, and identity among reported BLAST sequences for the target gene. These areas were analyzed by an oligonucleotide property calculator (Primer Premier 5.0), which uses the nearest-neighbor method to predict the amplicon Tm. After areas likely to produce amplicons with the desired Tm were selected, primers were designed using the Primer3 program (http://frodo.wi.mit.edu) and were synthesized by Belomed S.R.L. (Lima, Peru). These primers were then sequentially added to the mixture to determine their actual Tm as well as to determine whether nonspecific primer amplification occurred in the presence of other oligonucleotide primers. Primer concentrations were then optimized to produce melting curves of similar peak heights (area under the curve) between products and across a series of dilutions to simulate the various concentrations of template DNA extracted from the crude lysate preparation.
Table 2.
Primers for multiplex real-time PCR
| Pathogen | Gene | Orientationa | Primer (5′→3′) | Final concn (μM) | Amplicon size (bp) | Amplicon Tm (mean ± SD) | Reference or source |
|---|---|---|---|---|---|---|---|
| Salmonella spp. | invA | F | CATTTCTATGTTCGTCATTCCATTACC | 0.40 | 132 | 82.96 ± 0.05 | 11 |
| R | AGGAAACGTTGAAAAACTGAGGATTCT | ||||||
| Shigella spp. | ipaH | F | CGCGACGGACAACAGAATACACTCCATC | 0.20 | 108 | 85.56 ± 0.28 | This study |
| R | ATGTTCAAAAGCATGCCATATCTGTG | ||||||
| Campylobacter spp. | 16S rRNA | F | GGATGACACTTTTCGGAGC | 0.40 | 812 | 89.21 ± 0.24 | 10 |
| R | CATTGTAGCACGTGTGTC |
F, forward; R, reverse.
PCR conditions.
Initially, we evaluated previously reported multiplex assays (6–11) to determine whether they would work well in a real-time PCR. Nonspecific amplification or interference with new primers made most of the primers in these assays problematic. We sequentially eliminated primers and eventually were able to use several previously described primers in our system (Table 2). PCR was performed using a LightCycler 480 real-time PCR system (Roche Applied Science). Each multiplex PCR assay was performed with a final reaction mixture volume of 20 μl containing 0.5 U Phusion polymerase (Finnzymes OY, Espoo, Finland) in high-fidelity Phusion buffer with a final concentration of 200 μM deoxynucleotide triphosphates and 3 mM MgCl2. The primers were used at a final concentration of 0.2 to 0.4 μM (Table 2). SYBR green I (Cambrex Bio Science, Rockland, ME) was diluted as recommended by the manufacturer. The hot-start technique was used to prevent nonspecific amplification. The amplification cycles consisted of incubation at 98°C for 30 s, at 65°C for 30 s, at 72°C for 30 s, and at 72°C for 10 s. After 30 cycles, a melting curve was determined using SYBR green fluorescence with a ramp speed of 0.2°C/s between 72°C and 98°C, with a reading every 0.2°C. Melting peaks were automatically calculated by the software LightCycler 480 SW 1.5 (Roche Diagnostics) which, after subtracting background fluorescence from a set of water blanks, plotted the negative derivative of fluorescence with respect to temperature [−d(F)/dT versus T]. Representative strains of each species were analyzed by agarose gel electrophoresis (2.0% agarose gels) to ensure that no unwanted bands were seen and that the predicted product sizes were found.
Limit of detection and amplification efficiency in spiked stool samples.
We reactivated one strain of each pathogen onto MacConkey (Merck, Darmstadt, Germany), (Salmonella and Shigella) and chocolate agar (TSA [Oxoid; Basingstoke, Hampshire, United Kingdom] with 5% sheep blood) (Campylobacter) plates. After 37°C/18 h (Salmonella and Shigella) and 42°C/48 h (Campylobacter), we made 10-fold serial dilutions (equivalent to 100 to 107 CFU) and spiked 100 mg of stool sample from a healthy volunteer. The number of CFUs was determined by plating culture dilutions on MacConkey (Salmonella and Shigella) and chocolate agar (Campylobacter). Bacterial DNA was extracted from the stool samples with a Roche kit (High Pure PCR template preparation kit) and resuspended in 200 μl of Tris-EDTA (TE) buffer. Amplification efficiency (E) was estimated by using the slope of the standard curve and the formula E = (101/slope) − 1. A reaction with 100% efficiency generates a slope of −3.32.
RESULTS
We evaluated three enzymes: (i) Phusion hot-start DNA polymerase (Finnzymes, Finland), a Pyrococcus-like enzyme with a processivity-enhancing domain, (ii) Roche (FastStart Taq DNA polymerase), a chemically modified form of thermostable recombinant Taq, and (iii) Promega (GoTaq DNA polymerase). Of these, Phusion was the only enzyme that gave reliably reproducible amplification. The average melting temperatures (Tm) were 82.96 ± 0.05°C for the invA amplicon, 85.56 ± 0.28°C for ipaH, and 89.21 ± 0.24°C for 16S rRNA (Table 2). The spacing between peaks for each gene is shown in Fig. 1A. Remarkably few variations in intensity and Tm were observed among the different strains tested. On an individual gene basis, all genes tested were reliably amplified except for the 16S rRNA gene, which was detected in 39/40 strains expected to be positive. Among the other enteropathogens tested for cross-reaction, one strain could be considered false positive (1/34), although as an enteroinvasive E. coli (EIEC) strain it is expected to have the ipaH gene. The amplitudes of the melting curves were quite similar for all strains in each category as well. The individual peaks were symmetric, as shown in Fig. 1B, demonstrating the overlap and reproducibility of the assay for multiple strains. Analysis using agarose gel confirmed that the amplicons represented on the melting-curve graph were indeed of the correct molecular weights expected based on the primer sequences (Fig. 1C).
Fig 1.

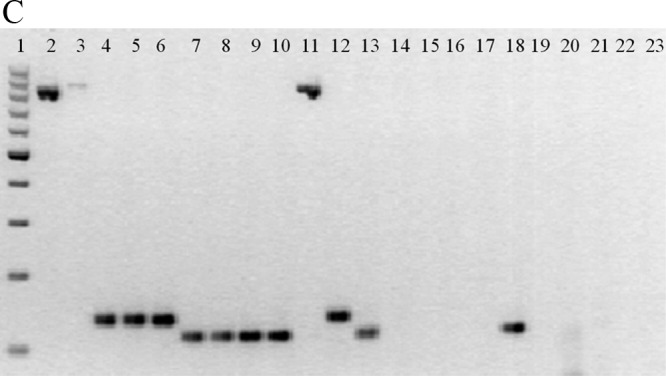
(A) Real-time PCR simultaneously detects three different genes. Data from individual tubes, each containing the ATCC strains S. enteritidis 13076, S. flexneri 12022, and C. jejuni subsp. jejuni 33560, are shown in a single graph so that the separation between individual amplicon melting curves is illustrated (from left to right, invA, ipaH, and 16S rRNA). The y axis (fluorescence) represents the negative derivative of fluorescence over temperature versus temperature. (B) Melting curves of DNA isolated from pure cultures of (top) Salmonella (n = 26), (center) Shigella (n = 49), and (bottom) Campylobacter (n = 41). Curves are superimposed to show the reproducibility within species. The y axis (fluorescence) represents the negative derivative of fluorescence over temperature versus temperature. In the top and bottom panels, the horizontal lines at the bottom represent negative controls. In the bottom panel, the lowest peak represents a positive strain. (C) Agarose gel (2%) of amplicons of representative strains from the multiplex real-time PCR. From left to right, 1, 100-bp molecular weight ladder; 2, C. coli; 3, C. jejuni; 4, S. enteritidis; 5, S. infantis; 6, Salmonella spp.; 7, S. boydii; 8, S. dysenteriae; 9, S. flexneri; 10, S. sonnei; 11, C. jejuni subsp. jejuni ATCC 33560; 12, S. enteritidis ATCC 13076; 13, S. flexneri ATCC 12022; 14, diffusely adherent E. coli; 15, enteroaggregative E. coli; 16, enteropathogenic E. coli; 17, enterotoxigenic E. coli; 18, enteroinvasive E. coli; 19, Shiga-like toxin producer E. coli; 20, E. coli K-12; 21, Pseudomonas aeruginosa; 22, Klebsiella pneumoniae; 23, Proteus mirabilis.
Mixed infections are common, especially in diarrheal disease. For this reason we simulated possible coinfections between these pathogens. We have not observed competition between the different sets of primers used to detect the genes involved in the virulence of these pathogens (Fig. 2).
Fig 2.

Mixed infection detected in a pool of colonies corresponding to S. enteritidis ATCC 13076 (invA), S. flexneri ATCC 12022 (ipaH), and C. jejuni subsp. jejuni ATCC 33560 (16S rRNA) (A) and another corresponding to S. enteritidis ATCC 13076 (invA) and S. flexneri ATCC 12022 (ipaH) (B).
The ability to detect Salmonella, Shigella, and Campylobacter in stool samples was tested by spiking serial dilutions of each enteropathogen into 100 mg of sample. The assay of stool samples detected the presence of these pathogens in the range of 103 to 107 CFU g−1 (Fig. 3A, B, and C), but accurately quantified values only from 104 to 107 CFU g−1. The standard plot showed that the regression coefficient was linear (R2 = 0.99, 0.98, and 0.99, respectively) over a 5-log dilution range, and the reaction efficiencies were 100.3%, 91.7%, and 99.0% for the invA, ipaH, and 16S rRNA genes, respectively (Fig. 4A, B, and C).
Fig 3.

In stool samples, the qPCR detected serial dilutions of 107 to 103 CFU g−1 of Salmonella (A), Shigella (B), and Campylobacter (C).
Fig 4.

In stool samples, the qPCR showed an efficiency of 100.3%, 91.7%, and 99.0% for the invA (A), ipaH (B), and 16S rRNA (C) genes, respectively, with correlation coefficients of 0.99, 0.98, and 0.99, respectively.
The total cost starting from the stool sample, including the DNA extraction and quantitative PCR (qPCR) analysis, was higher than the cost starting from culture ($6.0 versus $4.8); however, the time for obtaining a confirmed result was much less (4 h versus 24 to 72 h, depending of the pathogen).
DISCUSSION
Real-time PCR assays have been developed independently for the detection and quantification of some enteropathogens, Helicobacter pylori (12), Clostridium difficile (12), Campylobacter (13), Cryptosporidium (14), Salmonella (11), enteropathogenic E. coli (15), and enterohemorrhagic E. coli (16). Although the PCR conditions were standardized, when we tested our conventional PCR in the real-time thermocycler, we saw that the melting temperatures (Tm) of the products of Campylobacter spp. and Shigella spp. were similar and the peaks overlapped. We used BLAST software to examine the sequence of the ipaH gene for the most conserved region for all the strains of Shigella spp. and PrimerPremier 5.0 to analyze the new set of primers. The best target region was located between nucleotides 16 and 123, but the Tm for the PCR product overlapped with the invA gene amplified for Salmonella spp. We kept this region and solved the problem of overlapping with the “CG tails” strategy (17), adding 3 nucleotides (CGC) in front of the forward primer for Shigella spp. This multiplex approach showed higher analytical sensitivity than other methods described in previous studies (18–20) for detection of these three enteropathogens in stool samples. The efficiencies (90% to 110%) and the regression coefficient (almost 1.00) obtained for all the targets were in agreement with the minimum information for publication of quantitative real-time PCR experiments (MIQE) guidelines (21).
In this study, we first standardized a conventional PCR using primers that were previously reported for use against 16S rRNA (Salmonella spp. and Campylobacter spp.) (10, 18, 22) and ipaH (Shigella spp.) (7). Unfortunately, when we put all the primers in a single PCR, we obtained cross-reactions. Conventional PCR requires amplification in a thermocycler followed by product separation by gel electrophoresis. The cost, the time delay required to do gel analysis of PCR products, and the inability to analyze large numbers of strains are major impediments to this approach. In addition, while ethidium bromide is a relatively inexpensive reagent for DNA gel staining, it has human and environmental safety concerns. For these reasons, real-time fluorescence-based multiplex PCR has become an attractive technique. It offers the advantages of being a faster and more robust assay because it does not require post-PCR procedures to detect amplification products.
Our study had some limitations. First, although the sequences used as the targets in this multiplex PCR are from highly conserved regions of the genes, a weakness of this or any multiplex assay is that new variants of virulence genes could fail to amplify with the primers described. Second, this assay also identifies enteroinvasive E. coli (EIEC), since both Shigella spp. and EIEC have the ipaH gene. Third, it was not possible to compare the performance of the assay between cultures and stool samples because the latter had been stored for a long time and the DNA might have been degraded.
Nevertheless, this assay represents a simple, rapid, sensitive, and inexpensive system for the practical presumptive detection of these three enteropathogens. An advantage of this technique is that there is no need to run an electrophoresis gel to determine the presence of the amplicons, because each has a characteristic melting temperature (Tm) that is detected in the denaturation curve. Indeed, this approach also allows identification of possible mixed infections involving several of these pathogens. The cost of materials was under $6.00 per sample analyzed. Thus, the new real-time multiplex PCR provides reliable results within a short time and might be useful as an additional diagnostic tool whenever time is important in the diagnosis of enteropathogenic bacteria. Further studies should focus on the comparison of culture and qPCR results from stool samples to calculate the real sensitivity and specificity of this assay.
ACKNOWLEDGMENTS
This work was supported by a Public Health Service award (grants 1K01TW007405 to T.J.O. and R01-HD051716 to T.G.C.) from the National Institutes of Health and by Agencia Española de Cooperación Internacional para el Desarrollo (AECID), Spain, Programa de Cooperación Interuniversitaria e Investigación Científica con Iberoamérica (D/019499/08 and D/024648/09) (to J.R. and T.J.O).
Footnotes
Published ahead of print 12 June 2013
REFERENCES
- 1.Boschi-Pinto C, Velebit L, Shibuya K. 2008. Estimating child mortality due to diarrhoea in developing countries. Bull. World Health 86:710–717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Navaneethan U, Giannella RA. 2008. Mechanisms of infectious diarrhea. Nat. Clin. Pract. Gastroenterol. Hepatol. 5:637–647 [DOI] [PubMed] [Google Scholar]
- 3.Pfeiffer ML, DuPont HL, Ochoa TJ. 2012. The patient presenting with acute dysentery—a systematic review. J. Infect. 64:374–386 [DOI] [PubMed] [Google Scholar]
- 4.Pawlowski SW, Warren CA, Guerrant R. 2009. Diagnosis and treatment of acute or persistent diarrhea. Gastroenterology 136:1874–1886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guion CE, Ochoa TJ, Walker CM, Barletta F, Cleary TG. 2008. Detection of diarrheagenic Escherichia coli by use of melting-curve analysis and real-time multiplex PCR. J. Clin. Microbiol. 46:1752–1757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dutta S, Chatterjee A, Dutta P, Rajendran K, Roy S, Pramanik KC, Bhattacharya SK. 2001. Sensitivity and performance characteristics of a direct PCR with stool samples in comparison to conventional techniques for diagnosis of Shigella and enteroinvasive Escherichia coli infection in children with acute diarrhoea in Calcutta, India. J. Med. Microbiol. 50:667–674 [DOI] [PubMed] [Google Scholar]
- 7.Gómez-Duarte OG, Bai J, Newel E. 2009. Detection of E. coli, Salmonella spp., Shigella spp., Yersinia enterocolitica, Vibrio cholerae, and Campylobacter spp. enteropathogens by 3-reaction multiplex polymerase chain reaction. Diagn. Microbiol. Infect. Dis. 63:1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hien BT, Scheutz F, Cam PD, Serichantalergs O, Huong TT, Thu TM, Dalsgaard A. 2008. Diarrheagenic Escherichia coli and Shigella strains isolated from children in a hospital case-control study in Hanoi, Vietnam. J. Clin. Microbiol. 46:996–1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Islam MS, Hossain MS, Hasan MK, Rahman MM, Fuchs G, Mahalanabis D, Baqui AH, Albert MJ. 1998. Detection of Shigellae from stools of dysentery patients by culture and polymerase chain reaction techniques. J. Diarrhoeal Dis. Res. 16:248–251 [PubMed] [Google Scholar]
- 10.Logan JMJ, Edwards KJ, Saunders NA, Stanley J. 2001. Rapid identification of Campylobacter spp. by melting peak analysis of biprobes in real-time PCR. J. Clin. Microbiol. 39:2227–2232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pusterla N, Byrne BA, Hodzic E, Mapes S, Jang SS, Magdesian KG. 2010. Use of quantitative real time PCR for the detection of Salmonella spp. in fecal samples from horses at a veterinary teaching hospital. Vet. J. 186:252–255 [DOI] [PubMed] [Google Scholar]
- 12.Rinttilä T, Kassinen A, Malinen E, Krogius L, Palva A. 2004. Development of an extensive set of 16S rDNA-targeted primers for quantification of pathogenic and indigenous bacteria in faecal samples by real-time PCR. J. Appl. Microbiol. 97:1166–1177 [DOI] [PubMed] [Google Scholar]
- 13.Persson S, Olsen KE. 2005. Multiplex PCR for identification of Campylobacter coli and Campylobacter jejuni from pure cultures and directly on stool samples. J. Med. Microbiol. 54(Pt 11):1043–1047 [DOI] [PubMed] [Google Scholar]
- 14.Parr JB, Sevilleja JE, Samie A, Alcantara C, Stroup SE, Kohli A, Fayer R, Lima AA, Houpt ER, Guerrant RL. 2007. Detection and quantification of Cryptosporidium in HCT-8 cells and human fecal specimens using real-time polymerase chain reaction. Am. J. Trop. Med. Hyg. 76:938–942 [PMC free article] [PubMed] [Google Scholar]
- 15.Barletta F, Ochoa TJ, Mercado E, Ruiz J, Ecker L, Lopez G, Mispireta M, Gil AI, Lanata CF, Cleary TG. 2011. Quantitative real-time polymerase chain reaction for enteropathogenic Escherichia coli: a tool for investigation of asymptomatic versus symptomatic infections. Clin. Infect. Dis. 53:1223–1229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bellin T, Pulz M, Matusset A, Hempen HG, Gunzer F. 2001. Rapid detection of enterohemorrhagic Escherichia coli by real-time PCR with fluorescent hybridization probes. J. Clin. Microbiol. 36:370–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang J, Chuang K, Ahluwalia1 Pate MS, Umblas N, Mirel D, Higuchi R, Germer S. 2005. High-throughput SNP genotyping by single-tube PCR with Tm-shift primers. Biotechniques 39:885–893 [DOI] [PubMed] [Google Scholar]
- 18.Mokhtari W, Nsaibia S, Gharbi A, Aouni M. 2013. Real-time PCR using SYBR green for the detection of Shigella spp. in food and stool samples. Mol. Cell. Probes 27:53–59 [DOI] [PubMed] [Google Scholar]
- 19.Sommer D, Enderlein D, Antakli A, Schönenbrücher H, Slaghuis J, Redmann T, Lierz M. 2012. Salmonella detection in poultry samples. Comparison of two commercial real-time PCR systems with culture methods for the detection of Salmonella spp. in environmental and fecal samples of poultry. Tierarztl. Prax. Ausg. G Grosstiere Nutztiere 40:383–389 [PubMed] [Google Scholar]
- 20.Wiemer D, Loderstaedt U, von Wulffen H, Priesnitz S, Fischer M, Tannich E, Hagen RM. 2011. Real-time multiplex PCR for simultaneous detection of Campylobacter jejuni, Salmonella, Shigella and Yersinia species in fecal samples. Int. J. Med. Microbiol. 301:577–584 [DOI] [PubMed] [Google Scholar]
- 21.Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M, Mueller R, Nolan T, Pfaffl MW, Shipley GL, Vandesompele J, Wittwer CT. 2009. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin. Chem. 55:611–622 [DOI] [PubMed] [Google Scholar]
- 22.Lee SH, Jung BY, Rayamahji N, Lee HS, Jeon WJ, Choi KS, Kweon CH, Yoo HS. 2009. A multiplex real-time PCR for differential detection and quantification of Salmonella spp., Salmonella enterica serovar Typhimurium and Enteritidis in meats. J. Vet. Sci. 10:43–51 [DOI] [PMC free article] [PubMed] [Google Scholar]